

Abstract
microRNAs (miRNAs) are short non-coding RNA species, which act as potent gene expression regulators. Accurate identification of miRNA targets is crucial to understanding their function. Currently, hundreds of thousands of miRNA:gene interactions have been experimentally identified. However, this wealth of information is fragmented and hidden in thousands of manuscripts and raw next-generation sequencing data sets. DIANA-TarBase was initially released in 2006 and it was the first database aiming to catalog published experimentally validated miRNA:gene interactions. DIANA-TarBase v7.0 (http://www.microrna.gr/tarbase) aims to provide for the first time hundreds of thousands of high-quality manually curated experimentally validated miRNA:gene interactions, enhanced with detailed meta-data. DIANA-TarBase v7.0 enables users to easily identify positive or negative experimental results, the utilized experimental methodology, experimental conditions including cell/tissue type and treatment. The new interface provides also advanced information ranging from the binding site location, as identified experimentally as well as in silico, to the primer sequences used for cloning experiments. More than half a million miRNA:gene interactions have been curated from published experiments on 356 different cell types from 24 species, corresponding to 9- to 250-fold more entries than any other relevant database. DIANA-TarBase v7.0 is freely available.
INTRODUCTION
microRNAs (miRNAs) are short (∼23 nts) non-coding RNA species present in animals, plants and viruses. Recent research efforts have shed light into a series of novel miRNA regulatory roles (1–4). However, their most prominent function is still their extensive post-transcriptional gene expression regulation (5), which is observed in numerous physiological and pathological processes (6).
Accurate identification and cataloging of miRNA targets is crucial to understanding their function. To this end, numerous wet lab methodologies have been developed, enabling the validation of predicted miRNA interactions or the high-throughput screening and identification of novel miRNA targets (7). Currently available methodologies can elucidate different parts of the equation and are often used complementarily in investigative studies.
Experimental methods for the identification of miRNA:mRNA interactions
Experimental techniques are usually divided into low-yield and high-throughput methods, depending on their application scope and the number of obtained results per experiment. The most commonly used low-yield techniques are reporter genes, quantitative polymerase chain reaction (qPCR) and western blotting. Reporter genes are used for binding site validation, while qPCR and western blot or enzyme-linked immunosorbent assay (ELISA) assays are usually combined to identify interactions that induce mRNA decay and/or translation suppression.
The first high-throughput techniques that became available could be considered as an increased throughput/lower accuracy version of specific techniques (8). Microarrays can be utilized to identify possible miRNA:gene interactions, as a high-throughput version of qPCR and northern blotting, while quantitative proteomic techniques (e.g. pSILAC (9)) can be seen as a high-yield generalization of ELISA assays and western blots. Novel next-generation sequencing (NGS) experiments have offered a high-throughput/increased accuracy combination that has revolutionized the way we identify miRNA:gene interactions. These techniques are based on NGS sequencing of mRNA sites bound by the Argonaute (AGO) protein and are often accompanied by sequencing of small-RNAs, as well as complementary experiments, such as RNA-Seq and ribosome profiling (10,11).
HITS-CLIP was the first technique that offered for the first time a transcriptome-wide map of miRNA binding sites (10). However, the identified regions are usually wide and perplex the identification of the exact miRNA binding location, which is performed algorithmically. PAR-CLIP is a modified CLIP-Seq methodology, incorporating 4-thiouridine in the nascent RNAs, which are subsequently detected as T-to-C transition sites in the AGO:miRNA:RNA cross-linked regions (11). Compared to the results obtained by HITS-CLIP, the boundaries of the identified binding locations are sharper and significantly narrower, while T-to-C mutations close to the region occupied by the RNA-induced silencing complex (RISC) contribute to the identification of the exact miRNA recognition element (MRE) (12). Despite the accurate detection of the crosslinked region, these methods cannot directly reveal the specific miRNA participating in the interaction, which has to be bioinformatically identified. A more recent variant of the PAR-CLIP methodology (13) as well as the CLASH protocol (14) incorporate an extra ligation step, concatenating the miRNA to the mRNA binding region. The derived chimeric miRNA:mRNA fragments are subsequently sequenced and bioinformatically separated for the concurrent identification of the targeted mRNAs, binding sites and interacting miRNAs. The class of CLIP-Seq/CLASH experiments can reveal thousands of miRNA:gene interactions in each analysis and has significantly altered the scope and scale of relevant research projects.
