


Abstract
Whole-exome sequencing, which centres on the protein coding regions of disease/cancer associated genes, represents the most cost-effective method to-date for deciphering the association between genetic alterations and diseases. Large-scale whole exome/genome sequencing projects have been launched by various institutions, such as NCI, Broad Institute and TCGA, to provide a comprehensive catalogue of coding variants in diverse tissue samples and cell lines. Further functional and clinical interrogation of these sequence variations must rely on extensive cross-platforms integration of sequencing information and a proteome database that explicitly and comprehensively archives the corresponding mutated peptide sequences. While such data resource is a critical for the mass spectrometry-based proteomic analysis of exomic variants, no database is currently available for the collection of mutant protein sequences that correspond to recent large-scale genomic data. To address this issue and serve as bridge to integrate genomic and proteomics datasets, CMPD (http://cgbc.cgu.edu.tw/cmpd) collected over 2 millions genetic alterations, which not only facilitates the confirmation and examination of potential cancer biomarkers but also provides an invaluable resource for translational medicine research and opportunities to identify mutated proteins encoded by mutated genes.
INTRODUCTION
Cancer is a genetic disease, which arise as a consequence of genomic abnormalities stemming from somatically acquired mutations or inherited gene mutations. Genomic sequence can be altered on difference scales, which include single nucleotide variations (SNVs), small insertions and deletions (INDELs), rearrangements of genome segments and changes in the copy number of DNA fragments. Since specific mutations of cancer-associated genes are generally considered as DNA biomarkers for diagnosis and as molecular markers for therapeutic drug selection in clinical settings, several large-scale sequencing studies have been performed with the aim to further understand cancer genomics. To this end, the NCI Cancer Genome Atlas (TCGA) project has sequenced the genomes of over 10 000 tumour samples in 33 cancer types (https://tcga-data.nci.nih.gov/tcga/) (1). Sequencing data on the NCI-60 cell lines (2) as well as 947 cancer cell lines (3) also provide an extensive catalogue of cancer relevant variants as well as pharmacogenomics correlations between specific variants and anticancer agents. As mutations that alter the protein sequences have the most significant impact on protein stability and functionality, researchers have started to design multilayer experiments encompassing both genomic and proteomic methods in order to better understand the roles of coding variants in tumorigenesis and progression, as well as their clinical implications (4).
Sequence database search is the most widely used method for protein identification in the field of mass spectrometry-based proteomics (5,6). However, search results are directly affected by the specificity and completeness of sequence database—the mass-spectrometry search engine will likely miss the mutated peptides, which are not included in the reference databases. Recently, a mutated peptide database, named XMAn (7), was developed to translate disease- and cancer-related mutations from gene-level into mutated peptide sequences for proteomics search, based on known mutations gathered from widely accepted sources such as UniProt, IARC P53, OMIM and COSMIC (8–11). However, no existing mutated peptide database has been developed for global exploration of gene alterations discovered in large-scale sequencing studies such as those mentioned above, and efforts on mapping these mutations to protein sequences for further proteomics analysis are still very limited. Only two existing databases—Human Protein Mutant Database (HPMD) (12) and Human Cancer Proteome Variation Database (CanProVar) (13)—were designed for collecting mutated protein specific to colon cancer cell lines and single amino acid alternations from human cancer proteome, respectively. However, as the data sources were limited to specific cell lines or cancer types, it is unlikely that these resources archive the full repertoire of protein sequence variations derived from currently known genetic alterations. Moreover, these databases only annotate variant information in the header line without providing the mutated protein FASTA sequences. Therefore, existing peptide mass fingerprint search engines cannot directly assess these information portals, limiting their applicability in identifying mutant proteins derived from genetic variation databases (8,14) and cancer genome/exome sequencing studies (1,15,16).
