


Abstract
The InParanoid database (http://InParanoid.sbc.su.se) provides a user interface to orthologs inferred by the InParanoid algorithm. As there are now international efforts to curate and standardize complete proteomes, we have switched to using these resources rather than gathering and curating the proteomes ourselves. InParanoid release 8 is based on the 66 reference proteomes that the ‘Quest for Orthologs’ community has agreed on using, plus 207 additional proteomes from the UniProt complete proteomes—in total 273 species. These represent 246 eukaryotes, 20 bacteria and seven archaea. Compared to the previous release, this increases the number of species by 173% and the number of pairwise species comparisons by 650%. In turn, the number of ortholog groups has increased by 423%. We present the contents and usages of InParanoid 8, and a detailed analysis of how the proteome content has changed since the previous release.
INTRODUCTION
Orthologs are defined as genes in different species that diverged at their speciation event and, therefore, directly derive from a single gene in their last common ancestor (1). Paralogs, on the other hand, are genes separated by duplication and may be found either in the same or in different species. It is common that orthologs undergo duplications after the speciation event, generating multiple co-orthologs or inparalogs (2). In contrast, outparalogs are genes, either in the same or different species, that derive from a duplication event prior to a given speciation event. Outparalogs in different species are thus not orthologs. Orthology detection is a challenging task, partly because of the mentioned complications with gene duplications, but also because of gene loss, lateral gene transfer or other mechanisms that produce incongruent evolutionary patterns (3).
InParanoid is an algorithm designed with the aim to generate ortholog groups that include all inparalogs but no outparalogs. It is a graph-based method that starts with an exhaustive all-vs-all Basic Local Alignment Search Tool (BLAST) comparison of all protein sequences in two species and then applies a number of clustering rules to build ortholog groups (4). It operates on two species at a time, which may be considered a limitation, but also has advantages. The most typical orthology query is indeed pairwise, either one-to-one species queries, e.g. ‘find the fly ortholog to human gene X’, or one-to-many, e.g. ‘find all orthologs in any other species to human gene X’. Both queries are supported on the InParanoid website. An advantage of the pairwise nature of InParanoid is that the orthology assignments are not affected by other species. When building multi-species ortholog groups of more than two species, a compromise has to be made when conflicting signals are merged, and inevitably more errors are made, such as including outparalogs in ortholog groups. This typically happens when a gene in a distant species is orthologous to multiple inparalogs in two or more closely related species. From the point of view of the gene in the distant species, all genes are orthologous to it as they are related via a speciation event. However, the genes in the closely related species may not all be orthologous to each other, but instead make up separate ortholog groups, generated by duplications after the divergence from the distant species but before the closely related species split. This would make some of them outparalogs.
InParanoid has become popular for a multiple reasons. Aside from the website, which mainly contains eukaryotes, the software is available for stand-alone usage. It runs reasonably fast for a handful of species and has a good balance between false positives and false negatives, as testified in several benchmarks (5–7). On the online orthology benchmarks at http://orthology.benchmarkservice.org, InParanoid is generally ranked among the best methods, especially when looking at both coverage and accuracy. A drawback of InParanoid's pairwise approach, which it shares with most other ortholog inference methods, is however the quadratic runtime scaling with the number of species. With 273 species, this has become a tangible practical problem. In the future, more attention needs to be given to ways to reduce the computational burden.
We here present release 8 of InParanoid, for which the gathering of proteomes was radically different from previous releases. Thanks to the ‘Quest for Orthologs’ community efforts, we are now able to use pre-defined reference proteomes from UniProt, which allowed us to skip the time-consuming and error-prone step of curating proteomes from multiple sources. We analyze how this has affected the content of InParanoid, and also provide a number of use cases.
MATERIALS AND METHODS
A slightly modified version of the InParanoid 4.1 software (4,8) was used for computing InParanoid 8. The difference is in how the two BLAST (9) passes are run. In the distributed version 4.1, the two BLAST passes are run after each other—the first run to find all homologs between two species, and then a second run is launched per query sequence to make accurate alignments with only the homologs found in pass 1. We have not yet found a BLAST setting that simultaneously makes accurate alignments and efficiently avoids false low-complexity matches. The default composition-based score adjustment (10) in BLAST does the latter, and is used in pass 1, but it often truncates the alignments which may cause InParanoid to miss true orthologs (8), hence it is not used in pass 2.
