


Abstract
Chimeric RNAs that comprise two or more different transcripts have been identified in many cancers and among the Expressed Sequence Tags (ESTs) isolated from different organisms; they might represent functional proteins and produce different disease phenotypes. The ChiTaRS 2.1 database of chimeric transcripts and RNA-Seq data (http://chitars.bioinfo.cnio.es/) is the second version of the ChiTaRS database and includes improvements in content and functionality. Chimeras from eight organisms have been collated including novel sense–antisense (SAS) chimeras resulting from the slippage of the sense and anti-sense intragenic regions. The new database version collects more than 29 000 chimeric transcripts and indicates the expression and tissue specificity for 333 entries confirmed by RNA-seq reads mapping the chimeric junction sites. User interface allows for rapid and easy analysis of evolutionary conservation of fusions, literature references and experimental data supporting fusions in different organisms. More than 1428 cancer breakpoints have been automatically collected from public databases and manually verified to identify their correct cross-references, genomic sequences and junction sites. As a result, the ChiTaRS 2.1 collection of chimeras from eight organisms and human cancer breakpoints extends our understanding of the evolution of chimeric transcripts in eukaryotes as well as their functional role in carcinogenic processes.
INTRODUCTION
Chimeric RNAs may be produced by the joining of exons from different genes either through a complex splicing process or as the result of chromosome rearrangement (1–23). Thus, two loci on different chromosomes may produce chimeras through a genomic rearrangement event or through trans-splicing (21,24). Additionally, read-through transcription of two adjacent genomic loci may result in chimera synthesis (10,11,25–27). While many chimeras have been shown to be artifacts of the in vitro reverse transcription reaction (28–31), there is sufficient data demonstrating that some chimeras are translated into chimeric proteins (18). Here we establish an extended collection of putative chimeric transcripts whose existence are supported at different levels by experimental data, including tissue specific expression levels of chimeric RNAs and protein products (18,32).
Our ChiTaRS database of ‘Chimeric Transcripts and RNA-Seq data’ is a collection of chimeric transcripts identified by Expressed Sequence Tags (ESTs) and mRNAs from the GenBank (33), ChimerDB (26,34), dbCRID (35), TICdb (36) and other databases for humans, mouse and flies (37). Our pipeline for finding chimeric transcripts is shown on Supplementary Figure S1 (Supplementary Material). Here we present the updated ChiTaRS 2.1 database of more than 29 000 chimeric transcripts in eight organisms; the database incorporates major additions in content and functionality. The ChiTaRS database is currently used to study the identity and incidence of specific fusions of transcripts that may result in a chimeric RNA with novel biological function. In the original ChiTaRS database (32), there was some experimental data included, such as RNA-seq, and mass spectrometry identification of peptides formed by the translation of the chimeric RNA transcripts. In the current version, we extend the experimental data evidence and the organism coverage by chimeras from eight organisms: Homo sapiens, Mus musculus, Drosophila melanogaster, Rattus norvegicus, Bos taurus, Sus scrofa, Danio rerio and Saccharomyces cerevisiae. Furthermore, the new database version includes a novel type of particularly interesting sense–antisense chimeric transcripts, together with their experimental confirmation by the RNA-seq reads.
Cancer fusions resulting from chromosomal translocations, deletions or inversions are well characterized in cancer (38–48). Fusion proteins increase the complexity of the proteome in many types of cancers with the production of novel proteins (18). In other cases they can produce non-coding regulatory RNAs or interfere with other genomic regions (39–43). Although gene fusions can be detected by the RNA-seq technique, for many fusions the correct junction sequences have yet to be determined, and there are many inconsistencies between different databases, including the corresponding annotations in GenBank (33). Therefore, we have initiated a curation effort to collate information on cancer fusions from GenBank (33), UniProt (49), the Mitelman database (47,50) and the Atlas of Genetics and Cytogenetics in Oncology and Haematology (http://atlasgeneticsoncology.org/) and to run our chimeric transcript analytical methodology in order to determine the correct junction sites of these fusions. First, we automatically collected all the fusions from UniProt including their description and corresponding GenBank-ids and then we have verified those entries manually in order to find cancer breakpoints references in GenBank and other database. Next, we run our automatic procedure to identify chimeric junction sites for all the entries using the genomic sequence of the breakpoints. Finally, we produced the manual verification and identification of the junction sites for all 610 breakpoints from the Mitelman collection having the GenBank-id and for all 818 breakpoints without GenBank-id. Thus, ChiTaRS-2.1 incorporates the largest collection of cancer breakpoints and their junction sequences and it includes 1428 (about 800 new) annotated cancer fusions in different types of cancers. We added the corresponding fusion junction sites and the genomic sequences for all the breakpoints (See ‘Breakpoints’ and ‘Downloads’).
