


Abstract
The expression of almost all genes in animals is subject to post-transcriptional regulation by RNA binding proteins (RBPs) and microRNAs (miRNAs). The interactions between both RBPs and miRNAs with mRNA can be mapped on a whole-transcriptome level using experimental and computational techniques established in the past years. The combined action of RBPs and miRNAs is thought to form a post-transcriptional regulatory code. Here we present doRiNA 2.0, available at http://dorina.mdc-berlin.de. In this highly improved new version, we have completely reworked the user interface and expanded the database to improve the usability of the website. Taking into account user feedback over the past years, the input forms for both the simple and the combinatorial search function have been streamlined and combined into a single web page that will also display the search results. Especially, custom uploads is one of the key new features in doRiNA 2.0. To enable the inclusion of doRiNA into third-party analysis pipelines, all operations are accessible via a REST API. Alternatively, local installations can be queried using a Python API. Both the web application and the APIs are available under an OSI-approved Open Source license that allows research and commercial access and re-use.
INTRODUCTION
The complex life of any given RNA is largely determined through its interaction with other RNAs and RNA-binding proteins (RBPs). Every step of RNA maturation, turnover and localization is dependent or controlled through these interactions. Efficient experimental and computational methods have been proposed to characterize the RNA interactome on a global scale (1). The biological function of RNA molecules cannot be separated from their ability to bind to and interact with a wide space of chemical species, including small nucleic acids, and proteins. Local interaction sites are characterized by sequence and structure motifs, which define the binding conformation of the respective target RNA (2). Several concepts of post-transcriptional regulation exist in parallel and are thought to form the post-transcriptional regulatory code.
To date, the RNA interactome network is far from being understood, but with the help of data mining resources, progress can be made on deciphering this post-transcriptional regulatory code.
With this in mind, we have built the database of RNA interactions (doRiNA) to collect and integrate all available data on miRNA and RBP target sites from the public domain (3). Since the first appearance of doRiNA in 2012, new assays and algorithms have been developed to infer target sites of these post-transcriptional regulators. While cross-linking and immunoprecipitation (CLIP) techniques have become standard in mapping out RNA–protein interactions, little progress has been made on computational approaches for protein–RNA target prediction (4,5). Especially, HITS-CLIP (6), PAR-CLIP (7) and iCLIP (8) methods have become more established as witnessed by a growing number of publications from different research labs. More than 400 CLIP datasets have been deposited in GEO (9) alone at the time of writing. Moreover, novel approaches for direct mapping of miRNA–RNA interactions have recently emerged (crosslinking, ligation and sequencing of hybrids–CLASH). The CLASH method establishes inter-molecular ligations between a target (e.g. mRNA) and its regulator (e.g. miRNA). Helwak et al. (10) have pioneered this concept for human cells and Grosswendt et al. (11) report on a comprehensive analysis of old and new datasets where they discovered that RNA chimera of miRNAs and their respective target molecules shape spontaneously through an endogenous RNA ligase activity in cellular extracts.
Equally important, miRNA and RNA interactions are more accessible to computational predictions as a well-defined set of base pairing rules explains most of the known interactions accurately and generalizes well to new instances. The two leading programs, TargetScan (12) and PicTar (13) have seen several rounds of performance improvements and are considered as the gold standard in the field as reflected by thousands of citations (ISI Web of Science).
We were motivated by these recent developments to implement a new doRiNA framework using state-of-the-art web technology and including aforementioned miRNA targets and more RBP sites from the literature. We were especially interested in improving the user experience and to ensure that new datasets become immediately available in this rapidly expanding field of RNA biology.
To this end, we have built up an entirely new code base for doRiNA 2.0 that takes advantage of asynchronous client server communication, client side computing and a novel web front-end. Moreover, we have moved the backbone away from the classic CGI and MySQL setup and use a fast key-value cache and store (redis.io) instead as well as in-memory caching of frequent queries. In the later sections, we will present details on how frequent updates do not compromise speed and availability anymore. Additionally, custom data uploads has been a long requested feature that is now available in doRiNA 2.0. In summary, we report on doRiNA 2.0, an open source solution to data mining in post-transcriptional RNA biology, which is accessible through a modern web interface, a REST-like API and a Python API for local programming.