In order to improve computational throughput, we ran both BLAST passes in parallel and after both were done, extracted matches from pass 2 for homologs found in pass 1; see Figure 1. This can also save total real runtime as only two BLAST runs are launched instead of thousands of tiny runs per species comparison, which causes a lot of input/output (I/O) overhead. There are some drawbacks however: the pass 2 computation and results become much larger, and the infrastructure and work required to synchronize, supervise and load balance the increased number of computational jobs is considerable. We opted for this solution mainly because it offers a higher degree of parallelization.
Figure 1.
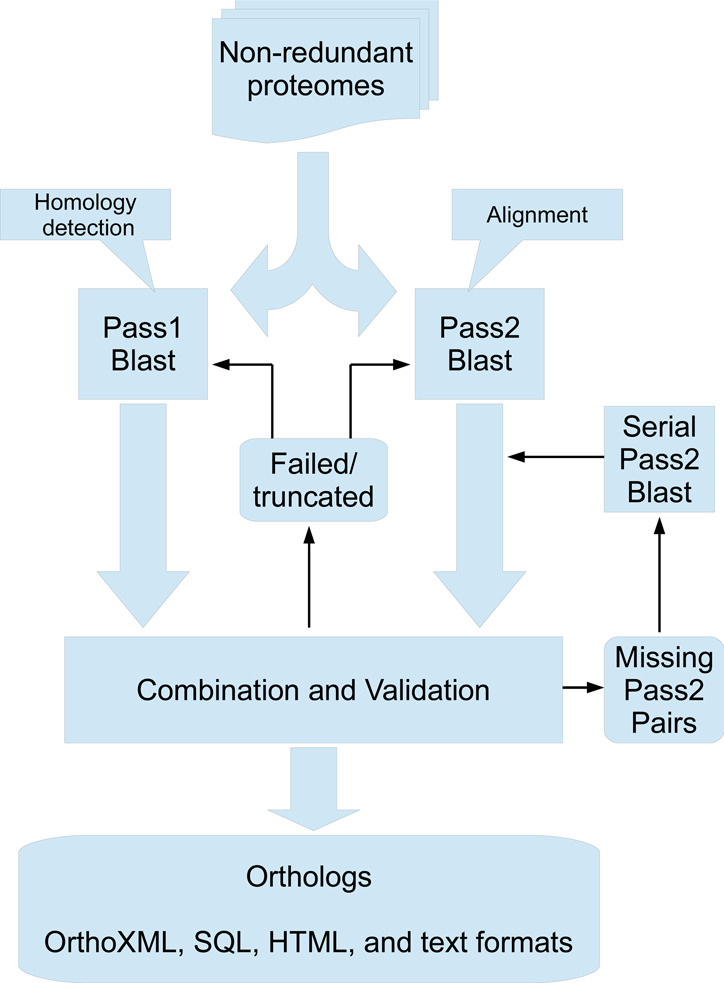
Workflow for the parallel 2-pass BLAST procedure used for generating InParanoid 8. BLAST runs are launched for all pairs of proteomes, running both passes in parallel. When both passes are finished, their outputs are validated by checking for truncation or failure to complete. Intra-proteome matches are checked against the proteome sequences to ensure inclusion of all genes. Pass 1 pairs are combined with pass 2 results such that only pairs accepted in pass 1 are kept, but with alignments from pass 2. A failed validation will either lead to a whole proteome rerun for failed/truncated results or individual serial pass2 reruns for pass1 pairs lacking pass2 results.
To speed up the parallel pass 2, the BLAST parameter z (effective database size) was changed from 1 to 5 000 000. This can reduce BLAST's ability to find matches, and it therefore happens at low frequency that homologs found in pass 1 are not reported in the parallel pass 2. To handle this, a quality control and repair step was added where missing pairs were rerun with a normal pass 2. It also catches other problems, such as failed or truncated runs, and repairs them too. Hence the quality control is currently more rigorous for the parallel method than for the serial. The computations were run on a Linux cluster with around 300 8-core nodes, and took in total about 113 core years of which nearly all was spent on running BLAST (blastall 2.2.18). Only about 0.5% of the computation time was spent on running the InParanoid orthology detection algorithm, implemented in Perl. Even though the procedure was modified, the parameters were effectively the same as by default. Compared to the previous release, the number of pairwise species comparisons increased by 650%, from 4950 to 37 128.