Cancer Mutant Proteome Database (CMPD) is designed to address this issue, aiming at improving the link between genomic and proteomics mutations. The mutated protein sequence collection was based on the exome or genome sequencing datasets from NCI-60 cell lines, 947 cancer cell lines from Cancer Cell Line Encyclopaedia project, and 5500 more cases from 20 TCGA cancer cohort studies (1,15–17). The identified genetic alterations (SNVs and INDELs) were converted to all possible mutated protein-coding sequences according to sample-specific transcript isoforms. A wide variety of databases, such as dbSNP (14), dbNFSP (18), ClinVar (19), COSMIC (8) and OMIM (10), has been integrated to our annotation database to facilitate compilation and exploration of associations between diseases and mutations and to eliminate any inconsistency in data format between annotation sources. Functional prediction results are pre-compiled from various prediction algorithms for evaluating the potential functional consequence of each non-synonymous mutation and for ranking the candidate list for further experimental validation.
To further enable the usability of CMPD data in mass spectrometry-based identification of genetic mutations at the protein level. FASTA-formatted sequences that comprise all plausible mutated tryptic peptides can be retrieved and appended to any FASTA-formatted protein database, which can then be accessed by search engines such as MASCOT (20) or SEQUEST (21). Considering the existing bias between samples or cell lines resulted from the number of expressed proteins or diverse genetic variation patterns, CMPD also provide custom filters for generating sample-specific FASTA databases to lower the signal-to-noise ratio. Taken together, CMPD can serve as a bridge between different systems biology platforms, facilitating functional interrogation of disease-associated gene variants from both genomic and proteomics mutation data. The applicability of CMPD to identifying mutated peptides was demonstrated by whole-exome sequencing and proteome resources of the COLO 205 cancer cell line.
MATERIALS AND METHODS
Construction of tryptic peptide database
A comprehensive list of gene mutations was extracted from large-scale cancer genomic sequencing projects including NCI-60 (2), CCLE (3), and TCGA (1,15–17). The Variant Call Format (VCF) as well as the Mutation Annotation Format (MAF) files corresponded to these sequencing projects can be downloaded from CellMiner (http://discover.nci.nih.gov/cellminer/), CCLE website (http://www.broadinstitute.org/ccle/home), and the MAF Dashboard (https://confluence.broadinstitute.org/display/GDAC/MAF+Dashboard) of the Broad Institute's Genome Data Analysis Center (GDAC), respectively. Gene information and FASTA-formatted sequence files at both protein and mRNA levels can be downloaded from ENSEMBL (22) and UCSC through BioMart (23) and UCSC Table Browser (24), respectively. Additional information on genes and proteins, such as HGNC gene symbol (25), identifier in external databases such as RefSeq (26) and UniProtKB (11), GO annotations (27) and disease descriptions, and KEGG pathway information (28), is available through the cross-reference functionality provided by BioMart (23). Moreover, ANNOVAR (29) was used to evaluate the overall consequences of SNVs and INDELs on corresponding transcripts. In addition, observed allele frequencies in the 1000 Genomes Project (30) and NHLBI Exome Sequencing Project (31), single nucleotide polymorphisms reported in dbSNP (14), somatic mutations categorized in COSMIC (8), medically important variants collected in ClinVar (19), and functional prediction results of all non-synonymous SNVs from popular algorithms (18,32–35) were compiled into an integrated SQLite database to facilitate variant-based prioritization. Non-synonymous coding variants and small INDELs identified by NCI-60, CCLE, and TCGA studies were introduced into protein sequences to create sequence pool of aberrant proteins. Since the majority of proteomic experiments use trypsin for proteolytic digestion, in silico digestion was applied to the mutant protein sequence pool to obtain theoretical tryptic peptides, allowing two additional ‘K’ or ‘R’ missed cleavages at both ends of the mutation sites.
Architecture of CMPD
CMPD includes two major components: a web interface for querying and retrieval of mutated protein sequences and a SOLite relational database for data storage. The web interface is built with PHP and JQuery (http://jquery.com), which allows users to query and explore the content of database. Dynamic tables and Summary charts are implemented using Google Chart API (https://developers.google.com/chart/) with instant search or filter functionalities.