In ChiTaRS-2.1, we also collected an additional type of chimeric RNA transcripts, the ‘read-though’ chimera, that begins upstream of gene 1 and ends at the termination site of adjacent gene 2. Such chimeras have been detected in various cancer and normal cells. Read-through chimeric transcripts are not included in other datasets like ChimerDB (26,34), TICdb (36) or dbCRID (35), and are thus unique to ChiTaRS-2.1. To view ‘Read-through’ chimeras we added a check-box on the ‘Full Collection’ page. All the entries in ChiTaRS-2.1 can be accessed from the UniProt Knowledgebase system (UniProtKB) that collates information on individual proteins from laboratories world-wide, including 2870 fusions proteins (and parental proteins) listed in UniProt (51). Chimeric RNAs and proteins have become a powerful tool for researchers over the past few years since they can be used as cancer markers as well as putative targets for the development of new drugs. Thus, the current ChiTaRS-2.1 database represents a basic starting point for identifying cancer fusions, for studying chimeric transcripts, for analyzing New-Generation-Sequencing results and for investigating the biological processes underlying the phenomenon of cancer fusions.
IMPROVEMENTS
Ten updates and improvements to the content and functionality of ChiTaRS are summarized in Table 1. Major improvements include: addition of chimeric transcripts from eight organisms, to the ability to compare and analyze chimeras from different organisms, links to PubMed references by means of an iHop online text-mining routine and a new category of chimeric transcripts: the sense–antisense chimeras.
Table 1. Major improvements as provided in the ChiTaRS-2.1 database.
| Features | ChiTaRS version 1.0 | ChiTaRS version 2.1 |
|---|---|---|
| Species | 3 species | 8 species |
| Number of chimeric transcripts | 16 261 | 29 164 |
| Chimeras validated by more than two RNA-seq reads spanning the junction site | 175 | 337 |
| Cancer breakpoints | 1286 | 1428 |
| Manually verified breakpoints | 456 | 1428 |
| UniProt cross-references | NA | 2229 |
| Sense–antisense chimeras | NA | 6044 |
| iHop cross-links | NA | 48 586 |
| Comparison and analysis of species | Not Available | Available |
| SpliceGraphs | 8000 | 8232 |
Updated database content
In the 2014 update, 29 164 chimeras and 1428 cancer breakpoints have been collected from eight organisms. The number of chimeras identified in each species is presented in Table 2. For all the 1428 cancer breakpoints produced by 1090 human genes, we have performed manual confirmation of their veracity using sequence information and experimental data from 6941 articles. In addition, 333 chimeric transcripts and their junction sites were confirmed by in-house RNA-seq including our previous results (19). Finally, four chimeric transcripts for the ATP1A1 gene, three from human and one from mouse, were extensively verified by means of RT-qPCR, PCR, cloning and sequencing procedures, in order to confirm their expression levels in six tissue samples from two organisms (human and mouse) (Supplementary Figure S2, Supplementary Material). Therefore, the ChiTaRS 2014 update includes experimental support for 337 transcripts, 1.9× more than in the original ChiTaRS database, which had support for 175 chimeras (Table 1).
Table 2. SAS chimeras identified in different organisms.
| Species | H. sapiens | M. musculus | D. melanogaster | R. norvegicus | B. taurus | D. rerio | S. cerevisiae | S. scrofa |
|---|---|---|---|---|---|---|---|---|
| Number of chimeric transcripts | 20 740 | 6224 | 2151 | 8 | 4 | 4 | 5 | 14 |
| Sense–antisense chimeras | 3998 | 1713 | 323 | 1 | 0 | 0 | 2 | 7 |
We identified chimeric transcripts from the GenBank (33) collection of ESTs and mRNAs for H. sapiens (UCSC reference genome: GRCh37/hg19), M. musculus (NCBI37/mm9) and D. melanogaster (BDGP R5/dm3) R. norvegicus (RGSC Rnor_6.0/rn6), B. taurus (Baylor College of Medicine HGSC Btau_4.6.1/bosTau7), D. rerio (Sanger Institute Zv9/danRer7), S. cerevisiae (SGD April 2011 sequence/sacCer3) and Sus Scrofa (Broad/Pig3). The ESTs and mRNA sequences were mapped to their corresponding reference genomic sequences using the UCSC BLAT program (52). We included a chimera if the first and the second sequence tracts of the chimera had a minimum identity of 95%, a minimum length of 50 nt, and if these two tracts could not be mapped linearly to the reference genome.