DATA
InParanoid takes as input complete proteomes with one representative protein sequence per gene, normally the splice form that gives the longest protein or the canonical form. In the past, a large effort was spent on collecting proteomes from various sources and parsing annotation to trace proteins to genes. This often introduced errors which could lead to incorrect orthology assignments. As this is a general problem in the orthology field, the ‘Quest for Orthologs’ (QFO; http://questfororthologs.org/) community has agreed on establishing and using standardized reference proteomes that are curated at the EBI (http://www.ebi.ac.uk/reference_proteomes). The QFO reference proteomes are a subset of the UniProt reference proteomes (11) and currently comprise 66 species. The UniProt reference proteomes are in turn a subset of the UniProt complete proteomes. For InParanoid 8 we used the QFO reference proteomes and added (i) all proteomes that were included in InParanoid 7 and are found in the UniProt complete proteomes, and (ii) additional eukaryotic proteomes from the UniProt complete proteomes, limited to one species for each genus not already covered. This resulted in 273 species, which constitutes a representative set of all completely sequenced (eukaryotic) species. It is a superset of the QFO reference proteomes and a subset of the UniProt complete proteomes. As in previous InParanoid releases it is strongly biased toward eukaryotes, with 246 eukaryotes, 20 bacteria and seven archaea. In total, 3 718 323 sequences were used as input.
INPARANOID CONTENT
Since the previous release, the number of species has increased by 173% from 100 to 273. Many of the new proteomes are smaller than the old ones, hence the increase in protein sequences is only 120%, from 1.7 to 3.7 million. The number of ortholog groups has increased by 423%, from 1.5 to 8.0 million, and the unique orthologous proteins by 141%, from 1.2 to 3.0 million. The discrepancy between these is due to the fact that the total number of ortholog groups grows quadratically with the number of species, while the unique orthologous proteins are limited to the number of total proteins. Still, the addition of new species in release 8 increased the average fraction of proteins that have an ortholog in another species from 0.74 to 0.81.
The average number of inparalogs per species across all ortholog groups is 1.41, slightly lower than in release 7 (1.46). Very closely related species still have an average of 1.00 inparalogs, but the highest average number for two species has increased to 10.94, about twice as high as in release 7, for Glycine max (soybean) when compared to Auricularia delicata (jelly fungus). We show in Figure 2 an example of an inparalog-rich ortholog group with soybean. A likely reason for the high number of inparalogs in soybean is increased ploidy from genome-wide duplications.
Figure 2.
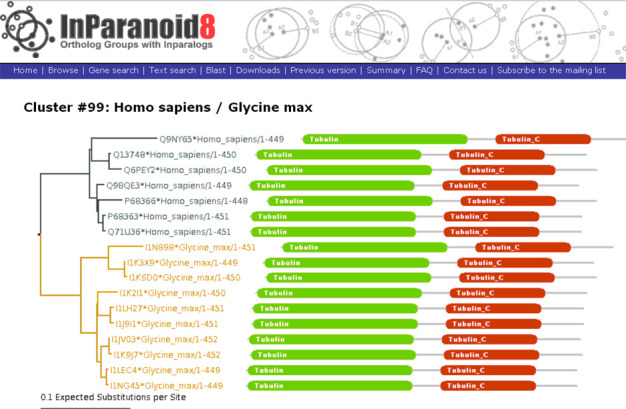
Example of online output when browsing InParanoid 8, showing the neighbor-joining tree and Pfam (20) domain architectures of the proteins in ortholog group 99 between human and soybean (Glycine max). All proteins have the same Pfam domain architecture—a Tubulin (green) and a Tubulin_C (red) domain. The tree indicates that these tubulin-alpha proteins have been duplicated many times independently in the two lineages since they diverged, giving rise to seven human and 10 soybean inparalogs. All human inparalogs are orthologous to all soybean inparalogs as they are all related via the inferred speciation event at the root of the tree.
