


Abstract
Recent technological innovations have ignited an explosion in virus genome sequencing that promises to fundamentally alter our understanding of viral biology and profoundly impact public health policy. Yet, any potential benefits from the billowing cloud of next generation sequence data hinge upon well implemented reference resources that facilitate the identification of sequences, aid in the assembly of sequence reads and provide reference annotation sources. The NCBI Viral Genomes Resource is a reference resource designed to bring order to this sequence shockwave and improve usability of viral sequence data. The resource can be accessed at http://www.ncbi.nlm.nih.gov/genome/viruses/ and catalogs all publicly available virus genome sequences and curates reference genome sequences. As the number of genome sequences has grown, so too have the difficulties in annotating and maintaining reference sequences. The rapid expansion of the viral sequence universe has forced a recalibration of the data model to better provide extant sequence representation and enhanced reference sequence products to serve the needs of the various viral communities. This, in turn, has placed increased emphasis on leveraging the knowledge of individual scientific communities to identify important viral sequences and develop well annotated reference virus genome sets.
INTRODUCTION
Recent outbreaks of Ebolavirus (1,2) and Middle East respiratory syndrome coronavirus (MERS-CoV) (3,4) clearly demonstrate the power of sequence analysis in viral surveillance, host reservoir identification and public health policy debate. As these viruses have filled media headlines, their genome sequences have spilled into international public databases. Such real time analysis promises to fundamentally alter our understanding of viral biology and significantly impact public health responses to viral disease, but it also places renewed emphasis on public research infrastructure that is necessary to support the storage and analysis of sequence data. This infrastructure includes primary databases that together comprise the International Nucleotide Sequence Database Collaboration (INSDC) (5), GenBank (6), European Molecular Biology Laboratory's European Bioinformatics Institute (EMBL-EBI) (7), and DNA Database of Japan (DDBJ) (8), and reference databases like the ViralZone Resource at the Swiss Institute of Bioinformatics (http://viralzone.expasy.org) (9) and the Viral Genome Resource at National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/genome/viruses/) (10). Whereas primary databases are archival repositories of sequence data, reference databases provide curated datasets that enable a number of activities, among them are transfer annotation to related genomes (11–13), sequence assembly and virus discovery (14–17), viral dynamics and evolution (18–20) and pathogen detection (14,21–23).
BACKGROUND AND HISTORY
The NCBI Viral Genomes Project was established in response to the growing need for a public, virus-specific, reference sequence resource (24). The project catalogs all complete viral genomes deposited in INSDC databases and creates so-called RefSeq records for each viral species. Each RefSeq is derived from an INSDC sequence record, but may include additional annotation and/or other information. Accessions for RefSeq genome records include the prefix ‘NC_’, allowing them to be easily differentiated from INSDC records. For example, the RefSeq genome record for Enterobacteria phage T4 has the accession NC_000866 but was derived from the INSDC record AF158101. Typically, the first genome submitted for a particular species is selected as a RefSeq, and once a RefSeq is created, other validated genomes for that species are indexed as ‘genome neighbors’. As such, the viral RefSeq data model is taxonomy centric, or more specifically, species centric, and all RefSeq records and genome neighbors are indexed at the species level. This model requires both the demarcation of individual viral species and the grouping of genome sequences into defined species.
GROWING NUMBER OF VIRAL GENOME SEQUENCES
The selection of RefSeq records and other validated genomes (‘genome neighbors’) includes several criteria. First, as a general rule all RefSeq records include annotation of genes and proteins. However, other validated genomes may not include annotation of sequence features or may only include partial annotation. Second, genome length is validated based on community-accepted standards. For some viruses, this means that the sequence must stretch the entire genome, and in other cases, when terminal sequences are hard to obtain, the sequence must cover the entire coding region of the virus. Third, patent sequences and synthetic sequences are not included as RefSeqs or validated genomes. Fourth, when RefSeq records are created for viral genomes comprised of multiple segments, a single genome set is represented by several RefSeq nucleotide records—one for each segment. Complete RefSeq genome sets or constellations are manually curated, and RefSeq records are not created unless all segments are present. In contrast, genome neighbor segments are not assembled into complete genome sets. Instead, each complete segment is added as a neighbor to the corresponding RefSeq segment. So in the case of Rotavirus A, segment 1 (NC_011503) has 1195 neighbor sequences, but segment 9 (NC_011503) has 2438.
