


Abstract
Microbiologists utilize ribosomal RNA genes as molecular markers of taxonomy in surveys of microbial communities. rRNA genes are often co-located as part of an rrn operon, and multiple copies of this operon are present in genomes across the microbial tree of life. rrn copy number variability provides valuable insight into microbial life history, but introduces systematic bias when measuring community composition in molecular surveys. Here we present an update to the ribosomal RNA operon copy number database (rrnDB), a publicly available, curated resource for copy number information for bacteria and archaea. The redesigned rrnDB (http://rrndb.umms.med.umich.edu/) brings a substantial increase in the number of genomes described, improved curation, mapping of genomes to both NCBI and RDP taxonomies, and refined tools for querying and analyzing these data. With these changes, the rrnDB is better positioned to remain a comprehensive resource under the torrent of microbial genome sequencing. The enhanced rrnDB will contribute to the analysis of molecular surveys and to research linking genomic characteristics to life history.
INTRODUCTION
In bacteria and archaea, the ribosomal RNA operon (rrn) typically codes for the 16S, 23S and 5S rRNAs. Together with a suite of proteins, these form ribosomes—the molecular machines responsible for catalyzing the mRNA-dependent polymerization of amino acids into protein. Unlike most bacterial and archaeal genes, the rRNA operon is frequently found in multiple copies, from 1–15 in bacteria and 1–4 in archaea (1). It has been suggested that rrn copy number is an index of microbial life histories, wherein rapid growth in response to favorable conditions and high translational power (copiotrophic life history traits) are positively correlated with rrn copy number (2,3), and oligotrophic organisms tend to have low copy number (4,5). Due to the central importance of ribosomal RNAs in the formation of peptide bonds (6), rRNA genes share regions of highly conserved sequence that are interspersed with more variable regions. These characteristics make the 16S gene a useful phylogenetic marker, key to our modern understanding of the evolutionary relationships among microbes.
The abundance of sequence data and knowledge about secondary structure has made the 16S gene the most popular target for culture-independent, sequence-based methods in microbiology. With the rapidly shrinking cost of sequencing, whole community 16S surveys have become a core tool in microbial ecology. Curated databases of aligned 16S sequences, including SILVA (7), the Ribosomal Database Project (RDP, 8) and Greengenes (9), have been developed to facilitate analysis of sequence data. Analysis pipelines usually produce estimates of per-taxon relative abundances based on the number of copies of 16S genes recovered in a sequence library.
Unfortunately, given the variable per-genome copy number of the 16S gene, a frequently recovered sequence may represent a high copy number taxon of lesser abundance, or a low copy number taxon of higher abundance. Inferences based on relative abundance of 16S genes may therefore not be representative of true community structure (10). This can be an important source of systematic bias in 16S surveys, along with differential DNA extraction and polymerase chain reaction amplification (11–13). Given knowledge of 16S gene copy number, molecular surveys can be corrected to remove this bias.
By mapping recovered sequences to available microbial genomes based on similarity in the 16S gene, the 16S copy number of the organism that contributed the sequence can be estimated and survey data adjusted accordingly. This general approach has been implemented in several software packages, including CopyRighter (14), pplacer and the picante R package (10), and incorporated into PICRUSt (15). The accuracy of these methods depends on a reference database of known 16S copy numbers mapped to a taxonomy or phylogeny. The rrnDB is a carefully curated, publicly available resource for copy number information, which can be easily integrated into existing correction methods.
Here we introduce an updated version of the rrnDB providing 16S copy number information derived from a new data source, a new website with expanded features, and mechanisms for maintaining concurrency with new genomes as they are published. At manuscript submission the database included 2635 bacterial records representing 1383 species, and 175 archaeal records representing 148 species. We foresee the new rrnDB contributing to improved copy number correction in metagenomic surveys. Further, the changes create a more robust platform for continued development as a resource supporting functional studies involving rrn copy number and life history strategies of bacteria and archaea. The new rrnDB is available on the WWW at the URL http://rrndb.umms.med.umich.edu/.