RESULTS AND DISCUSSION
Database statistics
Figure 1 shows the overview of CMPD. To generate this database, over 2 millions genetic alterations were retrieved from large-scale cancer genomics studies (1–3), which were subsequently annotated by using information from a variety of external databases. To facilitate functional interpretation of these alterations, only nucleotide sequence variants that alter the protein sequences are gathered by CMPD. The current version of CMPD contains 3 379 122 mutated protein sequences (including isoforms) with respect to 1 661 156 non-synonymous coding variants. Descriptions on the data sources and distribution of mutation types are summarized in Table 1. Supplementary Figure S1 is a Venn diagram illustrating the overlapping of protein-altering mutations between CMPD and widely accepted resources such as UniProt (11), COSMIC (8) and IARC TP53 (9). Since the mutation events collected in CMPD were gathered from cancer cell lines and TCGA cancerous samples, a large proportion of COSMIC mutations and all mutation events in IARC TP53 database were covered by CMPD database. As UniProt is dedicated to collect wild-type protein sequences with curated protein information for all species, human mutant proteins listed in UniProt variant database (http://www.uniprot.org/docs/humsavar) are related to diseases or polymorphisms. It is thus not surprising to see that just a few missense point mutations were overlapped with CMPD. Importantly, a large proportion of mutation events including missense, nonsense, and frame-shift mutations are not categorized in COSMIC, indicating that CMPD is equipped for identifying novel cancer biomarkers.
Figure 1.
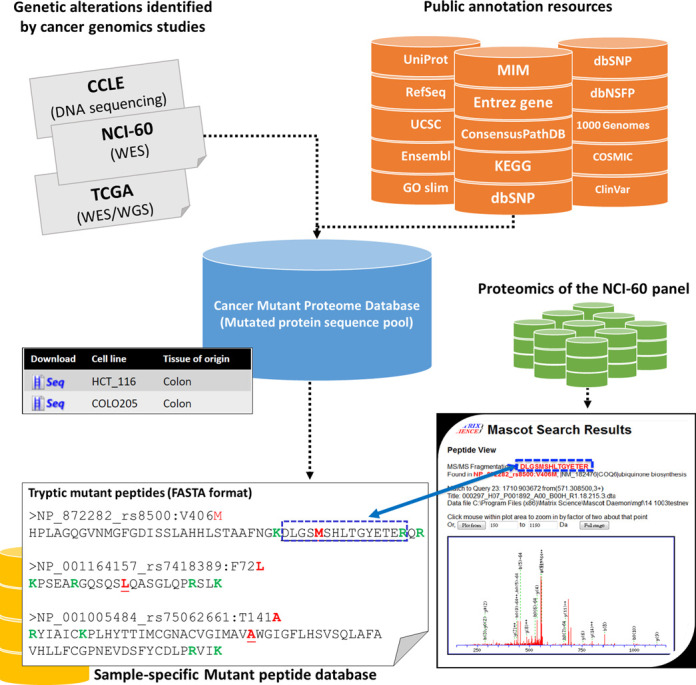
Overview of CMPD. Genetic alterations were gathered from large-scale cancer genomics studies such as NCI-60 WES, CCLE DNA sequencing, and TCGA WES/WGS projects. A wide variety of annotation sources were integrated in CMPD database to facilitate the functional interpretations of these alterations. The coding variants were introduced to protein sequences according the respective transcripts to generate mutant protein sequence collection. Sample-specific tryptic peptides with mutated amino acids can also be generated for proteomic searches.