An overview of available experimental techniques is presented in Table 1, along with short comments on their intended use, obtained results and expected throughput.
Table 1. Index of experimental techniques utilized for the identification of miRNA:gene interactions.
| Method | Throughput | Intended use |
|---|---|---|
| Reporter Genes (7) | Low | Validation of miRNA:UTR (or binding region) interaction |
| Northern Blotting (7) | Low | Relative effect of miRNA on mRNA levels |
| qPCR (7) | Low | Quantification of miRNA effect on mRNA levels |
| Western Blot (7) | Low | Relative assessment of miRNA effect on protein concentration |
| ELISA (7) | Low | Quantification of miRNA effect on protein concentration |
| 5′ RLM-RACE (7) | Low | Identification of cleaved mRNA targets |
| Microarrays (7) | High | High-throughput assessment of miRNA effect on mRNA expression |
| RNA-Seq (7) | High | High-throughput assessment of miRNA effect on mRNA expression |
| Quantitative Proteomics (e.g. pSILAC (9) | High | High-throughput assessment of miRNA effects on protein concentration |
| AGO-IP | High | Identification of enriched transcripts (miRNAs and mRNAs) in AGO immunoprecipitates |
| HITS-CLIP (10) | High | Sequencing of AGO binding regions on targeted transcripts |
| PAR-CLIP (11) | High | Sequencing of AGO binding regions on targeted transcripts |
| CLASH (14) / PAR-CLIP + Ligation (13) | High | Sequencing of AGO binding regions on targeted transcripts. Production of chimeric miRNA:mRNA reads for the identification of interacting pairs. |
| Biotin miRNA tagging (7) | High/Low | Pull-down of biotin-tagged miRNAs and estimation of bound transcript content using qPCR (Low yield), microarrays (High-throughput) and RNA-Seq (High-throughput) |
| IMPACT-Seq (25) | High | Pull-down of biotin-tagged miRNAs, identification of interacting pairs and binding regions. |
| PARE / Degradome-Seq (22) | High | High-throughput identification of cleaved mRNA targets |
| 3Life (24) | High | High-throughput reporter gene assay |
| miTRAP (23) | High | miRNA trapping by RNA baiting |
Databases of miRNA interactions
Low-yield and especially high-throughput experimental techniques have already identified hundreds of thousands of miRNA:gene interactions in different taxa, species, tissues, cell lines and experimental conditions. This wealth of information is fragmented and hidden in thousands of manuscripts, supplemental materials, figures and raw NGS data sets.
DIANA-TarBase was initially released in 2006 (15) and it was the first database aiming to catalog published experimentally validated miRNA:gene interactions. Since then, a handful of similar projects (16,17) index and map experimentally identified miRNA interactions utilizing manual article curation, in order to maintain a high quality level of database entries.
The sixth version of DIANA-TarBase (rel. December 2011) (8) inaugurated a new generation of such projects, incorporating for the first time novel methodologies, including CLIP-Seq experiments. The release of DIANA-TarBase v6.0 increased the then available target space by 16.5- to 175-fold, to 65 000 manually curated experimentally validated interactions. This radical increase in collected interactions was a prelude of the upcoming paradigm shift introduced by the new high-throughput methods. To our knowledge, miRTarBase (16) is the only similar database that has allocated resources to the curation of targets from such experiments. The last available version (v4.5, November 2013) hosts 51 460 entries derived from low-yield as well as high-throughput experiments. Other databases update less frequently or catalog significantly smaller sets of interactions. miRecords was first deployed in 2009 and focuses mostly on curating interactions from low-yield experiments (17). The last update of the database (April 2013) comprises 2705 interactions with 2028 derived from low-yield methodologies. There are also databases hosting CLIP-Seq sequencing results and/or that enable the online analysis of such data sets, such as StarBase (18) and CLIPZ (19). These databases differ significantly to the aforementioned repositories, since their aim is to catalog CLIP data sets and binding regions from any RNA binding protein.