Overall, most of the proteomes have not changed drastically in the number of sequences or inparalogs, as shown in Figures 3 and 4. If the number of inparalogs changed, then this is normally directly proportional to changes in the number of sequences, and a consequence of a major update in the genome project. We searched for species pairs in which a species had changed more than 1.5-fold in the number of inparalogs and found 28 cases of increase and 170 of decrease. However, all species except three only occurred once; these highly changed species are Brugia malayi, Branchiostoma floridae and Trypanosoma cruzi. Their relative change in the number of sequences is 48% increase, 44% decrease and 45% decrease. This correlates well with the overall change in the number of inparalogs: 45% increase, 45% decrease and 55% decrease. These three species are highlighted in Figure 3 to show that they are outliers. They show just how different a proteome can be defined in different resources; in InParanoid 7 they were taken from NCBI, JGI and GeneDB, respectively. For some species the number of sequences has changed drastically without affecting the overall number of inparalogs much. For example, the aphid Acyrthosiphon pisum has 227% more sequences but overall only 7% more inparalogs, indicating that the added sequences are highly species-specific or that the genome annotation now includes lower quality gene predictions.
Figure 3.
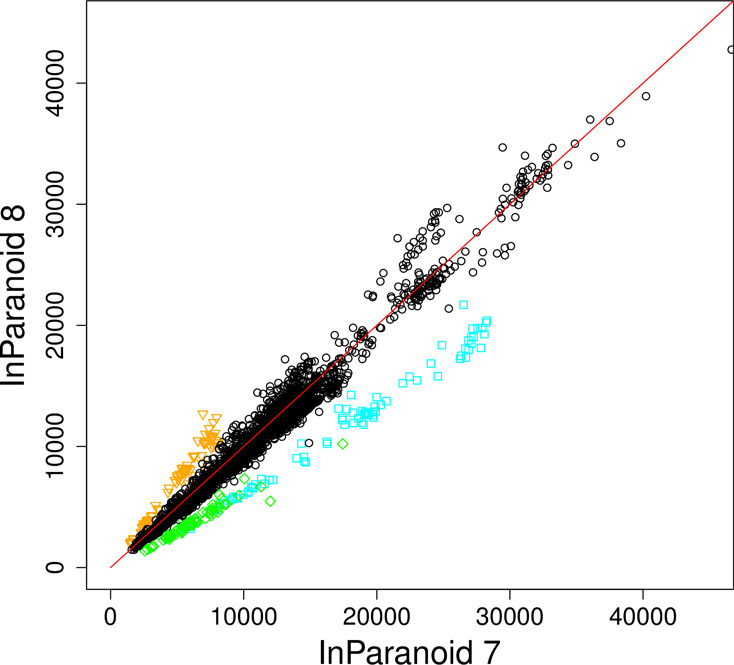
Scatterplot of the number of inparalogs between species pairs in InParanoid 8 and InParanoid 7, for the species common to both releases. The number of inparalogs has generally not changed much, with some exceptions that are highlighted in color (orange for B. malayi, green for T. cruzi and blue for B. floridae).
Figure 4.
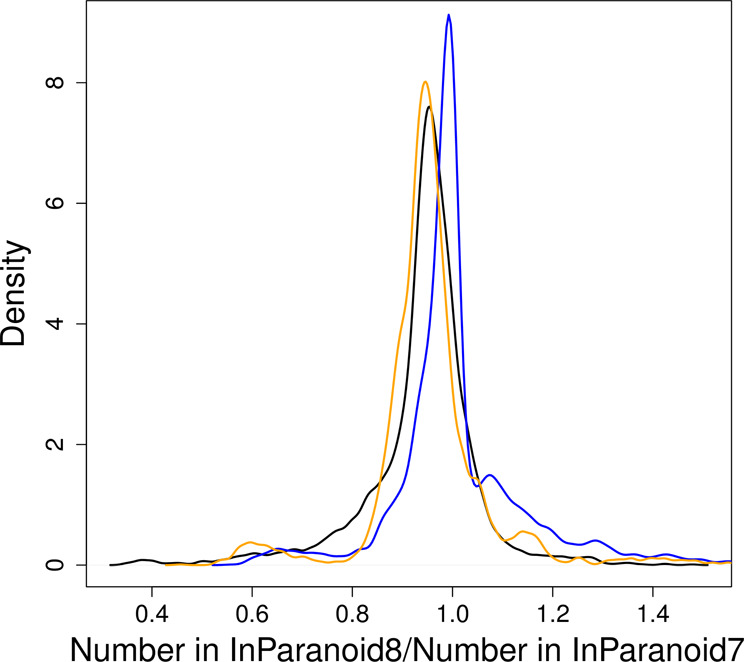
Distributions of the relative number of inparalogs (yellow), inparalogs per sequence (black) and sequences (blue), comparing InParanoid8 to InParanoid 7.