MATERIALS AND METHODS
The doRiNA 2 architecture
The doRiNA architecture was adapted around a core analysis library layer that can be included into third-party tools (see Figure 1). Access to the doRiNA 2 is provided through a user-friendly web interface, a REST API and a locally installable Python API, which all use the same web service. The doRiNA web server application utilizes this web service and processes the output for display in any modern web browser. Genome and regulator data are stored in standard file formats inside an on-disk directory structure to ease maintainability of curated datasets. Metadata and analysis results are stored in JSON format, which can be parsed easily in all major programming languages. The web service is implemented through a Python analysis library.
Figure 1.
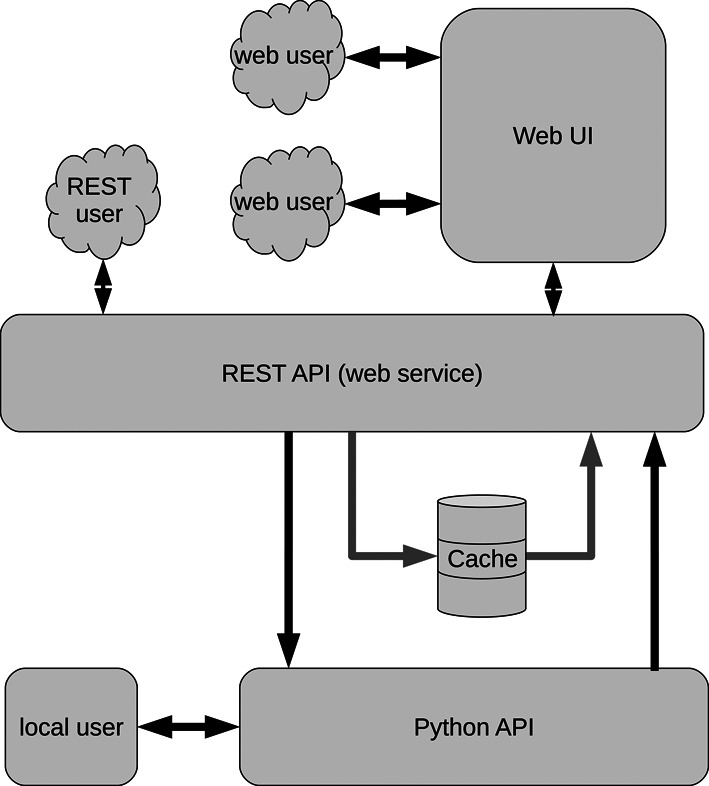
Overview of the modular doRiNA 2.0 architecture.
This analysis library handles all data analysis tasks. As genomic data is stored in GFF3 file format and regulators are stored in BED files, the Python interface (14) of BEDTools (15) is used to calculate the overlaps of the regulators of interest with the selected genome. Results are returned as pybedtools objects. To allow easy exploratory analysis and integration of the analysis library into pipelines not written in Python, a command line interface (CLI) script is provided with the library.
The web server is based on the Python-based Flask micro-framework (http://flask.pocoo.org). The web server provides a REST API that is accessed by the web application running inside the browser. The web application is written in JavaScript, using JQuery (http://jquery.com) and Knockout.js (http://knockoutjs.com) to control the single-page HTML5 user interface in a Model-View-Viewmodel architecture. Web server and web application exchange data asynchronously, passing JSON objects around via AJAX requests. The user interface assists the user in creating the query by providing search and completion options for genomes and regulators. Only control elements relevant to the current query options are made available, reducing the visual clutter of the interface while still allowing the user to fully customize their search. The web server processes incoming requests, scheduling them for analysis by the analysis library via a work-queue system based on Redis Queue (http://python-rq.org). After results are calculated, they are cached in a Redis in-memory database, allowing the web server to quickly return results for popular queries without having to repeat the analysis step. A server-side session handling takes care of removing uploaded regulator files.