Despite the evident advancements in the process of cataloging miRNA targets, the majority of studies examining miRNA regulatory networks and their effect on molecular pathways usually rely on in silico predictions, since they require increased numbers of interactions. Aim of TarBase v7.0, is to push the envelope further and to provide for the first time hundreds of thousands of high-quality manually curated experimentally validated miRNA:gene interactions, enhanced with the most detailed meta-data available to date. DIANA-TarBase currently indexes more than half a million entries, 9–250 times more than any other relevant database. All entries are accompanied with rich detailed meta-data that can also be used as search and filtering terms from the new application-like user interface. For instance, DIANA-TarBase v7.0 collects data regarding the experimental conditions, such as the exposition of cells to stressors, drugs or other agents, since these can alter miRNA regulatory networks. Another novel aspect of the database is its ability to include detailed information regarding the experimental methodologies utilized for the identification of each interaction, since experimental techniques cannot be considered as having equal information content.
METHODS AND RESULTS
Text mining pipeline for article selection
The selection of the most information-rich articles for manual curation is a complex process, since thousands of manuscripts published per year have ‘microRNA’ or ‘miRNA’ keywords in their abstract or title. DIANA-TarBase 6.0 introduced a text-mining assisted pipeline for identification of articles which would be subsequently subjected to manual curation. This pipeline has been significantly extended and enhanced, in order to be able to capture all the advancements in the experimental methodologies. The text mining algorithm has been iteratively fine-tuned based on the feedback of curators following the analysis of hundreds of manuscripts.
In brief, the subset of MedLine articles having the terms ‘microRNA’ or ‘miRNA’ (and variations) in their title, abstract, keywords or MeSH terms are selected for analysis by the text mining algorithm. Abstracts and publication meta-data are downloaded in XML format from MedLine and subjected to Named Entity Recognition. Gene mentions were initially identified using AIIAGMT (20). The pipeline was subsequently updated to utilize GNAT libraries and online services (21) for gene name tagging and normalization. An extensive in-house developed dictionary comprising all established, as well as novel experimental methodologies is utilized to recognize miRNAs, methods, important verbs and interaction terms. Sentences with a high probability for interaction (e.g. hosting gene, miRNA and interaction terms) are scored based on the methods found within the text. Highest scored articles will be forwarded for manual curation, as well as articles containing high-throughput methods relevant to miRNA function (e.g. AGO PAR-CLIP). The developed methodology has now been enhanced in order to be able to analyze freely available complete articles and meta-data from PubMed Central. This pipeline has diminished the probability of curators analyzing low or no interaction articles, which pose a significant overhead in manual curation processes.
Collected data
We have significantly increased the types of collected data and meta-data, in order to facilitate the extensive testing and validation of prediction algorithms, as well as to empower regulatory investigations with experimentally derived miRNA:gene interactions. Each interaction is now accompanied with detailed information regarding the performed experimental procedure, including tissue, cell type and condition. Furthermore, a more relaxed database schema permits the description of more complex experiments and interactions involving multiple cell types or even species (e.g. miRNA:gene interactions between the host and a viral miRNA or vice versa, experiments where a 3′UTR from one species is being tested using a cell type of a different species).
Until now, databases usually distinguished experimental protocols into basic categories (e.g. specific and high-throughput) or into a handful of major methodology classes (e.g. Sequencing, Proteomics, Blotting, etc.). The new database schema enables the characterization of each methodology with two identifiers: (i) a methodology class (12 classes) and (ii) a specific subtype (20 method subtypes). By utilizing twin-identifiers, it is now possible to distinguish two closely related methods that have different information content (e.g. biotin pull-down of miRNA targets + microarray transcript quantification versus biotin pull-down + qPCR transcript quantification).