DATABASE DESCRIPTION
Major improvements to the website and database include: expanded organism taxonomies to include both the National Center for Biotechnology Information (NCBI) and the RDP systems, new statistical summaries for 16S copy number in search results, downloadable copy number data optimized for use in copy number corrections in 16S molecular surveys, a new download area for sharing database contents, improved searching of records enabled by the availability of additional metadata, and additional links to related external resources.
Most records in the database are derived from annotations of published, completed genome sequences and these include estimates of both 16S gene and 23S gene copy number. About 8% of current records are based on data from experimental methods other than genome sequencing and are referred to as organism-based records. Some organism-based records may include data for either 16S or 23S gene copy number, but not both, depending on the experimental methods used. Counts of tRNA genes are present in most genome-based records, but the tRNA data are not quality controlled or curated by the rrnDB team. Data about 5S rRNA genes and internally transcribed spacers (ITS) are not present in the rrnDB starting with version 4.0.0.
Users can retrieve database entries by two different kinds of text searching, or by browsing a taxonomic hierarchy. ‘Search Record Annotations’ scans rrnDB record fields such as evidence, notes or references for a user-entered search phrase, and also supports retrieval of records by their 16S copy number. ‘Search Taxonomy’ is a taxonomic name scan, with substring matching, that takes advantage of rich metadata that are available as a result of having integrated the NCBI taxonomy database into rrnDB. This mode of searching can retrieve records using obsolete taxonomy names, synonyms, misspellings and others that may be found in the literature, including culture collection strain accessions. Substring searching of RDP taxonomy names is also available. ‘Browse Taxonomy’ is a way to retrieve records using pop-up selection lists that can be populated with taxonomic names from either the NCBI or the RDP systems.
Having NCBI and RDP taxonomies both in the system serves the different objectives and starting information that users may have when approaching the website. NCBI taxonomy is ubiquitous in many data sources and is a principal way that different resources are tied together. RDP taxonomy, being more rooted in phylogeny, is used to classify 16S sequences in molecular surveys.
Search results are returned on a separate web page in table format (Figure 1), one record per row, where each row is identified by a ‘Data source record id’ in the first column. We have adopted the ‘T number’ accessions of the Kyoto Encyclopedia of Genes and Genomes (KEGG, 16) as the data source record id for genome-based records, while for organism-based records a permutation of the ‘strain_id’ of the earlier rrnDB database is used. Each record is associated with an organism name originating from the data source. Another column is populated with the most recent organism name from the NCBI taxonomy database, and when assigned, the RDP genus is also shown. The organism names from NCBI and the data source are not always identical because data source names tend to lag behind changes in NCBI taxonomy. The copy numbers for 16S, 23S and tRNA genes of each record are in the table, which can be sorted by clicking on the header of most columns.
Figure 1.
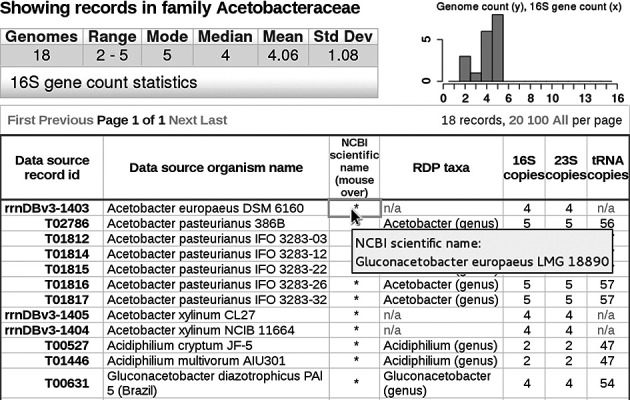
Screen shot of a ‘Browse Taxonomy’ search result for the family Acetobacteraceae using NCBI taxonomy. Statistics for 16S gene counts of all 18 records are shown in the upper-left table. The distribution of 16S counts among the records is shown in the histogram to the right. Summary data for the individual records are shown in the larger table below. Record ids that are prefixed with ‘rrnDBv3-’ were sourced from rrnDB v3.1.227. The other record ids are KEGG accessions. Data source organism names have been given higher visibility than NCBI names because they more often include strain designations. Viewing an NCBI name requires a mouse-hover over the table cell as shown for record rrnDBv3-1403. RDP taxonomy displayed in this table is limited to genus assignment. Each data source record id is hyperlinked to its corresponding record-detail web page. The records can be reordered by clicking on most column headers.