RBP target sites
We have added more than 67 new publicly available RBP datasets into doRiNA 2.0. (See supplementary Table I for new datasets). These new datasets include iCLIP, PAR-CLIP and HITS-CLIP experiments. In total, we have 100 RBP datasets for human (hg19), 30 for mouse (mm9) and 6 for Caenorhabditis elegans (ce6). We have chosen to directly use all RBP target sites as published rather than re-analyzing the sequencing experiments to be consistent with the published findings. Each RBP target file is converted to and input in BED format. If the RBP target sites were produced using a different assembly than hg19, mm9 or ce6, the files were lift-over to the respective assembly coordinates before uploading it to the database. Each RBP target site file is also paired with a corresponding JSON file containing metadata for the respective experiment such as method, references and description. Any novel RBP dataset can be deployed in the given directory structure and is immediately available after a cache refresh.
miRNA target sites
For miRNA target sites, we were previously relying on computational predictions only. In this update, we present both computational predictions and new experimental techniques that can identify miRNA and their targets by chimeric sequencing reads. For the computational predictions we use the most recent TargetScan6 and PicTar predictions. For experimental target site identification, we collected data from Hellwak et al. (10) and another RNA-ligation based method from Grosswendt et al. (11). PicTar and TargetScan target predictions are available for all species. Grosswendt et al. targets are available for mouse, human and C. elegans, and CLASH targets are only available for human. All miRNA target site information can be seamlessly integrated with RBP target sites if available.
Integration with the UCSC genome browser
As integration with the UCSC Genome Browser (16) was a popular feature in doRiNA 1, it is also included in doRiNA 2. However, instead of having to maintain a full local copy of the Genome Browser on our own servers, we take advantage of UCSC's TrackHub feature (17). This allows us to provide our doRiNA annotation tracks for joint visualization within the central UCSC Genome Browser instance and minimizes our resource and maintenance requirements.
Custom data upload
DoRiNA 2.0 enables users to upload their own data in the widely accepted BED format. The user decides at the beginning of a doRiNA session if he or she wants to upload a custom BED file. The contained features are displayed as additional regulator (uploaded custom regulator) on the web interface (see http://dorina.mdc-berlin.de/tutorials#custom-regulator) and can be used as any other datasets for search operations in doRiNA2.
Interfaces and APIs for access by third-party tools
Third-party tools may interact with doRiNA 2.0 on multiple layers. As shown in Figure 1, local users can utilize the Python API to run doRiNA queries on their own data, without needing to send any data over the network. The dorina core library, which is built on top of the pybedtools library (14), provides both the core analysis functions as well as a number of supporting functions to take care of data handling. The dorina library also provides a command line tool to quickly explore regulator interactions without having to create a custom script or using the web interface. The exact API is documented on the website (http://dorina.mdc-berlin.de/help#python-api).
In addition, the curated datasets from the public website along with the computational resources provided there can be used via an HTTP-based representational state transfer (REST) interface. This REST interface (http://dorina.mdc-berlin.de/docs#rest-api) has access to doRiNA on the same level as the web user interface. Moreover, clients of the REST API benefit from the additional features provided by the doRiNA 2 servers. These features include an in-memory result cache, drastically reducing the response times for popular queries.
RESULTS
Example applications using doRiNA2.0 web interface
The web interface of doRiNA2.0 can be used to query RBP and miRNA target sites with ease. Both RBPs and miRNAs are considered as regulators, and their targets are stored under that name convention in doRiNA2.0. Users first choose a genome assembly of interest and are then presented with a list of available regulators. The RBP target sites are classified by their experiment type (e.g. iCLIP or PAR-CLIP). miRNA target sites are classified by their prediction method. We currently offer both computational and experimental miRNA target site predictions: Computational predictions are based on TargetScan and PicTar predictions. Experimental predictions are collected from Helwak et al. (shown as CLASH miRNA:targets) and from Grosswendt et al. (shown as Grosswendt miRNA:targets).