Table 1. Data sources and database statistics.
| Data sources | NCI-60 panel | CCLE | TCGA |
|---|---|---|---|
| No. of cell lines or no. of samples | 61 cell lines | 947 cell lines | 5625 tumour samples (from 20 tumour types) |
| Variant identification approaches | Whole-exome sequencing | DNA sequencing (1651 cancer-associated genes) | Whole-exome sequencing or whole-genome sequencing |
| No. of coding variants | 63 288 | 64 433 | 1 533 435 |
| No. of SNVs (%) | 61 632 (97.38%) | 59 855 (92.89%) | 725 434 (47.30%) |
| No. of INDELs (%) | 1656 (2.62%) | 4578 (7.11%) | 808 001 (52.70%) |
| Mutant protein sequences deposited in CMPD | 135 286 | 291 662 | 2 952 174 |
| References | (2) | (3) | (1) |
| Links to raw data | Raw data at CellMinera | Raw data at CCLEb | TCGA Mutation Annotation Format (MAF) files downloadable from the MAF Dashboard of the Broad Institute's Genome Data Analysis Centerc |
Web interface
To facilitate the use of the CMPD resource, we have developed an intuitive, user-friendly interface for users to search, browse, prioritize and retrieve subset of mutant protein FASTA sequences. The web interface comprises three major components: (i) search; (ii) browse and (iii) download. As shown in Figure 2, users can search the database based on (a) chromosome names; (b) mutation types; (c) identifiers from HGNC gene symbol, UniProt, UCSC, RefSeq, dbSNP, and COSMIC and (d) keywords on GO terms, pathway or disease descriptions. Such functionality allows users to focus on relevant variants on the basis of their knowledge and interests.
Figure 2.

Major components of CMPD. CMPD comprises three major components: (i) search; (ii) browse and (iii) download. Users can search the database using chromosome names, gene symbols and keywords. In the ‘Browse’ page, mutation events are summarized as pie charts according to various annotation items. Hyperlinks to UCSC Genome Browser are also embedded in the result tables. Sample-specific mutated protein sequences can be obtained from the ‘Download’ pages.
The search results are returned as a human body map, summarizing mutation events according to their tissue types or locations on the human body. Detailed information is linked to dynamic HTML tables, which can be rendered into any custom format by sorting or modifying the displaying columns. The query results can also be rendered as summary pie charts according to features such as mutation types, protein domain, pathway and cytogenetic band. For instance, protein domains or pathways significantly enriched with deleterious mutations can be easily identified through information provided by the summary charts, expediting identification of disease markers and therapeutic targets. Moreover, all the search results can be exported to an Excel or tab-delaminated file for further investigation.
For browse function of CMPD, a summary table of tissue types and their original data sources is provided to users to efficiently access the mutation data of specific tissue, cell line or cancer type. The distribution of mutation events can be rendered as dynamic pie charts according to various annotation items such as gene symbol, mutation type and pathway. CMPD also provide instant filter to drill down mutations by keywords to facilitate target selection and information browsing. Furthermore, sample-specific mutations can be easily obtained by adding or removing sample names in the checkbox list and hyperlinks to UCSC Genome Browser are embedded in the resulting tables to coordinate CMPD mutation data with public resources.
As clinically relevant genomic mutations likely translate into aberrant protein sequences and structures, a sample-specific mutant protein database is a critical component of cancer studies focused on sequence-based biomarkers. Either the full length and tryptic mutant protien sequences of each cell line or cancer type can be downloaded as separated FASTA-formatted files. Protein alterations are properly annotated in the FASTA sequence format and according to functional consequences on each altered transcript. In addition, attributes such as gene symbol and description, RefSeq transcript and protein ID, mutation positions on both mRNA and protein sequences, and amino acids substitutions are included in the header line of each FASTA sequence. Users can use these attributions to obtain more information on the interested proteins. Detailed description on the header content can be found at http://cgbc.cgu.edu.tw/cmpd/help.php.