Each search generates a statistical summary of 16S gene counts for the retrieved records, and these are presented above the main table (Figure 1). Statistics include the record count for the result set, the minimum and maximum 16S copy numbers, and the mode, median, mean and standard deviation. A histogram with 15 bars (for copy number of 1–15) showing the relative distribution of 16S copy numbers in the search result, quickly communicates information about the search population. The histogram can be especially illuminating in certain cases, such as the 225 records of the family Enterobacteriaceae (Figure 2). This family shows a broad range of 1 to 9 for the minimum and maximum rrn copy numbers. The histogram reveals the distribution to be bimodal with peaks in the lower and middle regions of the copy number range. Sorting the result table on the ‘16S copies’ column and observation of organism names would reveal the low-copy-number peak to comprise insect-symbiotic organisms exclusively.
Figure 2.
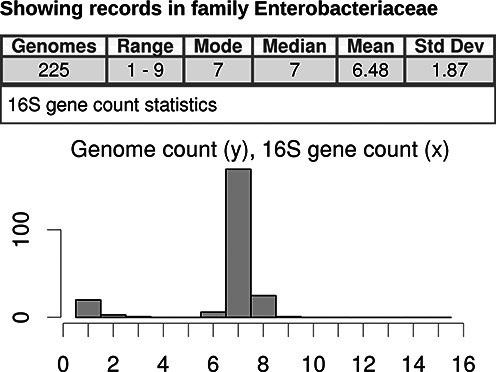
Screen shot showing the statistics and histogram portions of 225 records retrieved by the taxonomy browser for the family Enterobacteriaceae. The role of the histogram in clarifying search result statistics is apparent in this example. Although this figure does not show the individual records table like in Figure 1, it would be apparent from the organism names that insect-symbiotic bacteria comprise the low-16S cluster.
A detailed web page report about any record in a search result can be accessed by clicking on the data source record id of the corresponding table row. The detail reports include additional NCBI and RDP taxonomy information, the type of evidence supporting the rRNA gene counts, curator notes and hyperlinks to external KEGG, NCBI BioProject and NCBI taxonomy web pages. The linked-to external pages provide access to gene and genome sequences and annotations for users to wish to dig deeper. For organism-based records, we provide reference citations with links to NCBI PubMed entries.
For the purpose of adjusting organism abundance in molecular surveys, the mean 16S copy number for a taxon can be misleading if calculated from all genomes due to over-representation of some species. One way to correct for this potential source of bias is to calculate the mean of a taxon from the means of its sub-taxa. We have calculated these ‘pan-taxa statistics’ for all taxa, from genus to domain level, specifically to support copy number correction. The statistics are available for both RDP and NCBI taxonomies. The RDP development team has extended RDP Classifier to support adjustment of the relative abundance of each taxon. The newer version of the Classifier was trained with the rrnDB pan-taxa statistics and is available from RDP (http://rdp.cme.msu.edu/) and the RDP repository on SourceForge (http://sourceforge.net/projects/rdp-classifier/). The ‘Estimate’ feature of the rrnDB website is an on-line interface to the RDP Classifier, including 16S copy number adjustment of taxon abundance for user-uploaded 16S sequence files.