There are now 71 628 validated viral and viroid genome segments deposited within INSDC databases, not including influenza sequences, which are stored in a specialized database (11,25). This figure represents a nearly 9-fold increase since 2000 (Figure 1), and this rise reflects both steady increases in the number of novel viruses sequenced—as measured by the number of RefSeq genome segments—and a large increase in the number of genome neighbors, i.e. genome sequences belonging to viral species already represented by a RefSeq (Figure 1). As shown in Table 1, RefSeq genome segments are distributed among all viruses, but genome neighbor segments are concentrated among smaller, ssDNA, RNA, and retro-transcribing viruses. Although many of these neighbor genomes are concentrated among human pathogens, there are also several viruses of agricultural importance with high numbers of sequenced genomes (Table 2). While most of the viruses in Table 2 are well studied in the laboratory, many other sequenced viruses are not.
Figure 1.
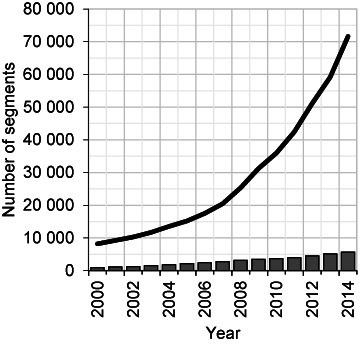
Number of validated virus and viroid segments. The numbers of validated virus and viroid segments available in INSDC databases are depicted by the black line, and the numbers of RefSeq virus and viroid segments by the gray columns. Data was calculated at the end of each year from 2000 to 2014, except for 2014 when data was calculated on September 15. INSDC influenza virus segments are not included.
Table 1. Number of RefSeq and total validated viral genome segments by genome typea.
| Virus genome type | RefSeq genome segments | Total genome segments | Total INSDC sequences |
|---|---|---|---|
| dsDNA viruses, no RNA stage | 1755 | 3023 | 115 911 |
| dsRNA viruses | 919 | 17 929 | 56 699 |
| ssDNA viruses | 669 | 6692 | 40 337 |
| ssRNA negative-strand viruses | 187 | 4384 | 478 791 |
| ssRNA positive-strand viruses, no DNA stage | 917 | 14 441 | 415 664 |
| Retro-transcribing viruses | 123 | 8614 | 727 762 |
Table 2. Viral species with more than 400 validated genomesa.
| Virus species | Number of genomes | Total INSDC sequences |
|---|---|---|
| Hepatitis B virus | 6058 | 74 342 |
| Dengue virus | 3478 | 15 169 |
| Human immunodeficiency virus 1 | 1825 | 561 866 |
| Hepatitis C virus | 1220 | 160 833 |
| Porcine circovirus 2 | 1212 | 3001 |
| Enterovirus A | 663 | 14 449 |
| JC polyomavirus | 495 | 4553 |
| Norwalk virus | 463 | 19 236 |
| Maize streak virus | 455 | 512 |
ADAPTING THE RefSeq DATA MODEL TO VIRUSES
The RefSeq data model for most organisms underscores the importance of very well annotated reference sequence records (26). Unfortunately, a minority of viral systems are experimentally well defined, so there is often little primary data on which to base genome annotations. In some cases, sequence homologies allow the transfer of annotation from experimentally defined to poorly characterized genomes (11–13). Yet, often genomes are annotated by purely ab initio processes (27–29). Given the difficulty of implementing a purely well annotated representation of viral genome sequences, the viral RefSeq model has evolved into a more flexible approach that includes both reference and representative sequences. Reference RefSeq records provide sources of well annotated sequence features, whereas representative records provide coverage of extant sequence variation. The comment ‘REVIEWED REFSEQ’ is added to RefSeq records to highlight those that include additional annotation, and as of this writing, there are 747 reviewed viral RefSeq records, including references for several human pathogens, such as human immunodeficiency virus 1 (NC_001802), Measles virus (NC_001498) and Poliovirus (NC_002058) and several other important viral systems such as Enterobacteria T4 (NC_000866), Enterobacteria T7 (NC_001604) and Tobacco mosaic virus (NC_001367). Reviewed viral RefSeq records can be retrieved from the Entrez Nucleotide database using the search term Viruses[Organism] AND srcdb_refseq[PROP] AND ‘reviewed’’[Filter] (quotes included).