Typically, users will pick their regulators of interest and may further limit the location of the target sites to one of the following: CDS (coding DNA sequence), 5′UTR, 3′UTR, introns or intergenic regions. In addition, users can opt for a complex query and define a second set of regulators (indicated as ‘set B’ in the user interface) that could be integrated with search results from the first set (‘set A’). Multiple set operations are possible for combining targets of regulator set A and regulator set B in the ‘combinatorial search’ pane:
AND: intersection of regulator target sites in set A and set B
OR: union of regulator target sites in set A and set B
XOR: regulator target sites that are found in set A or set B but not in both
NOT: regulator target sites that in set A but not in set B
Each set operation is automatically visualized by a Venn diagram to guide the user in interpreting the set operation. The user can additionally define distance or spacing constraints between target sites. Without a distance constraint, the search space is just confined to a target locus (transcript level in case of ‘any’), target sites may be spaced arbitrarily apart as long as they happen to fall into the same transcript.
Finally, users will get a list of target sites based on their selections of regulators and operations. These target sites can be browsed or downloaded as BED file and can be further visualized in the UCSC browser via our integrated Track Hubs feature.
Regulator centric searches
Mutual exclusive Srsf1 and Srsf2 binding on exons
The serine/arginine-rich proteins (SR proteins) are RNA-binding proteins that are associated with alternative splicing, and recent findings suggest that they are involved in both exon skipping and inclusion events (18). Furthermore, Pandit et al. suggest although Srsf1 and Srsf2 binding sites largely target the same exons, certain exons are regulated by only one of the two factors, such that there is mutually exclusive binding of Srsf1 or Srsf2. Exactly these exons can be easily identified with the help of doRiNA 2.0 (see Figure 2). First of all, we select Mouse mm9 as assembly of interest. Second, we select the Srsf1 HITS-CLIP experiment into ‘set A’ and the Srsf2 experiment into ‘set B’. Moreover, we limit the search region to CDS exons using the ‘Look for binding sites in region:’ pane. Lastly, we combine the two regulator sets using the XOR set operation for mutual exclusive binding. This query yields 44 376 sites, which could be redundant for overlapping transcripts, yet are either bound by Srsf1 (19 754 sites) or Srsf2 (24 622 sites) but not by both (see Figure 3).
Figure 2.
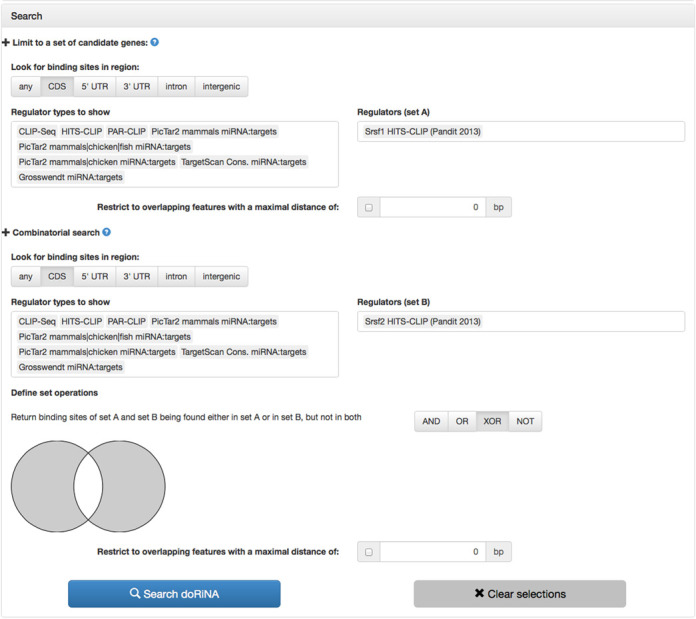
doRiNA 2.0 complex search interface. Search for mutual exclusive binding of Srsf1 and Srsf2 on coding exons without any additional constraints.