In ChiTaRS-2.1, we have added an analysis and comparison of the junction sites, rank and consistency between different chimeric transcripts (18) in all eight studied organisms. This new feature provides users the ability to study the evolution of chimeric transcripts and conservation of the junction sites for any chimera, including the 2337 chimeras conserved between human and mouse. A new improved interface allows users to ‘Compare and Analyze’ chimeras from different organisms (see a link at the Top Menu of the database webpage: http://chitars.bioinfo.cnio.es/). To illustrate the power of this new utility, we applied it to identify a putative chimera composed of RAD9A (RAD9A homolog A) and PPP1CA (protein phosphatase 1), present in both human and mouse ESTs (Figure 1A and B). In human, this chimera is encoded by the same strand as a read-through of the RAD9A and PPP1CA genes (Figure 1A). However, the transcript in mouse may be considered as sense–antisense (‘SAS’) chimera (see below), since the two genes incorporated in the chimeras are encoded by the opposite strands of the overlapping genes (Figure 1B). ChiTaRS-2.1 has the ‘Junction Search’ feature that may be applied for the junction sites analysis of all eight organisms using the alignment and the E-value found by the FASTA program (53). To conclude, our database provides unexplored datasets of evolutionarily conserved chimeric transcripts in eukaryotes and enables the study of their functional role in cellular processes.
Figure 1.
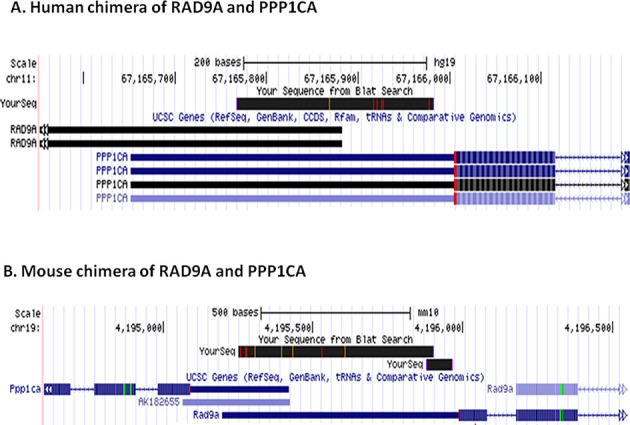
A putative chimera composed of RAD9A (RAD9A homolog A) and PPP1CA (protein phosphatase 1). (A) A chimera found among human ESTs. (B) A mouse chimera.
Sense-antisense chimeras
We identified a new class of fusion produced by the conjoining of exons from two different strands of the same open reading frame. We called this new type of chimera ‘SAS’ chimeras. These chimeras produce fusion transcripts incorporating both coding and non-coding exons of the same gene and are typically found in different types of cancers but also in normal cells. Novel SAS chimeras that have been found in any of the eight organisms in ChiTaRS-2.1 can be easily accessed by clicking a check-box (‘Sense-ANTIsense transcripts’) on the ‘Full Collection’ page. More than 6000 of chimeric RNA transcripts in humans that incorporate sense and antisense exons of the same open reading frame have been incorporated into ChiTaRS-2.1 (Table 2). Interestingly, junction sites of SAS chimeras have been found to incorporate palindromic sequences, and might be produced by exon–exon slippage during the transcription process (Figure 2). Thus, the palindromic motifs have been found in more than 60% of junction sites for human (Figure 2A), mouse (Figure 2B) and fly (Figure 2C) chimeras.
Figure 2.

The most frequent junction motifs of SAS chimeras are incorporate palindromic sequences. (A) Two palindromic motifs found for human SAS chimeras. (B) Motifs of the mouse SAS chimeras. (C) Motifs of the fly SAS chimeras.