The viral RefSeq model has traditionally focused on one RefSeq genome per species (24). Yet, biology and taxonomic criteria vary among viral species, and the one RefSeq per species model does not always sufficiently capture important sequence variants. This phenomenon is underscored in viral systems that undergo horizontal gene transfer where the genetic diversity within an otherwise closely related group of viruses cannot be captured with a single reference genome (29–30). Moreover, some viral communities are developing well defined subspecies classification such as the genotyping schemes for hepatitis B virus and hepatitis C virus (31–33). These genotyping schemes can provide an important framework for the interpretation of genome sequence data (34), and more communities are expected to develop genotyping schemes in the coming years. Finally, there are cases when the best characterized viral isolate is a laboratory variant, and it may be important to create multiple RefSeq records in order to provide both experimentally annotated references and sufficient sequence representation of circulating isolates. Together these cases highlight the need for both reference genome sequences that capture the best possible annotation and representative genome sequences that capture important intraspecies variation or define subspecies categories. Therefore the viral RefSeq model has expanded to include both reference and representative genome sequences to better serve community needs.
DATA CURATION AND COMMUNITY COLLABORATIONS
Taxonomy
The rising pace of viral discovery has a number of implications for data processing by the Viral Genomes Group. Viral taxonomy within the NCBI Taxonomy database is based on the list of valid species names and classifications provided by the International Committee for the Taxonomy of Viruses (ICTV) (35,36). When the Viral Genomes Project was initiated, there were many more viral species recognized by the ICTV than viral RefSeq genome sequence records (Figure 2). However, as the rate of viral genome sequencing has increased over the past decade, so too has the pace of viral discovery. As a result many RefSeqs are made from viruses clearly distinct from existing ones but without official taxonomy designation. Taxonomy also affects the interpretation of genome sequence data, and technical difficulties encountered when sequencing the termini of some ssRNA and ssDNA viruses often lead to differing community standards for ‘complete genomes’ (37). This means that some difficult to sequence genomes are considered complete if they include the entire coding region but are missing some terminal sequence. Improved methods may eventually resolve this issue (38), but in the meantime it would be useful for communities to define completeness standards with regard to current technology.
Figure 2.
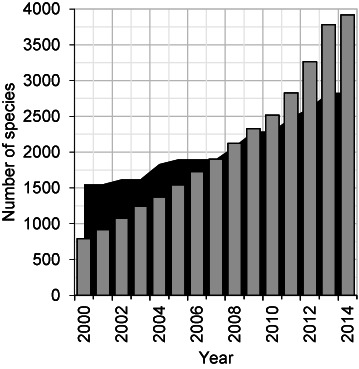
Number of ICTV species and viral RefSeq genomes. The numbers of viral species in the NCBI Taxonomy database that are represented by a RefSeq genome record are depicted by the gray bars, and the number of viral species recognized by the ICTV by the black area. Data was calculated at the end of each year from 2000 to 2014, except for 2014 when data was calculated on September 15. The number of ICTV recognized viral species for each year shown was derived from http://www.ictvonline.org/taxonomyReleases.asp.
In addition to manual selection based on genome length, the taxonomy of both RefSeq genome records and INSDC genome neighbor records are validated. Indeed, given that many novel virus genome sequences are submitted before analysis by the ICTV (see Figure 2), validation of taxonomy assignment is a major facet of curation. Taxonomy is important to the overall usability of NCBI viral genome resources, and when properly implemented, creates a framework for groups of related sequences. Using standards established by individual ICTV study sections (36) and published reports, the taxonomy of each viral genome is validated and updated as necessary. Newly submitted viral genomes without official ICTV assignment are placed with ‘uncharacterized’ taxonomy bins that are easily distinguished from those recognized by the ICTV. Often little information is included in the INSDC sequence record and a growing number of sequences do not include any linked publications. Using sequence analysis and comparative genomics, every attempt is made to place new genomes into a family (i.e. the ‘uncharacterized’ bin associated with a specific family) or lower order classification bin. However, some genomes are very distinct from previously characterized ones and only higher order classification is possible.
Sequence annotations
Reference viral RefSeq records are generally curated by biologists using in-house annotation tools and the scientific literature as guides. A panel of Viral Genome Advisors from outside NCBI bolsters curation efforts by offering expert guidance or taking responsibility for specific RefSeq records themselves. This approach is used for the maintenance of Adenovirus and Herpesvirus RefSeq records (39) and could be extended to other virus genomes (29). These efforts considered, the growing number of viral genomes submitted to INSDC databases and the rapid pace of scientific discovery make maintenance of up-to-date references difficult. Therefore collaboration with scientific communities is critical to providing accurate annotation. Sometimes these collaborative efforts are directed at curating a single RefSeq record, and all of the reviewed RefSeq records mentioned in the previous section were curated in collaboration with experts from individual viral communities. Other times these collaborations are more extensive and touch many sequence records. For example, overlapping gene annotations were corrected on RefSeq records from 14 virus families (Arteriviridae, Arteriviridae, Bunyaviridae, Caliciviridae, Circoviridae, Disistroviridae, Flavoviridae, Luteoviridae, Paramixovridae, Parvoviridae, Picornaviridae, Potyviridae, Reoviridae, Togaviridae) as directed by experimental or predictive analysis (40,41).