The website includes an ‘About rrnDB’ web page describing the database, a ‘Manual’ web page describing how to use the various features, and a contact email address for users to ask questions, suggest improvements or alert the curators about problems. A ‘Downloads’ web page provides access to tab-delimited tables of versioned rrnDB data as well as the pan-taxa tables. All of the software resources used in the project are freely available under open-source licenses and have strong community support.
DATA SOURCES
Genome-based records in the rrnDB are ultimately derived from the NCBI RefSeq collection. The specific data files that we process are acquired from KEGG and carry additional annotation created by KEGG. In particular the rrnDB makes use of KEGG ‘K numbers’, which apply consistent labeling to orthologous genes across multiple genomes to compute the 16S and 23S rRNA and tRNA gene counts of genomes. The use of K numbers to count rRNA gene copy numbers traverses problems caused by inconsistent labeling and annotation errors in sources upstream of KEGG.
KEGG source data are accessed by us through paid academic subscription to the KEGG data via their FTP site (http://www.kegg.jp/kegg/download/). We expect to bring new and updated genomes into the rrnDB with increased frequency using the KEGG data source. The amount of KEGG data that are necessary to share in order to run the rrnDB website is negligible and well within the terms of the KEGG academic license; therefore, all data made available through the rrnDB are presented without restriction for non-commercial use.
The NCBI taxonomy for bacteria and archaea is fully integrated into the rrnDB so as to support the taxonomy browsing and searching functions of the website. The integrated taxonomy also supports the computation of statistics from the rrnDB records aggregated at any node of the NCBI taxonomic tree. The NCBI taxonomy data will be updated together with each update of KEGG genomes. NCBI taxonomy data are freely available at the NCBI FTP site (http://www.be-md.ncbi.nlm.nih.gov/taxonomy/).
Records of the rrnDB are also mapped to the taxonomy system used by the Ribosomal Database Project (8). Genomes of the rrnDB are mapped to RDP taxonomy using the RDP Classifier tool (17), where each 16S rRNA gene sequence that is classified at a genus bootstrap score of 0.8 or more contributes to the genome's RDP taxonomy. We have been able to map ∼94% of the genome-based records to one or more RDP genera. A genome can map to multiple RDP taxonomies if the genome has multiple 16S genes and a degree of sequence dissimilarity among them. The only genome having been assigned dual RDP taxonomy in rrnDB v4.2.2 is that for Thermoanaerobacterium saccharolyticum DSM 571 (KEGG T01299), which mapped to the genus Thermohydrogenium as well as to itself. Divergence of 16S sequences within Thermoanaerobacterium strains has been described before (18).
The rrnDB holds 216 organism-based records that use rrn copy number estimates from various empirical methods (19). Twenty-five of these records have 23S gene counts but not 16S counts, and for the purpose of computing 16S copy number statistics we presume that their 16S and 23 gene copy numbers are equal.
DATA CURATION
Maintaining genome-based resources involves a trade-off between human curation of records, which is laborious but leads to improved data quality, and machine processing of records, which has higher throughput but can compromise data quality. When updating the rrnDB, a series of automated quality control (QC) tests are applied to identify genomes that may have problems in annotations that can affect the rrnDB. Problematic genomes are held back until the annotations are corrected at their source, or until the genomes can be manually curated. At present our QC pipeline probably retains some genomes that should be allowed through; however, given the increasing number and phylogenetic breadth of published genomes, conservative curation is preferable for most analyses. Our QC pipeline will improve over time as we examine held-back genomes and adjust the QC rules. In addition, our QC strategy does not eliminate human curation, though it does reduce it dramatically compared to earlier versions of the rrnDB.