A new field has been introduced to the database schema for marking interactions derived from chimeric reads from CLASH or modified CLIP-Seq experiments. These interactions have higher information content, since miRNA and mRNA sequences reside on the same read, enabling the accurate identification of both actors, as well as the exact site of the interaction. Even though these high-quality interactions are currently limited, the new database schema enables their detailed cataloging.
Specific attention was paid on archiving the exact binding site of each interaction, since such information is crucial for testing target prediction algorithms or for identifying regulatory regions on a transcript (e.g. deciphering the effect of a variant on a 3′UTR region). The curation pipeline was extended with tools and techniques that enabled the curators to identify targeted regions using any relevant information available within the manuscript or supplemental material (genomic/transcript coordinates, cloning primers, mutation sites, etc.). Any experimental information used by the curators for the identification of the targeted regions is kept within the database. Binding sites were also identified by analyzing an extensive array of CLIP-Seq methods. By including binding-site level data into the database, TarBase v7.0 can present positive/negative results from experimental validations of distinct binding sites on the same transcript.
Analysis of raw data
In cases where raw high-throughput data were available, curators obtained the relevant data sets, commented each library with extensive metadata and marked them for reanalysis in order to maintain optimal quality standards for all identified interactions. Specifically, data available from online repositories or supplemental materials of 1 CLASH, 31 PAR-CLIP and 122 HITS-CLIP libraries were analyzed and included in the database.
The CLIP-Seq analysis has been performed using an in-house developed pipeline. Regions formed by at least five overlapping reads were included to the analysis. For PAR-CLIP data, peaks containing adequate T-to-C (sense strand) or A-to-G (antisense strand) incorporation were selected. At least two transitions in the same position for peaks with less than 50 reads were required, while for the remaining regions we applied the threshold of >5%, as indicated by Hafner et al. (11). For all CLIP-Seq data sets having replicates, a peak had to be present in at least two replicates in order to be considered as valid. Where available, top expressed miRNAs were retrieved from the original publication. In all other instances, we analyzed publically available small-RNA-Seq libraries derived from the relevant cell lines. miRNA:gene interactions were inferred using a CLIP-peak-guided MRE search algorithm considering the miRNA:mRNA binding type, binding free energy, MRE conservation and AU flanking content.
Changes over 50% were utilized as a threshold for microarray and biotin pull-down experiments. In cases where replicates were available, an interaction had to be present in at least two replicates, in order to be included to the database.
Database statistics
The database comprises more than half a million interactions spanning 24 species, a 9- to 250-fold increase compared to TarBase 6.0 and other manually curated databases, including miRTarBase and miRecords. The database encompasses interactions derived from the widest variety of experiments to date, which are performed utilizing 28 different experimental techniques, 356 cell types and 59 different tissues. Results from all methodologies presented in Table 1 and their variations are included to the database. The number of targets derived from major method classes is depicted in Figure 1. Importantly, we have paid significant attention to curate articles utilizing highly specific low-yield techniques, such as reporter genes, as well as state-of-the-art methodologies, such as CLIP-Seq and CLASH experiments. The updated database contains more than 7500 interactions derived from specific techniques (4-fold increase versus TarBase v6.0) and more than 500 000 interactions derived from high-throughput experiments (9-fold increase versus TarBase v6.0). Specifically, DIANA-TarBase v7.0 incorporates data derived from 154 CLIP-Seq/CLASH data sets, as well as more than a hundred other high-throughput data sets including Degradome-Seq (22), AGO-IP (7), biotin pull-down (7), miTRAP (23), 3′Life (24) and IMPACT-Seq (25).
Figure 1.