A new emphasis has been placed on initiating annotation collaborations at the beginning of a large genome sequencing program so that reference annotations, isolate naming schemes and other standards can be established prior to sequence submission (42–44). These collaborations often include members of the UniProt Viral Protein Annotation Program (45) (9), and/or curators from sequencing centers and other databases (46) in addition to members of the relevant viral communities and effectively ensure both well annotated references and consistently annotated INSDC sequence records. Such arrangements underscore the extensive impact of viral genome annotation issues—from public databases to sequencing centers to individual researcher communities—and were formalized within the Viral Genome Annotation Working Group, which brings together stakeholders and provides a forum for the discussion of annotation issues (29,47). In addition to protein annotation and isolate naming issues, this group is working to define standards for viral genome sequence data.
Viral host type
As the number of viral sequences has risen, so has the demand for curated metadata describing sequences. The Viral Genomes Group has implemented two models designed to capture and standardize metadata. In the first model exemplified by the Virus Variation Resource, host, isolation country and other important metadata are parsed from individual sequence records, mapped against vocabulary lists and standardized (25,48). Sequences can then be searched using these standardized metadata terms. Currently, only a small subset of viral sequences are included in the Virus Variation Resource, including those for influenza, dengue and West Nile viruses, but the ultimate goal is to expand this semi-automated model to include more viruses.
The second model captures and standardizes host information for all viruses, and whenever a new RefSeq record is created, a manually curated ‘viral host’ property is assigned to the relevant species within the NCBI Taxonomy database. The property defines higher order, biologically relevant taxonomic host groups—algae, archaea, bacteria, diatom, environment, fungi, human, invertebrates, plants, protozoa and vertebrates—and enable sorting and selection of sequences within the NCBI Taxonomy (http://www.ncbi.nlm.nih.gov/taxonomy) and Viral Genomes Resource. For example searching the NCBI Taxonomy database with the term ‘vhost fungi’[Properties] (quotes included) will return a list of taxonomy groups comprised of viruses that infect fungi. Users can then select the ‘Genome’ database from ‘Find related data’ link on the Taxonomy search page to view all viral genomes associated with viruses retrieved from the search. In cases where a virus infects multiple types of organisms, multiple terms are assigned, for example ‘invertebrates, plants’. To search NCBI Taxonomy for viruses that infect multiple hosts simply include ‘AND’ between search terms, for example ‘vhost invertebrates’[Properties] AND ‘vhost plants’[Properties] (quotes included). The current distribution of assigned viral host terms is shown in Figure 3.
Figure 3.
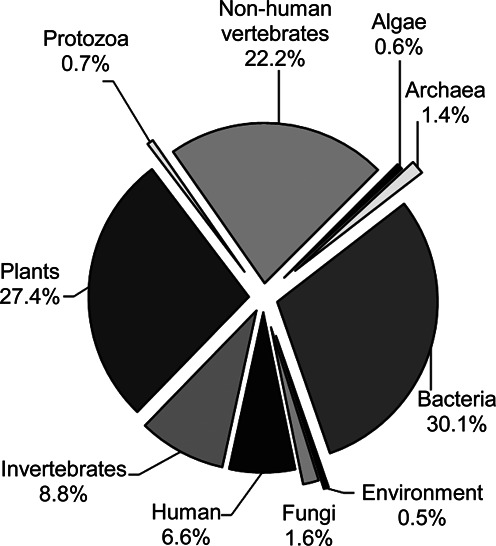
Distribution of host types among viral RefSeq genomes. The distribution of viral host types assigned to RefSeq genomes is depicted for algae, archaea, bacteria, environment, fungi, human, invertebrates, plants, protozoa and vertebrates host groups. Diatom is not shown.
RESOURCES AND TOOLS
The NCBI Viral Genome Resource can be accessed at www.ncbi.nlm.nih.gov/genome/viruses/. On this home page, users will find ftp links where users can download accession list of all viral and viroid genomes (RefSeq and genome neighbors) and the complete viral and viroid RefSeq dataset. Perhaps the central features of the resource are the viral and viroid genome browsers. These tables list all viral and viroid species represented by a reference sequence and include links to genome neighbor sequences. Users can navigate to specific taxonomic groups and sort the table by viral host type. Once a dataset has been defined by taxonomy and host types, users can download the resultant table, the list of RefSeq accessions in the table, or a list that includes RefSeq and genome neighbor accessions as well as taxonomy and viral host information.