As before, we generated an orthology-based phylogenetic tree by UPGMA clustering of pairwise species distances derived from shared ortholog content. The distances were calculated as 1 minus the fraction of orthologous proteins, averaged over both directions (12). This ‘orthophylogram’ is too large to be shown as a figure but can be accessed on the InParanoid website in the Download directory in Newick or PDF format. It mostly corresponds to the established taxonomy, but we noted a few exceptions. For instance, the northern greater galago (Otolemur garnettii), which is taxonomically classified as a primate, does not cluster with other primates but instead with various domesticated and wild non-primate mammals. This could reflect a problem with its genome sequence, but we note that it shares physical characteristics with e.g. cats and rodents.
The gray short-tailed opossum (Malus domestica), a marsupial which we previously felt was misplaced inside the placental mammalian lineage, has now been joined by another marsupial, the Tasmanian devil (Sarcophilus harrisii). This adds support to the scenario that the marsupials branched out from the placentals.
Macaque monkey (Macaca mulatta) is still oddly placed, clearly outside the other primates, indicating that the annotation of this genome still needs to be improved (8).
The InParanoid orthologs are available four formats. We recommend using the OrthoXML (13) format as this is structured, flexible and resistant to errors. We also provide legacy text, HTML and SQL formats. The SQL format is probably easiest to parse for simple purposes, but lacks additional information available in other formats such as annotations, alternative seed orthologs, statistics, etc.
INPARANOID USAGE
InParanoid is used widely, testified by a total of 1977 citations to all InParanoid papers (Google Scholar, September 18, 2014). The InParanoid website http://InParanoid.sbc.su.se receives on average about 1500 sessions per month (Google Analytics). The InParanoid software is one of the few downloadable fully automatic ortholog-inference programs and has become popular in many applications that require orthologs. It is made available through a webpage (linked from the InParanoid homepage) that sends a copy by email; we have received 1609 download requests from August 2011 to August 2014. Some examples of InParanoid uses, either using the database directly or running the software, are listed below.
Woods et al. (14) compared phenotype-based gene groups between species, looking for groups with significant enrichment of orthologs identified by InParanoid. They thus connected disease phenotypes in human with other phenotypes in chicken, zebrafish, Escherichia coli and Caenorhabditis elegans, and called such pairs ‘orthologous phenotypes’. Furthermore, diseases were connected with each other by looking for shared orthologous phenotypes. They report a number of compelling examples and used the method to predict novel candidate disease genes.
Karányi et al. (15) built a database called FSRD of 1985 fungal stress response proteins, and used InParanoid to identify their orthologs in 28 species including human and plant pathogens and other fungi.
Ciomborowska et al. (16) used InParanoid orthologs to predict whether human genes are generated by retroposition. They found 20 new human candidate retrogenes that lack introns, yet their C. elegans orthologs have introns.
Hoeppner and Poole (17) used InParanoid in their pipeline to identify orthologous snoRNA-bearing host genes across 44 eukaryote genomes. Using Dollo parsimony, they reconstructed the pattern of snoRNA conservation across the eukaryote tree and showed that dozens of snoRNAs are traceable to the Last Eukaryotic Common Ancestor (LECA).
Finally, we note that the orthophylogram generated from InParanoid is somewhat unique as it provides quantitative distances between species based on their entire proteomes. These distances have been used to calculate phylogenetic profile scores in the FunCoup database (18).
FUTURE PERSPECTIVES
The main challenge for the future is the quadratically scaling computational complexity of InParanoid. If we could find a way to retain high accuracy with just one BLAST pass, this would save about 50% of the computation and greatly simplify the procedure. Another possibility is to use pre-calculated homology data, but that would require developing a whole new pipeline to ensure high quality, which may not be achievable. A third solution would be to develop an incremental updating scheme that only reruns BLAST on new and modified sequences. A fourth scenario is to move to linearly scaling Hieranoid (19). It is however still unclear whether Hieranoid's multi-species approach, which has to compromise when merging conflicting orthology evidences, will be able to match InParanoid's quality.