We hypothesize that SAS chimeric transcripts may function as antisense transcripts that inhibit the expression of one (or both) of the parent genes. Evidence for such an antisense role of chimeric transcripts in genomic translocation is typified by two studies of the TEL/ETV6 gene (54). A chromosomal translocation in a myelodysplastic syndrome (MDS) patient, fusing the sense strand of the TEL/ETV6 gene on 12p13 to the antisense strand of Thousand-And-One amino acid protein Kinase 1 (TAOK1) gene on 17q11, results in a chimeric transcript that acts as an antisense RNA on wild-type TAOK1 mRNA. This antisense is likely to be clinically relevant, since down regulation of WT-TAOK1 protein expression is associated with weaken patient response to chemotherapy (54). A second report showed that translocation of t(12;17)(p13;p12-p13) in secondary acute myeloid leukemia (AML) results in fusion of TEL/ETV6 and the antisense strand of PER1. Expression of the chimeric transcript containing antisense sequences to PER1 was confirmed in this case; it reduced the expression level of the WT-PER1 protein and affected the overall response of a patient to the chemotherapy drugs (55). Therefore, the SAS chimeras in ChiTaRS-2.1 is a unique collection that allows to study the effect of antisense transcripts in cancers. In ChiTaRS-2.1, there are 69 SAS chimeras confirmed by RNA-seq reads spanning the junction sites (see ‘Full Collection’).
New RNA-seq evidence for the expression of chimeras
To establish the veracity of all the chimeric transcripts in ChiTaRS-2.1, we produced RNA-seq libraries of three human cancer cell lines: MCF7 (breast cancer), LNCAP (prostate cancer), VCAP (prostate cancer) and one fly cell line MBN (timorous blood Drosophila cell line). The datasets have 85 million (M) paired-end reads of 50 nt per sample. The reads mapping to the template chimeras was carried out following the previously described procedure (18). For the MCF7, LNCAP, VCAP and MBN cell lines, we required at least five RNA-seq reads covering the chimeric junction site with only a maximum of two mismatches allowed (Table 2). This requirement is more restricted than one used in our previous studies (18) in order to decrease a number of artifacts. As a result, we confirmed the presence of 333 chimeras: 297 in human, 8 in mouse, 28 in fly (see ‘Full Collection’). These 297 chimeras include 175 previously reported cases, 89 new ones expressed in MCF7, VCAP and LNCAP, and 69 SAS chimeras confirmed by RNA-seq reads. Interestingly, an inter-chromosomal fusion, NDUFAF2-MAST4, in VCAP, identified previously by ChimeraScan (56), was identified in our sample, since we detected five junction-spanning paired-end reads for this chimera. Such examples in the database demonstrate that our methodology is sufficiently sensitive for the analysis of the expression of putative chimeras. We analyzed all the chimeras expressed in MCF7 (118 transcripts), finding that they include known cancer breakpoints, sense–antisense chimeric transcripts and read-through chimeras from our new database ChiTaRS-2.1 (see ‘Full Collection’). The chimeras are generally highly expressed in comparison to a normal breast tissue (Supplementary Material, Supplementary Figure S2, in reads assigned per kilobase of target per million mapped reads (RPKM), P < 0.05). As such, the new version of ChiTaRS contains the highest number of chimeric transcripts known today and the largest collection of experimental evidences for the expression of chimeras. All the datasets in ChiTaRS-2.1 can be retrieved from ‘Downloads’.
Functionality improvements
To improve the data access and analyses of the information on chimeric transcripts contained in ChiTaRS, a new interface with enhanced query capacity and support information have been added (Figure 3). Every ChiTaRS-2.1 entry is associated to a genomic position in the UCSC browser, which appears in a new pop-up window and includes downloadable files incorporating all the transcription start/stop sites, the genomic, chromosomal and strand location (Figure 3). Publications associated with each of the two genes in every chimera can be easily accessed using an automated PubMed search, and all the retrieved references can be downloaded using the ‘Save Text’ option (See ‘Full Collection’ and Figure 3). To improve the visual association of chimeric transcripts with gene function, we have added a link to the iHOP family (57–60) of web services (www.ihop-net.org/) for every gene in the ChiTaRS 2.1 database. The iHOP, Information Hyperlinked over Proteins (57), engine provides information on gene function, potential gene–gene relation in networks of genes, as an intuitive way of screening the millions of abstracts in PubMed for relevant publications (Figure 3). This improvement provides users with an easy means of exploring and combining information for each parental gene of a chimera.
Figure 3.