Several specialized viral resources and tools are also linked through the Viral Genomes Resource home page. These include specialized resources for influenza, dengue and West Nile and other viruses that are part of the Virus Variation Resource (http://www.ncbi.nlm.nih.gov/genome/viruses/variation/) (25,48,49). The link to the Retrovirus Resource (http://www.ncbi.nlm.nih.gov/genome/viruses/retroviruses) provides access to the Retrovirus Genotyping Tool and HIV-1, Human Interaction Database (50,51). These tools are designed to assist retroviral researchers in the identification and classification of sequences and to document HIV-1 and human protein and replication interactions through a searchable interface. Finally, there is a link to the Pairwise Sequence Comparison Tool (PASC) (http://www.ncbi.nlm.nih.gov/sutils/pasc), a Blast-based tool with graphical output that can be used to establish taxonomic classification criteria of some viruses and classify viruses with newly sequenced genomes (52,53).
Both RefSeq records and other genomes for species are linked throughout NCBI resources and can be used in a variety of operations. Among these, the RefSeq dataset can be used to reduce the redundancy of Blast searches (http://blast.ncbi.nlm.nih.gov/Blast.cgi) (54), providing fewer, higher quality sequences within search results. To restrict nucleotide Blast searches to include only viral RefSeq genomes, employ the ‘Choose Search Set’ options in the Blast search interface (55): Select ‘Reference genomic sequences (RefSeq_genomic)’ in the database field and enter ‘Viruses’ in the ‘Organism’ field text box. For protein Blast searches, the viral RefSeq protein set can be used by selecting ‘Reference proteins’ (RefSeq_proteins) in the database field and entering ‘Viruses’ in the ‘Organism’ field text box. Data derived from viral RefSeqs are also used to support a number of other databases including Gene (56) and Protein Clusters (57).
Each species that includes a RefSeq can be found in the Genome database (http://www.ncbi.nlm.nih.gov/genome) (56). This resource can be searched by taxonomy names, and retrieved genome records include links to all RefSeqs for that species. Each individual genome record also includes links to neighbor sequences for that species under ‘Related information’, and these can be viewed by selecting the ‘Other genomes for species’ option. These links display all genome neighbor records in the nucleotide database where they can be viewed and/or downloaded. Genome neighbor records can also be retrieved from multiple genome records using the ‘Find related data’ options, allowing the user to search for an entire viral family or similar and then retrieve all genome neighbor records defined by the original search criteria. Simply select ‘Nucleotide’ in ‘Database’ pull down menu and ‘Other genomes for species’ from the ‘Option’ pull down menu to return all genome neighbors for all the species listed in the search results.
FUTURE DIRECTIONS
As the sequencing revolution continues to gather steam, and the rate of viral genome sequencing increases, reference databases will be pressed to serve growing community needs. Meeting these will require further collaboration with individual viral communities and across public databases. Data models will also need to shift to better represent the extant sequence universe and provide better standardized sequence annotation.
Once annotated, large-scale genome sequence data will need to be presented in ways that facilitate human data sorting and discovery operations. This will require semi-automated metadata capture and standardization, as well as innovative interfaces and tools that leverage metadata in discovery operations. Many of these approaches and processes are currently being tested within the NCBI Virus Variation Resource (25) where users can readily find sequences based on specific, standardized sequence descriptors, greatly improving the accessibility and utility of viral sequence data. While currently limited to a handful of human pathogens, our intent is to expand the Virus Variation data model to include more viruses from more viral communities. This should open up a number of possibilities and will support the aggregation and retrieval of sequences based on community-defined criteria like genotypes or complete genome sets as is currently possible for influenza virus sequences (11,25).
The growing cloud of viral genome sequences also poses significant barriers to the maintenance of reference genome records. The pace of experimental discovery and the number and breadth of viral genomes make it increasingly difficult to provide well annotated, up-to-date reference sequences. To counter, we must leverage community knowledge and activities against the goal of better RefSeq viral resources and must collaborate with viral communities to maintain well annotated reference sequences, develop community-accepted gene and protein naming standards and define community-established subspecies classification schemes. Though collaborations have been initiated within some communities (29,42–44,47), they need to be scaled to include more groups. As a public resource, we serve a range of communities—from the public health to the basic research—and rely on them to both better inform our mission and help support it. Only by engaging our stakeholders and working together on shared goals can we provide the rigorous resources necessary to support viral sequence data activities.
Acknowledgments
We would like to thank Vyacheslav Chetvernin, Boris Fedorov, Sergey Resenchuck, Igor Tolstoy, Tatiana Tatusova and Jim Ostell for their development and support.