Figure 3.
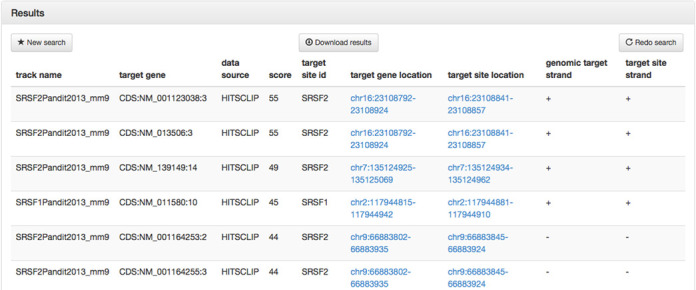
Complex search results for Srsf1 XOR Srsf2 binding.
Local API access
A repetition of the previous web interface example could be accomplished with the local Python API as well and would look like this:

Regulator centric, with custom regulator file
Genic regions that form circular RNAs and overlap with Ago sites in HEK293 cells
Circular RNAs have emerged as a potential regulatory RNA species in recent years (19, 20). The discovery that circRNAs act as ‘sponges’ for miRNAs highlights one of the potentially many regulatory roles of circular RNAs. Circular RNAs are typically generated from primary transcripts that also give rise to linear transcript isoforms. We will demonstrate the utility of our custom BED file upload function with a circular RNA use case. We retrieved the BED file of all detected circles in HEK293 cells from the study of Memczak et al. (21). This dataset may be conveniently obtained using the following URL (http://www.circbase.org/download/hsa_hg19_Memczak2013_HEK293.bed)
Our idea is to test the custom regulator set (circRNA loci) for overlap with Argonaute binding sites, which would be indicative of a putative sponge function. To this end, we composed a search query as shown in Figure 4: For set A, we select all circRNAs that are <100 bp apart from an AGO2 target site as defined by Kishore et al. (22). For set B, we select all circRNAs that are less than 100 bp apart from an AGO1-4 target site as defined by Hafner et al. (7). The union of the two sets is reported as result (OR operation). Evidently some circles overlap with others and so do AGO sites. For a better characterization, we download these results and find that 889 circles are <100 bp apart from 566 PAR-CLIP and 606 CLIP-Seq AGO target sites.
Figure 4.
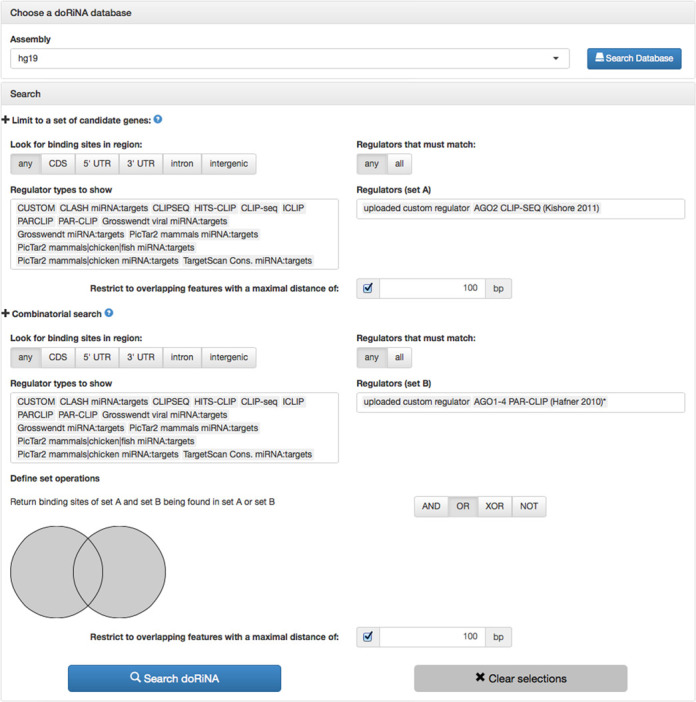
Complex search with custom ‘regulator’ file in BED format. Circular RNA loci in HEK293 cells have been added to the search interface as ‘uploaded custom regulator’. This query looks for overlaps of circular RNA loci and AGO binding sites.