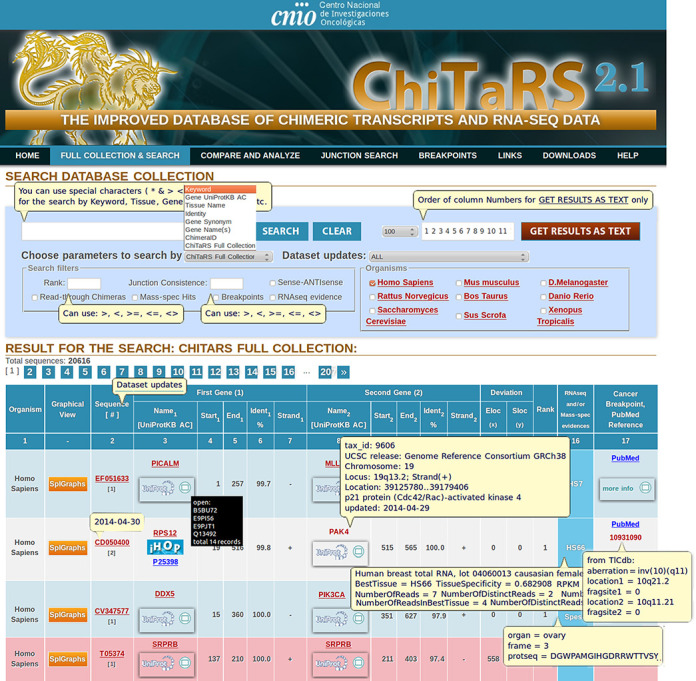
A new interface with enhanced query capacity and support information has been added to the ChiTaRS-2.1 database.
CONCLUSIONS AND PERSPECTIVES
The current update of the ChiTaRS-2.1 database represents a 1.9-fold increase of chimeric transcripts as compared to the initial ChiTaRS release, and includes a significant extension of specific research-oriented features. ChiTaRS-2.1 provides extensive experimental evidence for chimeras and cancer fusions, and this information can be considered instrumental for planning new experiments or for the analysis of large scale RNAseq experiments. The database will be updated every six months to include the growing number of chimeras published. International projects like ICGC and TCGA will benefit from this database and on all incremental additions to the database, for improving the process of chimera identification and validation in cancer research. To conclude, the ChiTaRS-2.1 database is designed to advance the field of Cancer Research as well as our understanding of the phenomenon of chimeric transcripts and its evolution in eukaryotes.
AVAILABILITY
The ChiTaRS-2.1 content will be continuously maintained and updated every six months. The database is now publicly accessible at http://chitars.bioinfo.cnio.es/ and the old version of the database is accessible at http://chitars-old.bioinfo.cnio.es/.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors would like to thank the following contributors whose work makes the ChiTaRS-2.1 possible: MPLabs LTD for the website design and its support, Kovid BioAnalytics LTD for the manual verification of all the cancer breakpoints in ChiTaRS-2.1, the ACGT Inc. for the RT-qPCR, cloning and sequencing experiments, the Genomics Unit at CNIO for RNA extractions, library preparation and the RNA sequencing results. We thank our users for their consistent support and valuable feedback and our outstanding group for their priceless discussions and suggestions.
Footnotes
Present addresses:
Alessandro Gorohovski, National Technical University of Ukraine (KPI), Kiev 03056, Ukraine.
Dunja Vucenovic, Department of Molecular biology, Faculty of Science, University of Zagreb, Zagreb, Croatia.
FUNDING
Miguel Servet (FIS: CP11/00294) [to M.F.-M. for staff scientists]. Funding for open access charge: NHGRI-NIH ENCODE [HG00455-04]; Blueprint European Union project [282510]; Spanish Government [BIO2007-66855]; Spanish National Bioinformatics Institute (INB-ISCIII), Genecode/ENCODE NHGRI-NIH [HG00455-04].
Conflict of interest statement. None declared.