FUNDING
This research was supported by the Intramural Research Program of the National Institutes of Health, National Library of Medicine. Funding for open access charge: Intramural Research Program of National Institutes of Health, National Library of Medicine.
Conflict of interest statement. None declared.
REFERENCES
- 1.Baize S., Pannetier D., Oestereich L., Rieger T., Koivogui L., Magassouba N., Soropogui B., Sow M.S., Keita S., De Clerck H., et al. Emergence of Zaire Ebola virus disease in Guinea—preliminary report. N. Engl. J. Med. 2014;371:1418–1425. doi: 10.1056/NEJMoa1404505. [DOI] [PubMed] [Google Scholar]
- 2.Gire S.K., Goba A., Andersen K.G., Sealfon R.S., Park D.J., Kanneh L., Jalloh S., Momoh M., Fullah M., Dudas G., et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345:1369–1372. doi: 10.1126/science.1259657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haagmans B.L., Al Dhahiry S.H., Reusken C.B., Raj V.S., Galiano M., Myers R., Godeke G.J., Jonges M., Farag E., Diab A., et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect. Dis. 2014;14:140–145. doi: 10.1016/S1473-3099(13)70690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cotten M., Watson S.J., Kellam P., Al-Rabeeah A.A., Makhdoom H.Q., Assiri A., Al-Tawfiq J.A., Alhakeem R.F., Madani H., AlRabiah F.A., et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study. Lancet. 2013;382:1993–2002. doi: 10.1016/S0140-6736(13)61887-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karsch-Mizrachi I., Nakamura Y., Cochrane G., International Nucleotide Sequence Database Collaboration. The International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 2012;40:D33–D37. doi: 10.1093/nar/gkr1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benson D.A., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2014;42:D32–D37. doi: 10.1093/nar/gkt1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brooksbank C., Bergman M.T., Apweiler R., Birney E., Thornton J. The European Bioinformatics Institute's data resources 2014. Nucleic Acids Res. 2014;42:D18–D25. doi: 10.1093/nar/gkt1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kosuge T., Mashima J., Kodama Y., Fujisawa T., Kaminuma E., Ogasawara O., Okubo K., Takagi T., Nakamura Y. DDBJ progress report: a new submission system for leading to a correct annotation. Nucleic Acids Res. 2014;42:D44–D49. doi: 10.1093/nar/gkt1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masson P., Hulo C., De Castro E., Bitter H., Gruenbaum L., Essioux L., Bougueleret L., Xenarios I., Le Mercier P. ViralZone: recent updates to the virus knowledge resource. Nucleic Acids Res. 2013;41:D579–D583. doi: 10.1093/nar/gks1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pruitt K.D., Tatusova T., Maglott D.R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bao Y., Bolotov P., Dernovoy D., Kiryutin B., Tatusova T. FLAN: a web server for influenza virus genome annotation. Nucleic Acids Res. 2007;35:W280–W284. doi: 10.1093/nar/gkm354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang S., Sundaram J.P., Stockwell T.B. VIGOR extended to annotate genomes for additional 12 different viruses. Nucleic Acids Res. 2012;40:W186–W192. doi: 10.1093/nar/gks528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S., Sundaram J.P., Spiro D. VIGOR, an annotation program for small viral genomes. BMC Bioinformatics. 2010;11:451. doi: 10.1186/1471-2105-11-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borozan I., Watt S.N., Ferretti V. Evaluation of alignment algorithms for discovery and identification of pathogens using RNA-Seq. PLoS One. 2013;8:e76935. doi: 10.1371/journal.pone.0076935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaynor A.M., Nissen M.D., Whiley D.M., Mackay I.M., Lambert S.B., Wu G., Brennan D.C., Storch G.A., Sloots T.P., Wang D. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007;3:e64. doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holtz L.R., Finkbeiner S.R., Zhao G., Kirkwood C.D., Girones R., Pipas J.M., Wang D. Klassevirus 1, a previously undescribed member of the family Picornaviridae, is globally widespread. Virol. J. 2009;6:86. doi: 10.1186/1743-422X-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dutilh B.E., Cassman N., McNair K., Sanchez S.E., Silva G.G., Boling L., Barr J.J., Speth D.R., Seguritan V., Aziz R.K., et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014;5:4498. doi: 10.1038/ncomms5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cotten M., Petrova V., Phan M.V., Rabaa M.A., Watson S.J., Ong S.H., Kellam P., Baker S. Deep sequencing of norovirus genomes defines evolutionary patterns in an urban tropical setting. J. Virol. 