A real-example on using CMPD
As a proof-of-principle experiment, LC–MS files of the COLO-205 cell proteome downloaded from previous study (http://wzw.tum.de/proteomics/NCI60/) (4) were searched against the mutated tryptic peptide database generated by CMPD using Mascot search engine. The search result identified 10 matching peptides specific for their relevant mutant proteins (Table 2), providing strong evidence that CMPD could be used in linking genetic mutations at the protein level.
Table 2. Genetic mutations identified in protein level.
| No. | Mutated tryptic peptide | Gene symbol | Mutation | Protein accession | Description |
|---|---|---|---|---|---|
| 1 | QFEESQGRTSSK | TG | R2530Q | NP_003226 | Thyroglobulin precursor |
| 2 | DLGSMSHLTGYETER | COQ6 | V406M | NP_872282 | Ubiquinone biosynthesis monooxygenase COQ6 isoform a |
| 3 | ALREMVSNMSGPSGEEEAK | SPERT | K302E | NP_001273270 | Spermatid-associated protein isoform 2 |
| 4 | MTBGDPSVISVNGTDFTFR | ADCK5 | A496T | NP_777582 | Uncharacterized aarF domain-containing protein kinase 5 |
| 5 | GPEGAMGLPGMRGPPGPGCK | COL4A4 | S1403P | NP_000083 | Collagen alpha-4(IV) chain precursor |
| 6 | LMARDSTR | SPTBN4 | G1331S | NP_066022 | Spectrin beta chain, non-erythrocytic 4 isoform sigma1 |
| 7 | MNBDLRISCMSKPPAPNPTTPR | MAP2K3 | P11T | NP_002747 | Dual specificity mitogen-activated protein kinase kinase 3 isoform A |
| 8 | TIHSEQAVFDIYYPTEQVTIQVLPPKSAIK | ALCAM | N258S | NP_001230209 | CD166 antigen isoform 2 precursor |
| 9 | WEDQENESVQYGRNMSSMAYSLYLFTR | CTNNAL1 | I593S | NP_001273903 | Alpha-catulin isoform b |
| 10 | MABKVTLTGDTEDEDSASTSNSLKR | SULT1C4 | D5E | NP_006579 | Sulfotransferase 1C4 |
CONCLUSIONS
With the advancements of both next-generation sequencing and proteomics technologies in recent years, considerable efforts have been devoted to connecting transcriptomics and proteomics data for the identification of potential biomarkers for biological studies and cancer therapies. However, inconsistent file formats derived from different omics studies have complicated subsequent comparative studies. Moreover, existing mutant protein databases are constrained by specific cancer types (36) or out-dated data sources (13), and unable to generate sample-specific or customized protein database. To address these research needs, we create a comprehensive mutant proteome database, named CMPD, which incorporates extensive mutation data from many large-scale cancer genomics studies, cross-referenced to various annotation sources to facilitate target selection and information browsing. In brief, CMPD serves as a bridge between genomic data and proteomic studies, providing a fully integrated account of mutations at DNA, RNA and protein levels.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank Prof. Yu-Sun Chang and Dr Shu-Jen Chen for their comments and suggestions for this manuscript.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Ministry of Education, Taiwan [EMRPD1D0841 to P.J.H., EMRPD1D0671 to P.T., EMRPD1D0671 to B.T., EMRPD1D0811 to L.H.]; Ministry of Science and Technology, Taiwan [MOST 103-2632-B-182-001]. Funding for open access charge: Ministry of Science and Technology, Taiwan [MOST 103-2632-B-182-001].
Conflict of interest statement. None declared.