REFERENCES
- 1.Birney E., Stamatoyannopoulos J.A., Dutta A., Guigó R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guigó R., Flicek P., Abril J.F., Reymond A., Lagarde J., Denoeud F., Antonarakis S., Ashburner M., Bajic V.B., Birney E., et al. EGASP: the human ENCODE Genome Annotation Assessment Project. Genome Biol. 2006;7(Suppl. 1):S1–S31. doi: 10.1186/gb-2006-7-s1-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffin T.J., Gygi S.P., Ideker T., Rist B., Eng J., Hood L., Aebersold R. Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol. Cell. Proteomics. 2002;1:323–333. doi: 10.1074/mcp.m200001-mcp200. [DOI] [PubMed] [Google Scholar]
- 5.Velculescu V.E., Zhang L., Zhou W., Vogelstein J., Basrai M.A., Bassett D.E., Hieter P., Vogelstein B., Kinzler K.W. Characterization of the yeast transcriptome. Cell. 1997;88:243–251. doi: 10.1016/s0092-8674(00)81845-0. [DOI] [PubMed] [Google Scholar]
- 6.Cirulli E.T., Singh A., Shianna K.V., Ge D., Smith J.P., Maia J.M., Heinzen E.L., Goedert J.J., Goldstein D.B., (CHAVI), C.f.H.A.V.I. Screening the human exome: a comparison of whole genome and whole transcriptome sequencing. Genome Biol. 2010;11:R57. doi: 10.1186/gb-2010-11-5-r57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finta C., Zaphiropoulos P.G. Intergenic mRNA molecules resulting from trans-splicing. J. Biol. Chem. 2002;277:5882–5890. doi: 10.1074/jbc.M109175200. [DOI] [PubMed] [Google Scholar]
- 8.Kapranov P., Drenkow J., Cheng J., Long J., Helt G., Dike S., Gingeras T.R. Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. 2005;15:987–997. doi: 10.1101/gr.3455305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Segni G., Gastaldi S., Tocchini-Valentini G.P. Cis- and trans-splicing of mRNAs mediated by tRNA sequences in eukaryotic cells. Proc. Natl. Acad. Sci. U.S.A. 2008;105:6864–6869. doi: 10.1073/pnas.0800420105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akiva P., Toporik A., Edelheit S., Peretz Y., Diber A., Shemesh R., Novik A., Sorek R. Transcription-mediated gene fusion in the human genome. Genome Res. 2006;16:30–36. doi: 10.1101/gr.4137606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parra G., Reymond A., Dabbouseh N., Dermitzakis E.T., Castelo R., Thomson T.M., Antonarakis S.E., Guigó R. Tandem chimerism as a means to increase protein complexity in the human genome. Genome Res. 2006;16:37–44. doi: 10.1101/gr.4145906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romani A., Guerra E., Trerotola M., Alberti S. Detection and analysis of spliced chimeric mRNAs in sequence databanks. Nucleic Acids Res. 2003;31:e17. doi: 10.1093/nar/gng017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell P.J., Stephens P.J., Pleasance E.D., O'Meara S., Li H., Santarius T., Stebbings L.A., Leroy C., Edkins S., Hardy C., et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ortiz de Mendíbil I., Vizmanos J.L., Novo F.J. Signatures of selection in fusion transcripts resulting from chromosomal translocations in human cancer. PLoS One. 2009;4:e4805. doi: 10.1371/journal.pone.0004805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H., Wang J., Mor G., Sklar J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science. 2008;321:1357–1361. doi: 10.1126/science.1156725. [DOI] [PubMed] [Google Scholar]
- 16.Li H., Wang J., Ma X., Sklar J. Gene fusions and RNA trans-splicing in normal and neoplastic human cells. Cell Cycle. 2009;8:218–222. doi: 10.4161/cc.8.2.7358. [DOI] [PubMed] [Google Scholar]
- 17.Edgren H., Murumagi A., Kangaspeska S., Nicorici D., Hongisto V., Kleivi K., Rye I.H., Nyberg S., Wolf M., Borresen-Dale A.L., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011;12:R6. doi: 10.1186/gb-2011-12-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frenkel-Morgenstern M., Lacroix V., Ezkurdia I., Levin Y., Gabashvili A., Prilusky J., Del Pozo A., Tress M., Johnson R., Guigo R., et al. Chimeras taking shape: potential functions of proteins encoded by chimeric RNA transcripts. Genome Res. 2012;22:1231–1242. doi: 10.1101/gr.130062.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frenkel-Morgenstern M., Valencia A. Novel domain combinations in proteins encoded by chimeric transcripts. Bioinformatics. 2012;28:i67-i74. doi: 10.1093/bioinformatics/bts216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asmann Y.W., Necela B.M., Kalari K.R., Hossain A., Baker T.R., Carr J.M., Davis C., Getz J.E., Hostetter G., Li X., et al. Detection of redundant fusion transcripts as biomarkers or disease-specific therapeutic targets in breast cancer. Cancer Res. 2012;72:1921–1928. doi: 10.1158/0008-5472.CAN-11-3142. [DOI] [PubMed] [Google Scholar]