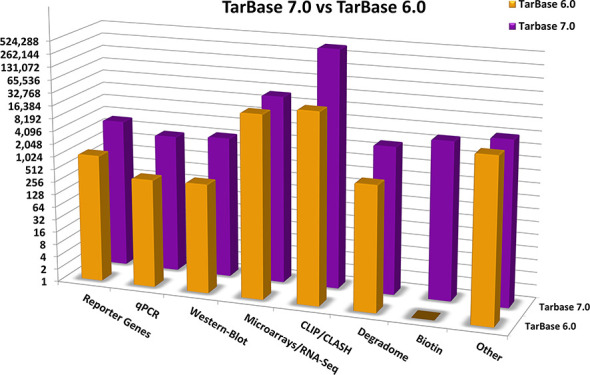
Entries per methodology for TarBase v7.0 and TarBase v6.0. The y-axis (number of entries) is in log2 scale and each mark signifies doubling of available entries.
INTERFACE
Querying the database
The database query can be performed by entering any combination of miRNA and/or gene names or supported identifiers (ENSEMBL (26) gene IDs for genes and miRBase (27) MIMAT accessions for miRNAs). If genes and miRNAs are concurrently provided, TarBase will return all indexed interactions of the selected miRNAs with any of the provided genes.
The interface (Figure 2) is designed around the new database schema, in order to cater to users extended meta-data regarding each interaction. Users can easily identify positive or negative experimental results, the utilized experimental methodology, experimental conditions including cell/tissue type and treatment. The new interface provides also advanced information ranging from the binding site location, as identified experimentally as well as in silico, to the primer sequences used for cloning experiments.
Figure 2.
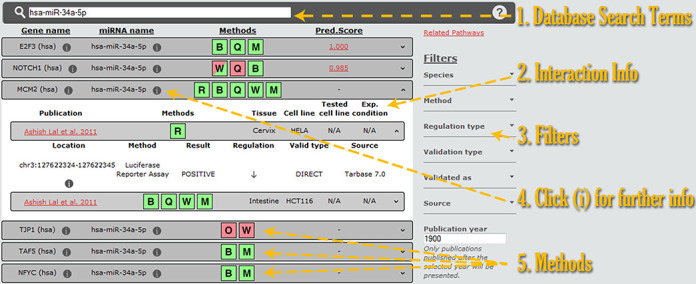
Screen-shot depicting the DIANA-TarBase v7.0 interface. Users can enter the query terms in the simple search box (1). Interaction information is presented below (2), while further details are accessible by expanding the result panel or by selecting the information links (4). All results are color-coded, with green and red showing positive and negative experimental outcomes, respectively (5). Mixed results are presented using both colors. Users can filter the query results using any combination of the filtering options (3).
Since January 2014, DIANA-TarBase has been integrated to the official ENSEMBL (26) distribution. All TarBase entries having binding site coordinates can be explored directly from the ENSEMBL genome browser. Each DIANA-TarBase entry has a link pointing to the relevant browser view and coordinates, facilitating user interaction with both databases.
This version is also seamlessly incorporated to other DIANA-Tools. The DIANA-TarBase v7.0 user can easily perform a pathway analysis for the miRNA (s) under investigation, identify their predicted targets or examine if they have been identified experimentally or in silico to target long non-coding RNAs, using DIANA-miRPath v2.0 (28), microT-CDS (29) and LncBase (30), respectively.
Advanced searching and filtering
TarBase v7.0 supports advanced real-time search and filtering. All relevant options have been incorporated in the main result screen, in order to enable users to easily filter and query the database. The provided search and filtering options include: miRNA/gene combinations, species, experimental methodology class and subtype, type of regulation and validation, selection of positive or negative experimental results, year of publication and DIANA-microT-CDS threshold for interactions which are also predicted in silico. As in the previous version, DIANA-TarBase also integrates interactions from the latest available versions of external databases, including miRTarBase and miRecords. Users can easily filter results and include/exclude external sources or data derived from previous TarBase versions. All statistics provided within the manuscript do not include external or integrated interactions.