2014;88:11056–11069. doi: 10.1128/JVI.01333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dennis A.F., McDonald S.M., Payne D.C., Mijatovic-Rustempasic S., Esona M.D., Edwards K.M., Chappell J.D., Patton J.T. Molecular epidemiology of contemporary G2P[4] human rotaviruses cocirculating in a single U.S. community: footprints of a globally transitioning genotype. J. Virol. 2014;88:3789–3801. doi: 10.1128/JVI.03516-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reyes A., Semenkovich N.P., Whiteson K., Rohwer F., Gordon J.I. Going viral: next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012;10:607–617. doi: 10.1038/nrmicro2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kostic A.D., Ojesina A.I., Pedamallu C.S., Jung J., Verhaak R.G., Getz G., Meyerson M. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat. Biotechnol. 2011;29:393–396. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q., Jia P., Zhao Z. VirusFinder: software for efficient and accurate detection of viruses and their integration sites in host genomes through next generation sequencing data. PLoS One. 2013;8:e64465. doi: 10.1371/journal.pone.0064465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naccache S.N., Federman S., Veeraraghavan N., Zaharia M., Lee D., Samayoa E., Bouquet J., Greninger A.L., Luk K.C., Enge B., et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 2014;24:1180–1192. doi: 10.1101/gr.171934.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bao Y., Federhen S., Leipe D., Pham V., Resenchuk S., Rozanov M., Tatusov R., Tatusova T. National Center for Biotechnology Information Viral Genomes Project. J. Virol. 2004;78:7291–7298. doi: 10.1128/JVI.78.14.7291-7298.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brister J.R., Bao Y., Zhdanov S.A., Ostapchuck Y., Chetvernin V., Kiryutin B., Zaslavsky L., Kimelman M., Tatusova T.A. Virus Variation Resource—recent updates and future directions. Nucleic Acids Res. 2014;42:D660–D665. doi: 10.1093/nar/gkt1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pruitt K.D., Tatusova T., Brown G.R., Maglott D.R. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mills R., Rozanov M., Lomsadze A., Tatusova T., Borodovsky M. Improving gene annotation of complete viral genomes. Nucleic Acids Res. 2003;31:7041–7055. doi: 10.1093/nar/gkg878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kattenhorn L.M., Mills R., Wagner M., Lomsadze A., Makeev V., Borodovsky M., Ploegh H.L., Kessler B.M. Identification of proteins associated with murine cytomegalovirus virions. J. Virol. 2004;78:11187–11197. doi: 10.1128/JVI.78.20.11187-11197.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brister J.R., Le Mercier P., Hu J.C. Microbial virus genome annotation-mustering the troops to fight the sequence onslaught. Virology. 2012;434:175–180. doi: 10.1016/j.virol.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawrence J.G., Hatfull G.F., Hendrix R.W. Imbroglios of viral taxonomy: genetic exchange and failings of phenetic approaches. J. Bacteriol. 2002;184:4891–4905. doi: 10.1128/JB.184.17.4891-4905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pourkarim M.R., Amini-Bavil-Olyaee S., Kurbanov F., Van Ranst M., Tacke F. Molecular identification of hepatitis B virus genotypes/subgenotypes: revised classification hurdles and updated resolutions. World J. Gastroenterol. 2014;20:7152–7168. doi: 10.3748/wjg.v20.i23.7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simmonds P., Bukh J., Combet C., Deleage G., Enomoto N., Feinstone S., Halfon P., Inchauspe G., Kuiken C., Maertens G., et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 33.Smith D.B., Bukh J., Kuiken C., Muerhoff A.S., Rice C.M., Stapleton J.T., Simmonds P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanwar S., Dusheiko G. Is there any value to hepatitis B virus genotype analysis. Curr. Gastroenterol. Rep. 2012;14:37–46. doi: 10.1007/s11894-011-0233-5. [DOI] [PubMed] [Google Scholar]
- 35.Federhen S. The NCBI Taxonomy database. Nucleic Acids Res. 2012;40:D136–D143. doi: 10.1093/nar/gkr1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jancovich J.K., Chinchar V.G., Hyatt A., Miyazaki T., Williams T., Zhang Q.Y. In: Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. San Diego, CA: Elsevier Academic Press; 2012. [Google Scholar]
- 37.Maan S., Rao S., Maan N.S., Anthony S.J., Attoui H., Samuel A.R., Mertens P.P. Rapid cDNA synthesis and sequencing techniques for the genetic study of bluetongue and other dsRNA viruses. J. Virol. Methods. 2007;143:132–139. doi: 10.1016/j.jviromet.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 38.Alfson K.J., Beadles M.W., Griffiths A. A new approach to determining whole viral genomic sequences including termini using a single deep sequencing run. J. Virol. Methods. 2014;208:1–5. doi: 10.1016/j.jviromet.2014.07.023. [DOI] [PubMed] [Google Scholar]