The initial QC tests identify genomes that are missing some annotations, or in some cases all annotations, for 16S or 23S rRNA genes. The tests count the number of genes that are assigned the K numbers K01977 (16S rRNA) and K01980 (23S rRNA). It is at this stage that the 16S and 23S counts that enter the rrnDB are also computed for genomes that pass QC. For a genome to pass, it must have at least one 16S gene and one 23S gene that is annotated, and the count of annotated 16S and 23S genes must be equal. To increase confidence in 16S counts at this stage, we perform the tests using two different KEGG data source files that should give identical counts. A genome is held back until the next update if the redundant data sources do not agree.
Nine percent of genomes that have entered our QC pipeline have been held back by the above tests; however, more than half of those passed within four months later, during a subsequent update from a new KEGG release. The condition that 16S and 23S gene counts must be equal is admittedly a blunt tool. Cases of rRNA operons missing the 16S rRNA gene, which would cause unequal 16S and 23S counts, have been demonstrated in some bacteria (20). Again, our QC pipeline will become more refined as we examine the individual cases of genomes that are held back.
Further annotation-based testing is designed to detect genomes containing duplicate annotations for what is essentially the same 16S gene. As of this writing the duplication test has discovered two genomes where a 16S or 23S gene had been annotated twice, but with a 1- to 8-base offset between the endpoint coordinates of the duplicates.
Sequence-based quality control steps examine the 16S rRNA gene sequences of all genomes for evidence suggesting that any of them may not be a valid 16S gene sequence. This is done by aligning the putative 16S gene test sequences to the SILVA SSU reference set using the SINA Aligner (21). Any gap in the multiple alignment that is present in every test sequence is removed, then an estimated phylogeny is constructed using FastTree (22). The midpoint-rooted tree has revealed DNA sequences showing unexpectedly long, deep branches suggesting potential annotation problems with those sequences. BLAST similarity searches of the suspect sequences against the NCBI number database are then conducted. Sequences showing low-scoring 16S hits, or only hits to non-16S genes, are taken as justification to hold the genome for examination by a curator. Nine genomes have been held back by the sequence-based criteria.
A final QC test looks for genomes having 16S copy number counts that are outside of the usual range displayed by other genomes of its species. Any species group that shows a difference of three or more between the lowest and highest 16S copy number, is manually examined for genomes that are candidates for having annotation errors affecting 16S gene counts. We have used the database to assess 16S copy-number variability in single-species aggregates of records (Figure 3). For 301 species that are represented by at least two records, 77% are invariant within the species for 16S copy number. An additional 16% of species vary by only one 16S copy. Only 3% of species vary by more than two 16S copies and the maximum variability was five (one species represented by two genomes). Seven genomes have been held back by these criteria.
Figure 3.

Histogram showing 16S copy number variability in 301 species aggregates of the rrnDB records. Only species that are represented by at least two records are counted in this display. Fully 77% of the species show zero variance in 16S gene copy number count among the comprising records. Sixteen percent of the species vary by only one copy, and only 3% of species show a copy number spread of three or more.
FUTURE DEVELOPMENT
One goal of research in the Schmidt lab group has been to understand the physiological and evolutionary implications of rrn redundancy. That goal has spurred the development of internal resources that have found their way into every major revision of the rrnDB since its introduction in 2001. Most recently we have begun to integrate the higher-order functional ontologies of the KEGG database into our research database systems. A goal for development of the rrnDB is to extend that access to the integrated functional and copy number data to the broader community. To a large extent, the creation of that capacity was the reason why we chose KEGG as a data source for the new rrnDB.
Acknowledgments
The authors thank Jim Cole, Qiang Wang and Benli Chen of the Ribosomal Database Project for helpful discussions and for incorporating rrnDB 16S copy number data into RDP Classifier.