CONCLUSION
DIANA-TarBase has been completely redesigned in every aspect for its seventh version. The utilized database has been significantly extended with richer meta-data and detailed information for each interaction, while the interface now supports advanced real-time querying and result filtering. During the past few years NGS methodologies have revolutionized almost every aspect of biological research. Novel NGS-based high-throughput miRNA target identification techniques have enabled the identification of thousands of interactions present in specific cell types or experimental conditions. By analyzing more than 150 raw NGS data sets and extracting interactions from hundreds of meticulously curated articles, DIANA-TarBase v7.0 is the first relevant database to break the barrier of 100 000 entries by indexing more than half a million interactions in 24 species, 9–250 times more than any other manually curated database. This wealth of information can be utilized for exploratory studies, enforcing or even at cases substituting in silico predicted interactions.
Acknowledgments
We would like to thank Serafeim Chatzopoulos, Penny Georgiou and Filopoimin Lykokanellos for the IT support, as well as Simos Kazantzidis, Evangelos Karatzas and Anaxagoras Fotopoulos for their contribution to the initial version of the database.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
‘TOM’, ‘ARISTEIA’ Action of the ‘OPERATIONAL PROGRAMME EDUCATION AND LIFELONG LEARNING’, General Secretariat for Research and Technology, Ministry of Education, Greece, European Social Fund (ESF) and National Resources; ‘MEDA’, Development Grants For Research Institutions – KRIPIS, General Secretariat for Research and Technology, Ministry of Education, Greece, European Regional Development Fund. Funding for open access publication charges: TOM’, ‘ARISTEIA’ Action of the ‘OPERATIONAL PROGRAMME EDUCATION AND LIFELONG LEARNING’,General Secretariat for Research and Technology, Ministry of Education, Greece, European Social Fund (ESF) and National Resources.
Conflict of interest statement. None declared.
REFERENCES
- 1.Das S., Ferlito M., Kent O.A., Fox-Talbot K., Wang R., Liu D., Raghavachari N., Yang Y., Wheelan S.J., Murphy E., et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res. 2012;110:1596–1603. doi: 10.1161/CIRCRESAHA.112.267732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gagnon K.T., Li L., Chu Y., Janowski B.A., Corey D.R. RNAi Factors Are Present and Active in Human Cell Nuclei. Cell Reports. 2014;6:211–221. doi: 10.1016/j.celrep.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang V., Place R.F., Portnoy V., Wang J., Qi Z., Jia Z., Yu A., Shuman M., Yu J., Li L.-C. Upregulation of Cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Research. 2012;40:1695–1707. doi: 10.1093/nar/gkr934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsui M., Chu Y., Zhang H., Gagnon K.T., Shaikh S., Kuchimanchi S., Manoharan M., Corey D.R., Janowski B.A. Promoter RNA links transcriptional regulation of inflammatory pathway genes. Nucleic Acids Research. 2013;41:10086–10109. doi: 10.1093/nar/gkt777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huntzinger E., Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 6.Vlachos I.S., Hatzigeorgiou A.G. Online resources for miRNA analysis. Clin Biochem. 2013;46:879–900. doi: 10.1016/j.clinbiochem.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Thomson D.W., Bracken C.P., Goodall G.J. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011;39:6845–6853. doi: 10.1093/nar/gkr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vergoulis T., Vlachos I.S., Alexiou P., Georgakilas G., Maragkakis M., Reczko M., Gerangelos S., Koziris N., Dalamagas T., Hatzigeorgiou A.G. TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res. 2012;40:D222–229. doi: 10.1093/nar/gkr1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selbach M., Schwanhausser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 10.Chi S.W., Zang J.B., Mele A., Darnell R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jr, Jungkamp A.C., Munschauer M., et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kishore S., Jaskiewicz L., Burger L., Hausser J., Khorshid M., Zavolan M. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods. 2011;8:559–564. doi: 10.1038/nmeth.1608. [DOI] [PubMed] [Google Scholar]