DISCUSSION AND CONCLUSIONS
In the recent years, a large number of high-throughput methods became available to profile RNA–RNA and RNA–protein interactions in post-transcriptional regulation. A major challenge in deriving new biologically meaningful insights from this information is the sheer size of the available data. Ever since its introduction in 2012, doRiNA has assisted biologists in meeting this challenge by providing an easy to use interface to available target site data for both miRNA and RBP regulators. Building on numerous feedbacks from our users, our new release, doRiNA 2.0, adds more data types and sets, supports the integration of private data, updates the user interface to Web 2.0 technology and supports seamless integration into third-party tools that might be part of more complex analyses.
Comparison to related work
The previously published databases AURA (23) and starBase (24) have different scopes. Aura focuses only on untranslated regions yet integrates RBP and miRNA target site information. However, RBPs do not only bind to 5′ and 3′ UTRs. In doRiNA, we try to include all available RBP sites regardless of their position within the gene locus. An important example in this regard is Rbfox2, a regulator of alternative splicing, which preferentially binds to intronic regions (25). From this viewpoint, doRiNA presents itself as a more general resource. Equally important, doRiNA possesses unique query capabilities that can be combined with private binding site data through our dynamic web front end. Complex searches of this kind are not possible with Aura. The response times of doRiNA are considerably faster and the functionality of doRiNA is accessible via REST and a Python API.
StarBase aims to be a comprehensive resource for all RNA interactions that involve proteins and RNAs as regulators. Moreover, StarBase tries to broaden the search space of possible interaction targets by including transcripts from pseudogenes, circular RNAs and lncRNAs. While we are using the most up-to-date gene annotation, we take up a more conservative view on the regulator side as we only consider published data ‘as is’. We do not perform any reanalysis and leverage the work of primary authors by integrating their data with other datasets within doRiNA. Hence, doRiNA2 results may only partially overlap with StarBase.
Clearly, it is not possible to perform combinatorial and distant-constraint queries in StarBase. Secondly, REST or Python APIs are also not available to end-users.
In summary, doRiNA 2 improves the previously available version in several important aspects. The curated set of regulators was extended significantly, adding more than 60 RBP datasets, TargetScan miRNA sites and novel miRNA target site predictions from RNA ligation-based methods. The user interface was reworked to make the large number of datasets more accessible, while also allowing users to provide their own regulator data. Architecture changes to the server side both allow for easier addition of newly curated datasets and provide an API that can be used to better integrate with doRiNA from third-party software.
AVAILABILITY
The doRiNA database is available at http://dorina.mdc-berlin.de under an OSI-approved Open Source license. There are no access restrictions for academic or commercial use. Source code of the analysis library and the web server are available at https://github.com/dieterich-lab/dorina. We kindly ask researchers to cite the doRiNA manuscript if they include results in their publications. The users can also get help by visiting the user forum https://groups.google.com/d/forum/dorina_discussion or sending e-mails directly to dorina_discussion@googlegroups.com. Please contact the corresponding authors if you want to see your dataset included.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
First of all, we would like to thank Dan Muntaneau for his support in setting up the web service for doRiNA 2.0. We are very grateful to all primary authors of data tracks who assisted us in collecting the relevant target site information.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Max Planck Society [to K.B. and C.D.]. Helmholtz Association[to R.W., M,L. , N.R. and A.A.]. Funding for open access charge: Berlin Institute for Medical Systems Biology Max Delbrück Centre Berlin-Buch; Helmholtz Association.