- 21.Gingeras T.R. Implications of chimaeric non-co-linear transcripts. Nature. 2009;461:206–211. doi: 10.1038/nature08452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maher C.A., Palanisamy N., Brenner J.C., Cao X., Kalyana-Sundaram S., Luo S., Khrebtukova I., Barrette T.R., Grasso C., Yu J., et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc. Natl. Acad. Sci. U.S.A. 2009;106:12353–12358. doi: 10.1073/pnas.0904720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maher C.A., Kumar-Sinha C., Cao X., Kalyana-Sundaram S., Han B., Jing X., Sam L., Barrette T., Palanisamy N., Chinnaiyan A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Djebali S., Lagarde J., Kapranov P., Lacroix V., Borel C., Mudge J.M., Howald C., Foissac S., Ucla C., Chrast J., et al. Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS One. 2012;7:e28213. doi: 10.1371/journal.pone.0028213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prakash A., Tomazela D.M., Frewen B., Maclean B., Merrihew G., Peterman S., Maccoss M.J. Expediting the development of targeted SRM assays: using data from shotgun proteomics to automate method development. J. Proteome Res. 2009;8:2733–2739. doi: 10.1021/pr801028b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim P., Yoon S., Kim N., Lee S., Ko M., Lee H., Kang H., Kim J. ChimerDB 2.0–a knowledgebase for fusion genes updated. Nucleic Acids Res. 2010;38:D81–D85. doi: 10.1093/nar/gkp982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denoeud F., Kapranov P., Ucla C., Frankish A., Castelo R., Drenkow J., Lagarde J., Alioto T., Manzano C., Chrast J., et al. Prominent use of distal 5′ transcription start sites and discovery of a large number of additional exons in ENCODE regions. Genome Res. 2007;17:746–759. doi: 10.1101/gr.5660607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Houseley J., Tollervey D. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS One. 2010;5:e12271. doi: 10.1371/journal.pone.0012271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McManus C.J., Duff M.O., Eipper-Mains J., Graveley B.R. Global analysis of trans-splicing in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 2010;107:12975–12979. doi: 10.1073/pnas.1007586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu C.S., Yu C.Y., Chuang C.Y., Hsiao M., Kao C.F., Kuo H.C., Chuang T.J. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014;24:25–36. doi: 10.1101/gr.159483.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu C.Y., Liu H.J., Hung L.Y., Kuo H.C., Chuang T.J. Is an observed non-co-linear RNA product spliced in trans, in cis or just in vitro. Nucleic Acids Res. 2014;42:9410–9423. doi: 10.1093/nar/gku643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frenkel-Morgenstern M., Gorohovski A., Lacroix V., Rogers M., Ibanez K., Boullosa C., Andres Leon E., Ben-Hur A., Valencia A. ChiTaRS: a database of human, mouse and fruit fly chimeric transcripts and RNA-sequencing data. Nucleic Acids Res. 2013;41:D142–D151. doi: 10.1093/nar/gks1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benson D.A., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2014;42:D32–D37. doi: 10.1093/nar/gkt1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim N., Kim P., Nam S., Shin S., Lee S. ChimerDB–a knowledgebase for fusion sequences. Nucleic Acids Res. 2006;34:D21–D24. doi: 10.1093/nar/gkj019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kong F., Zhu J., Wu J., Peng J., Wang Y., Wang Q., Fu S., Yuan L.L., Li T. dbCRID: a database of chromosomal rearrangements in human diseases. Nucleic Acids Res. 2011;39:D895–D900. doi: 10.1093/nar/gkq1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Novo F.J., de MendÃbil I.O., Vizmanos J.L. TICdb: a collection of gene-mapped translocation breakpoints in cancer. BMC Genomics. 2007;8:33. doi: 10.1186/1471-2164-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X., Zhao L., Jiang H., Wang W. Short homologous sequences are strongly associated with the generation of chimeric RNAs in eukaryotes. J. Mol. Evol. 2009;68:56–65. doi: 10.1007/s00239-008-9187-0. [DOI] [PubMed] [Google Scholar]