- 39.Davison A.J. Herpesvirus systematics. Vet. Microbiol. 2010;143:52–69. doi: 10.1016/j.vetmic.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sabath N., Wagner A., Karlin D. Evolution of viral proteins originated de novo by overprinting. Mol. Biol. Evol. 2012;29:3767–3780. doi: 10.1093/molbev/mss179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rancurel C., Khosravi M., Dunker A.K., Romero P.R., Karlin D. Overlapping genes produce proteins with unusual sequence properties and offer insight into de novo protein creation. J. Virol. 2009;83:10719–10736. doi: 10.1128/JVI.00595-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matthijnssens J., Ciarlet M., McDonald S.M., Attoui H., Banyai K., Brister J.R., Buesa J., Esona M.D., Estes M.K., Gentsch J.R., et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG) Arch. Virol. 2011;156:1397–1413. doi: 10.1007/s00705-011-1006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuhn J.H., Bao Y., Bavari S., Becker S., Bradfute S., Brister J.R., Bukreyev A.A., Chandran K., Davey R.A., Dolnik O., et al. Virus nomenclature below the species level: a standardized nomenclature for natural variants of viruses assigned to the family Filoviridae. Arch. Virol. 2013;158:301–311. doi: 10.1007/s00705-012-1454-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuhn J.H., Bao Y., Bavari S., Becker S., Bradfute S., Brister J.R., Bukreyev A.A., Cai Y., Chandran K., Davey R.A., et al. Virus nomenclature below the species level: a standardized nomenclature for laboratory animal-adapted strains and variants of viruses assigned to the family Filoviridae. Arch. Virol. 2013;158:1425–1432. doi: 10.1007/s00705-012-1594-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.UniProt Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pickett B.E., Sadat E.L., Zhang Y., Noronha J.M., Squires R.B., Hunt V., Liu M., Kumar S., Zaremba S., Gu Z., et al. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012;40:D593–D598. doi: 10.1093/nar/gkr859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brister J.R., Bao Y., Kuiken C., Lefkowitz E.J., Le Mercier P., Leplae R., Madupu R., Scheuermann R.H., Schobel S., Seto D., et al. Towards Viral Genome Annotation Standards, Report from the 2010 NCBI Annotation Workshop. Viruses. 2010;2:2258–2268. doi: 10.3390/v2102258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Resch W., Zaslavsky L., Kiryutin B., Rozanov M., Bao Y., Tatusova T.A. Virus variation resources at the National Center for Biotechnology Information: dengue virus. BMC Microbiol. 2009;9:65. doi: 10.1186/1471-2180-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bao Y., Bolotov P., Dernovoy D., Kiryutin B., Zaslavsky L., Tatusova T., Ostell J., Lipman D. The influenza virus resource at the National Center for Biotechnology Information. J. Virol. 2008;82:596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rozanov M., Plikat U., Chappey C., Kochergin A., Tatusova T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004;32:W654–W659. doi: 10.1093/nar/gkh419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu W., Sanders-Beer B.E., Katz K.S., Maglott D.R., Pruitt K.D., Ptak R.G. Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res. 2009;37:D417–D422. doi: 10.1093/nar/gkn708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bao Y., Chetvernin V., Tatusova T. PAirwise Sequence Comparison (PASC) and its application in the classification of filoviruses. Viruses. 2012;4:1318–1327. doi: 10.3390/v4081318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bao Y., Chetvernin V., Tatusova T. Improvements to pairwise sequence comparison (PASC): a genome-based web tool for virus classification. Arch. Virol. 2014;159:3293–3304. doi: 10.1007/s00705-014-2197-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boratyn G.M., Camacho C., Cooper P.S., Coulouris G., Fong A., Ma N., Madden T.L., Matten W.T., McGinnis S.D., Merezhuk Y., et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013;41:W29–W33. doi: 10.1093/nar/gkt282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T.L. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2014;42:D7–D17. doi: 10.1093/nar/gkt1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klimke W., Agarwala R., Badretdin A., Chetvernin S., Ciufo S., Fedorov B., Kiryutin B., O'Neill K., Resch W., Resenchuk S., et al. The National Center for Biotechnology Information's Protein Clusters Database. Nucleic Acids Res. 2009;37:D216–D223. doi: 10.1093/nar/gkn734. [DOI] [PMC free article] [PubMed] [Google Scholar]