FUNDING
National Institutes of Health [M0099549 to T.M.S.]; National Science Foundation's Long-Term Ecological Research Program [DEB 1027253 to T.M.S]; Department of Energy Office of Science Graduate Fellowship Program [DOE SCGF to B.R.K.R., in part] by the American Recovery and Reinvestment Act of 2009, administered by ORISE-ORAU [DE-AC05–06OR23100]. Funding for open access charge: The National Science Foundation's Long-Term Ecological Research Program [DEB 1027253 to T.M.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Klappenbach J.A., Saxman P.R., Cole J.R., Schmidt T.M. rrndb: the ribosomal RNA operon copy number database. Nucleic Acids Res. 2001;29:181–184. doi: 10.1093/nar/29.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klappenbach J.A., Dunbar J.M., Schmidt T.M. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 2000;66:1328–1333. doi: 10.1128/aem.66.4.1328-1333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dethlefsen L., Schmidt T.M. Performance of the translational apparatus varies with the ecological strategies of bacteria. J. Bacteriol. 2007;189:3237–3245. doi: 10.1128/JB.01686-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eichorst S.A., Breznak J.A., Schmidt T.M. Isolation and characterization of soil bacteria that define Terriglobus gen. nov., in the phylum Acidobacteria. Appl. Environ. Microbiol. 2007;73:2708–2717. doi: 10.1128/AEM.02140-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavicchioli R., Ostrowski M., Fegatella F., Goodchild A., Guixa-Boixereu N. Life under nutrient limitation in oligotrophic marine environments: an eco/physiological perspective of Sphingopyxis alaskensis (formerly Sphingomonas alaskensis) Microb. Ecol. 2003;45:203–217. doi: 10.1007/s00248-002-3008-6. [DOI] [PubMed] [Google Scholar]
- 6.Schuwirth B.S., Borovinskaya M.A., Hau C.W., Zhang W., Vila-Sanjurjo A., Holton J.M., Cate J.H.D. Structures of the bacterial ribosome at 3.5 A resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 7.Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole J.R., Wang Q., Fish J.A., Chai B., McGarrell D.M., Sun Y., Brown C.T., Porras-Alfaro A., Kuske C.R., Tiedje J.M. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeSantis T.Z., Hugenholtz P., Larsen N., Rojas M., Brodie E.L., Keller K., Huber T., Dalevi D., Hu P., Andersen G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kembel S.W., Wu M., Eisen J.A., Green J.L. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput. Biol. 2012;8:e1002743. doi: 10.1371/journal.pcbi.1002743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinto A.J., Raskin L. PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS One. 2012;7:e43093. doi: 10.1371/journal.pone.0043093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan S., Cohen D.B., Ravel J., Abdo Z., Forney L.J. Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS One. 2012;7:e33865. doi: 10.1371/journal.pone.0033865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan J.L., Darling A.E., Eisen J.A. Metagenomic sequencing of an in vitro-simulated microbial community. PLoS One. 2010;5:e10209. doi: 10.1371/journal.pone.0010209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angly F.E., Dennis P.G., Skarshewski A., Vanwonterghem I., Hugenholtz P., Tyson G.W. CopyRighter: a rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction. Microbiome. 2014;2:1–13. doi: 10.1186/2049-2618-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langille M.G.I., Zaneveld J., Caporaso J.G., McDonald D., Knights D., Reyes J.A., Clemente J.C., Burkepile D.E., Vega Thurber R.L., Knight R., et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Q., Garrity G.M., Tiedje J.M., Cole J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Větrovský T., Baldrian P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One. 2013;8:e57923. doi: 10.1371/journal.pone.0057923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee Z.M., Bussema C., 3rd, Schmidt T.M. rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res. 2009;37:D489–D493. doi: 10.1093/nar/gkn689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz J.J., Gazumyan A., Schwartz I. rRNA gene organization in the Lyme disease spirochete, Borrelia burgdorferi. J. Bacteriol. 1992;174:3757–3765. doi: 10.1128/jb.174.11.3757-3765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pruesse E., Peplies J., Glöckner F.O. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–1829. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price M.N., Dehal P.S., Arkin A.P. FastTree 2—approximately maximum—likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]