REFERENCES
- 1.Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A., et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abaan O.D., Polley E.C., Davis S.R., Zhu Y.J., Bilke S., Walker R.L., Pineda M., Gindin Y., Jiang Y., Reinhold W.C., et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013;73:4372–4382. doi: 10.1158/0008-5472.CAN-12-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gholami A.M., Hahne H., Wu Z., Auer F.J., Meng C., Wilhelm M., Kuster B. Global proteome analysis of the NCI-60 cell line panel. Cell Rep. 2013;4:609–620. doi: 10.1016/j.celrep.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 5.Fournier F., Joly Beauparlant C., Paradis R., Droit A. rTANDEM, an R/Bioconductor package for MS/MS protein identification. Bioinformatics. 2014;30:2233–2234. doi: 10.1093/bioinformatics/btu178. [DOI] [PubMed] [Google Scholar]
- 6.Tabb D.L., Fernando C.G., Chambers M.C. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007;6:654–661. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang X., Lazar I.M. XMAn: a Homo sapiens mutated-peptide database for the MS analysis of cancerous cell states. J. Proteome Res. 2014 doi: 10.1021/pr5004467. doi:10.1021/pr5004467. [DOI] [PubMed] [Google Scholar]
- 8.Forbes S.A., Bindal N., Bamford S., Cole C. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2010;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petitjean A., Mathe E., Kato S., Ishioka C., Tavtigian S.V., Hainaut P., Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 10.Amberger J., Bocchini C., Hamosh A. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®) Hum. Mutat. 2011;32:564–567. doi: 10.1002/humu.21466. [DOI] [PubMed] [Google Scholar]
- 11.Consortium U. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathivanan S., Ji H., Tauro B.J., Chen Y.S. Identifying mutated proteins secreted by colon cancer cell lines using mass spectrometry. J. Proteomics. 2012;76:141–149. doi: 10.1016/j.jprot.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 13.Li J., Duncan D.T., Zhang B. CanProVar: a human cancer proteome variation database. Hum. Mutat. 2010;31:219–228. doi: 10.1002/humu.21176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherry S.T. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koboldt D.C., Fulton R.S., McLellan M.D., Schmidt H., Kalicki-Veizer J., McMichael J.F., Fulton L.L., Dooling D.J., Ding L., Mardis E.R., et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muzny D.M., Muzny D.M., Bainbridge M.N., Bainbridge M.N., Chang K., Chang K., Dinh H.H., Dinh H.H., Drummond J.A., Drummond J.A., et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X., Jian X., Boerwinkle E. dbNSFP v2. 0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 2013;34:2393–2402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2013;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perkins D.N., Pappin D.J., Creasy D.M., Cottrell J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 21.Eng J.K., McCormack A.L., Yates J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 22.Flicek P., Amode M.R., Barrell D., Beal K. Ensembl 2014. Nucleic Acids Res. 2013;42:D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kasprzyk A. BioMart: driving a paradigm change in biological data management. Database (Oxford) 2011;2011:bar049. doi: 10.1093/database/bar049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mangan M.E., Williams J.M., Kuhn R.M., Lathe W.C. The UCSC genome browser: what every molecular biologist should know. Curr. Protoc. Mol. Biol. 2014;107:19.9.1–19.9.36. doi: 10.1002/0471142727.mb1909s107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray K.A., Daugherty L.C., Gordon S.M., Seal R.L., Wright M.W., Bruford E.A. Genenames.org: the HGNC resources in 2013. Nucleic Acids Res. 2013;41:D545–D552. doi: 10.1093/nar/gks1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M., et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The Gene Ontology Consortium. The Gene Ontology in 2010: extensions and refinements. Nucleic Acids Res. 2009;38:D331–D335. doi: 10.1093/nar/gkp1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanehisa M., Goto S., Sato Y., Furumichi M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.1000 Genomes Project Consortium. Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu W., O'Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J., et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sim N.L., Kumar P., Hu J., Henikoff S., Schneider G., Ng P.C. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–W457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 34.Chun S., Fay J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X., Slebos R.J.C., Wang D., Halvey P.J., Tabb D.L., Liebler D.C., Zhang B. Protein identification using customized protein sequence databases derived from RNA-Seq data. J. Proteome Res. 2012;11:1009–1017. doi: 10.1021/pr200766z. [DOI] [PMC free article] [PubMed] [Google Scholar]