Acknowledgments
This project was supported by BILS (Bioinformatics Infrastructure for Life Sciences). We thank Alan Sousa da Silva, EBI, for providing non-redundant eukaryotic UniProt complete proteomes.
FUNDING
Funding for open access charge: Swedish Research Council [2011-6020].
Conflict of interest statement. None declared.
REFERENCES
- 1.Fitch W.M. Distinguishing homologous from analogous proteins. Syst. Zoolog. 1970;19:99–113. [PubMed] [Google Scholar]
- 2.Sonnhammer E.L., Koonin E.V. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 2002;18:619–620. doi: 10.1016/s0168-9525(02)02793-2. [DOI] [PubMed] [Google Scholar]
- 3.Sonnhammer E.L., Gabaldon T., Sousa da Silva A.W., Martin M., Robinson-Rechavi M., Boeckmann B., Thomas P.D., Dessimoz C. Big data and other challenges in the quest for orthologs. Bioinformatics. 2014;30:2993–2998. doi: 10.1093/bioinformatics/btu492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Remm M., Storm C.E., Sonnhammer E.L. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 2001;314:1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- 5.Hulsen T., Huynen M.A., de Vlieg J., Groenen P.M. Benchmarking ortholog identification methods using functional genomics data. Genome Biol. 2006;7:R31. doi: 10.1186/gb-2006-7-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen F., Mackey A.J., Vermunt J.K., Roos D.S. Assessing performance of orthology detection strategies applied to eukaryotic genomes. PloS One. 2007;2:e383. doi: 10.1371/journal.pone.0000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altenhoff A.M., Dessimoz C. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput. Biol. 2009;5:e1000262. doi: 10.1371/journal.pcbi.1000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostlund G., Schmitt T., Forslund K., Kostler T., Messina D.N., Roopra S., Frings O., Sonnhammer E.L. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010;38:D196–D203. doi: 10.1093/nar/gkp931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 10.Yu Y.K., Altschul S.F. The construction of amino acid substitution matrices for the comparison of proteins with non-standard compositions. Bioinformatics. 2005;21:902–911. doi: 10.1093/bioinformatics/bti070. [DOI] [PubMed] [Google Scholar]
- 11.The UniProt Consortium T.U. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berglund A.C., Sjolund E., Ostlund G., Sonnhammer E.L. InParanoid 6: eukaryotic ortholog clusters with inparalogs. Nucleic Acids Res. 2008;36:D263–D266. doi: 10.1093/nar/gkm1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmitt T., Messina D.N., Schreiber F., Sonnhammer E.L. Letter to the editor: SeqXML and OrthoXML: standards for sequence and orthology information. Brief. Bioinform. 2011;12:485–488. doi: 10.1093/bib/bbr025. [DOI] [PubMed] [Google Scholar]
- 14.Woods J.O., Singh-Blom U.M., Laurent J.M., McGary K.L., Marcotte E.M. Prediction of gene-phenotype associations in humans, mice, and plants using phenologs. BMC Bioinformatics. 2013;14:203. doi: 10.1186/1471-2105-14-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karanyi Z., Holb I., Hornok L., Pocsi I., Miskei M. FSRD: fungal stress response database. Database. 2013;2013:bat037. doi: 10.1093/database/bat037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciomborowska J., Rosikiewicz W., Szklarczyk D., Makalowski W., Makalowska I. “Orphan” retrogenes in the human genome. Mol. Biol. Evol. 2013;30:384–396. doi: 10.1093/molbev/mss235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoeppner M.P., Poole A.M. Comparative genomics of eukaryotic small nucleolar RNAs reveals deep evolutionary ancestry amidst ongoing intragenomic mobility. BMC Evol. Biol. 2012;12:183. doi: 10.1186/1471-2148-12-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmitt T., Ogris C., Sonnhammer E.L. FunCoup 3.0: database of genome-wide functional coupling networks. Nucleic Acids Res. 2014;42:D380–D388. doi: 10.1093/nar/gkt984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schreiber F., Sonnhammer E.L. Hieranoid: hierarchical orthology inference. J. Mol. Biol. 2013;425:2072–2081. doi: 10.1016/j.jmb.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 20.Finn R.D., Bateman A., Clements J., Coggill P., Eberhardt R.Y., Eddy S.R., Heger A., Hetherington K., Holm L., Mistry J., et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]