- 13.Grosswendt S., Filipchyk A., Manzano M., Klironomos F., Schilling M., Herzog M., Gottwein E., Rajewsky N. Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol Cell. 2014;54:1042–1054. doi: 10.1016/j.molcel.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helwak A., Tollervey D. Mapping the miRNA interactome by cross-linking ligation and sequencing of hybrids (CLASH) Nat Protoc. 2014;9:711–728. doi: 10.1038/nprot.2014.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sethupathy P., Corda B., Hatzigeorgiou A.G. TarBase: A comprehensive database of experimentally supported animal microRNA targets. RNA. 2006;12:192–197. doi: 10.1261/rna.2239606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu S.D., Tseng Y.T., Shrestha S., Lin Y.L., Khaleel A., Chou C.H., Chu C.F., Huang H.Y., Lin C.M., Ho S.Y., et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42:D78–85. doi: 10.1093/nar/gkt1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao F., Zuo Z., Cai G., Kang S., Gao X., Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37:D105–110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J.-H., Liu S., Zhou H., Qu L.-H., Yang J.-H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Research. 2014;42:D92-D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khorshid M., Rodak C., Zavolan M. CLIPZ: a database and analysis environment for experimentally determined binding sites of RNA-binding proteins. Nucleic Acids Res. 2011;39:D245–252. doi: 10.1093/nar/gkq940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith L., Tanabe L., Ando R., Kuo C.-J., Chung I.-F., Hsu C.-N., Lin Y.-S., Klinger R., Friedrich C., Ganchev K., et al. Overview of BioCreative II gene mention recognition. Genome Biology. 2008;9:S2. doi: 10.1186/gb-2008-9-s2-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hakenberg J., Gerner M., Haeussler M., Solt I., Plake C., Schroeder M., Gonzalez G., Nenadic G., Bergman C.M. The GNAT library for local and remote gene mention normalization. Bioinformatics. 2011;27:2769–2771. doi: 10.1093/bioinformatics/btr455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.German M.A., Luo S., Schroth G., Meyers B.C., Green P.J. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat Protoc. 2009;4:356–362. doi: 10.1038/nprot.2009.8. [DOI] [PubMed] [Google Scholar]
- 23.Braun J., Misiak D., Busch B., Krohn K., Hüttelmaier S. Rapid identification of regulatory microRNAs by miTRAP (miRNA trapping by RNA in vitro affinity purification) Nucleic Acids Research. 2014;42:e66. doi: 10.1093/nar/gku127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolter J.M., Kotagama K., Pierre-Bez A.C., Firago M., Mangone M. 3′LIFE: a functional assay to detect miRNA targets in high-throughput. Nucleic Acids Res. 42:2015, e132. doi: 10.1093/nar/gku626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan S.M., Kirchner R., Jin J., Hofmann O., McReynolds L., Hide W., Lieberman J. Sequencing of Captive Target Transcripts Identifies the Network of Regulated Genes and Functions of Primate-Specific miR-522. Cell Rep. 2014;8:1225–1239. doi: 10.1016/j.celrep.2014.07.023. [DOI] [PubMed] [Google Scholar]
- 26.Flicek P., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2014. Nucleic Acids Research. 2014;42:D749-D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Research. 2014;42:D68-D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vlachos I.S., Kostoulas N., Vergoulis T., Georgakilas G., Reczko M., Maragkakis M., Paraskevopoulou M.D., Prionidis K., Dalamagas T., Hatzigeorgiou A.G. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012;40:W498–504. doi: 10.1093/nar/gks494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paraskevopoulou M.D., Georgakilas G., Kostoulas N., Vlachos I.S., Vergoulis T., Reczko M., Filippidis C., Dalamagas T., Hatzigeorgiou A.G. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;41:W169–173. doi: 10.1093/nar/gkt393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paraskevopoulou M.D., Georgakilas G., Kostoulas N., Reczko M., Maragkakis M., Dalamagas T.M., Hatzigeorgiou A.G. DIANA-LncBase: experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Res. 2013;41:D239–245. doi: 10.1093/nar/gks1246. [DOI] [PMC free article] [PubMed] [Google Scholar]