Conflict of interest statement. None declared.
REFERENCES
- 1.Dieterich C., Stadler P.F. Computational biology of RNA interactions. Wiley Interdiscip. Rev. RNA. 2013;4:107–120. doi: 10.1002/wrna.1147. [DOI] [PubMed] [Google Scholar]
- 2.Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X., Gueroussov S., Albu M., Zheng H., Yang A., et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anders G., Mackowiak S.D., Jens M., Maaskola J., Kuntzagk A., Rajewsky N., Landthaler M., Dieterich C. doRiNA: a database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2012;40:D180–D186. doi: 10.1093/nar/gkr1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agostini F., Zanzoni A., Klus P., Marchese D., Cirillo D., Tartaglia G.G. catRAPID omics: a web server for large-scale prediction of protein-RNA interactions. Bioinformatics. 2013;29:2928–2930. doi: 10.1093/bioinformatics/btt495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paz I., Kosti I., Ares M., Jr, Cline M., Mandel-Gutfreund Y. RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014;42:W361–W367. doi: 10.1093/nar/gku406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darnell R.B. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA. 2010;1:266–286. doi: 10.1002/wrna.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jr., Jungkamp A.C., Munschauer M., et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konig J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M., et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Helwak A., Kudla G., Dudnakova T., Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grosswendt S., Filipchyk A., Manzano M., Klironomos F., Schilling M., Herzog M., Gottwein E., Rajewsky N. Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol. Cell. 2014;54:1042–1054. doi: 10.1016/j.molcel.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedman R.C., Farh K.K., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lall S., Grun D., Krek A., Chen K., Wang Y.L., Dewey C.N., Sood P., Colombo T., Bray N., Macmenamin P., et al. A genome-wide map of conserved microRNA targets in C. elegans. Curr. Biol. 2006;16:460–471. doi: 10.1016/j.cub.2006.01.050. [DOI] [PubMed] [Google Scholar]
- 14.Dale R.K., Pedersen B.S., Quinlan A.R. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics. 2011;27:3423–3424. doi: 10.1093/bioinformatics/btr539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karolchik D., Barber G.P., Casper J., Clawson H., Cline M.S., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42:D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raney B.J., Dreszer T.R., Barber G.P., Clawson H., Fujita P.A., Wang T., Nguyen N., Paten B., Zweig A.S., Karolchik D., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 2014;30:1003–1005. doi: 10.1093/bioinformatics/btt637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pandit S., Zhou Y., Shiue L., Coutinho-Mansfield G., Li H., Qiu J., Huang J., Yeo G.W., Ares M., Jr., Fu X.D. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell. 2013;50:223–235. doi: 10.1016/j.molcel.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilusz J.E., Sharp P.A. Molecular biology. A circuitous route to noncoding RNA. Science. 2013;340:440–441. doi: 10.1126/science.1238522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansen T.B., Jensen T.I., Clausen B.H., Bramsen J.B., Finsen B., Damgaard C.K., Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 21.Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M., et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 22.Kishore S., Jaskiewicz L., Burger L., Hausser J., Khorshid M., Zavolan M. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat. Methods. 2011;8:559–564. doi: 10.1038/nmeth.1608. [DOI] [PubMed] [Google Scholar]
- 23.Dassi E., Malossini A., Re A., Mazza T., Tebaldi T., Caputi L., Quattrone A. AURA: Atlas of UTR Regulatory Activity. Bioinformatics. 2012;28:142–144. doi: 10.1093/bioinformatics/btr608. [DOI] [PubMed] [Google Scholar]
- 24.Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jangi M., Boutz P.L., Paul P., Sharp P.A. Rbfox2 controls autoregulation in RNA-binding protein networks. Genes Dev. 2014;28:637–651. doi: 10.1101/gad.235770.113. [DOI] [PMC free article] [PubMed] [Google Scholar]