- 38.Puente X.S., Pinyol M., Quesada V., Conde L., Ordóñez G.R., Villamor N., Escaramis G., Jares P., Beà S., González-Díaz M., et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Costa V., Angelini C., De Feis I., Ciccodicola A. Uncovering the complexity of transcriptomes with RNA-Seq. J. Biomed. Biotechnol. 2010:853916. doi: 10.1155/2010/853916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quesada V., Conde L., Villamor N., Ordóñez G.R., Jares P., Bassaganyas L., Ramsay A.J., Beà S., Pinyol M., Martínez-Trillos A., et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2012;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 41.Guffanti A., Iacono M., Pelucchi P., Kim N., Soldà G., Croft L.J., Taft R.J., Rizzi E., Askarian-Amiri M., Bonnal R.J., et al. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics. 2009;10:163. doi: 10.1186/1471-2164-10-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi Y.L., Takeuchi K., Soda M., Inamura K., Togashi Y., Hatano S., Enomoto M., Hamada T., Haruta H., Watanabe H., et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. 2008;68:4971–4976. doi: 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- 43.Soda M., Choi Y.L., Enomoto M., Takada S., Yamashita Y., Ishikawa S., Fujiwara S., Watanabe H., Kurashina K., Hatanaka H., et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 44.Wang X.S., Prensner J.R., Chen G., Cao Q., Han B., Dhanasekaran S.M., Ponnala R., Cao X., Varambally S., Thomas D.G., et al. An integrative approach to reveal driver gene fusions from paired-end sequencing data in cancer. Nat. Biotechnol. 2009;27:1005–1011. doi: 10.1038/nbt.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kannan K., Wang L., Wang J., Ittmann M.M., Li W., Yen L. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proc. Natl. Acad. Sci. U.S.A. 2011;108:9172–9177. doi: 10.1073/pnas.1100489108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herai R.H., Yamagishi M.E. Detection of human interchromosomal trans-splicing in sequence databanks. Brief. Bioinform. 2010;11:198–209. doi: 10.1093/bib/bbp041. [DOI] [PubMed] [Google Scholar]
- 47.Mitelman F., Johansson B., Mertens F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 48.Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.UniProt Consortium. Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitelman F., Mertens F., Johansson B. Prevalence estimates of recurrent balanced cytogenetic aberrations and gene fusions in unselected patients with neoplastic disorders. Genes Chromosomes Cancer. 2005;43:350–366. doi: 10.1002/gcc.20212. [DOI] [PubMed] [Google Scholar]
- 51.Magrane M., Consortium U UniProt Knowledgebase: a hub of integrated protein data. Database. 2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karolchik D., Barber G.P., Casper J., Clawson H., Cline M.S., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42:D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pearson W.R., Lipman D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. U.S.A. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang M., Foo J., Gonen M., Guilhot J., Mahon F.X., Michor F. Selection pressure exerted by imatinib therapy leads to disparate outcomes of imatinib discontinuation trials. Haematologica. 2012;97:1553–1561. doi: 10.3324/haematol.2012.062844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murga Penas E.M., Cools J., Algenstaedt P., Hinz K., Seeger D., Schafhausen P., Schilling G., Marynen P., Hossfeld D.K., Dierlamm J. A novel cryptic translocation t(12;17)(p13;p12-p13) in a secondary acute myeloid leukemia results in a fusion of the ETV6 gene and the antisense strand of the PER1 gene. Genes Chromosomes Cancer. 2003;37:79–83. doi: 10.1002/gcc.10175. [DOI] [PubMed] [Google Scholar]
- 56.Iyer M.K., Chinnaiyan A.M., Maher C.A. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011;27:2903–2904. doi: 10.1093/bioinformatics/btr467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmann R., Valencia A. A gene network for navigating the literature. Nat. Genet. 2004;36:664. doi: 10.1038/ng0704-664. [DOI] [PubMed] [Google Scholar]
- 58.Hoffmann R., Valencia A. Implementing the iHOP concept for navigation of biomedical literature. Bioinformatics. 2005;21(Suppl. 2):ii252–ii258. doi: 10.1093/bioinformatics/bti1142. [DOI] [PubMed] [Google Scholar]
- 59.Hoffmann R., Krallinger M., Andres E., Tamames J., Blaschke C., Valencia A. Text mining for metabolic pathways, signaling cascades, and protein networks. Sci. STKE. 2005;2005:pe21. doi: 10.1126/stke.2832005pe21. [DOI] [PubMed] [Google Scholar]
- 60.Fernández J.M., Hoffmann R., Valencia A. iHOP web services. Nucleic Acids Res. 2007;35:W21–W26. doi: 10.1093/nar/gkm298. [DOI] [PMC free article] [PubMed] [Google Scholar]