


Abstract
Microbial genome sequencing projects produce numerous sequences of deduced proteins, only a small fraction of which have been or will ever be studied experimentally. This leaves sequence analysis as the only feasible way to annotate these proteins and assign to them tentative functions. The Clusters of Orthologous Groups of proteins (COGs) database (http://www.ncbi.nlm.nih.gov/COG/), first created in 1997, has been a popular tool for functional annotation. Its success was largely based on (i) its reliance on complete microbial genomes, which allowed reliable assignment of orthologs and paralogs for most genes; (ii) orthology-based approach, which used the function(s) of the characterized member(s) of the protein family (COG) to assign function(s) to the entire set of carefully identified orthologs and describe the range of potential functions when there were more than one; and (iii) careful manual curation of the annotation of the COGs, aimed at detailed prediction of the biological function(s) for each COG while avoiding annotation errors and overprediction. Here we present an update of the COGs, the first since 2003, and a comprehensive revision of the COG annotations and expansion of the genome coverage to include representative complete genomes from all bacterial and archaeal lineages down to the genus level. This re-analysis of the COGs shows that the original COG assignments had an error rate below 0.5% and allows an assessment of the progress in functional genomics in the past 12 years. During this time, functions of many previously uncharacterized COGs have been elucidated and tentative functional assignments of many COGs have been validated, either by targeted experiments or through the use of high-throughput methods. A particularly important development is the assignment of functions to several widespread, conserved proteins many of which turned out to participate in translation, in particular rRNA maturation and tRNA modification. The new version of the COGs is expected to become an important tool for microbial genomics.
INTRODUCTION
The constantly accelerating pace of microbial genome sequencing continues to flood public databases with sequences of deduced proteins, only a small fraction of which has been ever studied experimentally or could be studied in detail any time soon. The only feasible way to assign functions to these proteins is to predict them through computational analysis. The Clusters of Orthologous Groups of proteins (COGs) database, first created in 1997, has been a popular tool for functional annotation. Its success was based on several key factors. First, COGs relied on the analysis of complete microbial genomes (proteomes), which allowed reliable assignment of orthologs and paralogs for most genes using a simple approach based on the search of triangles of bidirectional best hits (1). This approach allowed both recognition of distant homologs and separation of closely related paralogs. Another key factor was the use of a family-based approach whereby the function(s) of the characterized member(s) of the protein family (COG) was harnessed to assign function(s) to the entire family and describe the range of the potential functions when there were more than one. Finally, the membership of the COGs and the functional annotation were subject to careful manual curation which aimed at assigning biological functions to each COG while avoiding annotation errors and overpredictions. In 2003, COGs have been incorporated into the NCBI's Conserved Domain Database (CDD; (2,3)). Subsequently, COG annotations were included into the SEED database (4) and the possibility to compare newly sequenced genomes against COGs had been provided by the Integrated Microbial Genomes (IMG) system at the DOE's Joint Genome Institute (5).
In contrast to the protein domain databases, such as Pfam, SMART or CDD (3,6–7), most entries in the COG database were full-length proteins, which offered a distinct perspective at the microbial protein content and its evolution. In some cases, splitting proteins into separate domains was deliberately avoided, allowing a better description of, for example, two-component response regulators, which either consist of a stand-alone phosphoacceptor receiver (REC) domain or combine this domain with a variety of DNA-binding, RNA-binding, ligand-binding or enzymatic domains (8,9). However, even assigning different types of response regulators to different COGs did not fully solve the problem of their classification, owing to the sheer number of these proteins encoded in nearly every microbial genome (8,9). Nevertheless, inclusion of full-length proteins has been a major advantage of the COG approach; in the current versions of CDD and InterPro, full-protein entries are provided by other databases, such as TIGRFAMs or Panther (3,10–12).
The COG database went through several updates, which gradually increased its genome coverage to 62 organisms, including 46 bacterial, 13 archaeal and three eukaryotic genomes (13–15), and has been widely used in the microbial genomic community. Gene assignment to COGs provided for a variety of comparative-genomic studies, and COG functional classification of the encoded proteins has been adopted as one of the required descriptors of newly sequenced genomes (16), in particular by the journal ‘Standards in Genome Science’ that is dedicated to the publication of new genome descriptions. However, the COGs have not been updated in full since 2003, which obviously rendered (almost) all COGs incomplete and many COG annotations obsolete. Certain COG names have been updated by the authors of this work on an ad hoc basis and these corrections have been included in the CDD (3). Furthermore, in the interim, the COG-making algorithm and software have been improved and several focused offshoots of the COG projects have been developed. These specialized versions of the COGs included clusters of orthologous genes for Cyanobacteria, Lactobacillaceae and, particularly, Archaea (17–20). The latter version of the COGs, named arCOGs, has been continuously updated and manually curated (19,20). Nevertheless, the incompleteness of the COG membership and the absence of up-to-date COG annotations have become major impediments to the use of this system in comparative genomics. A major extension of the COGs is implemented in the EggNOG database, with an increased number of genomes included and new clusters of orthologs (denoted NOGs, after Non-supervised Orthologous Groups); however, EggNOG is completely automatic, without manual supervision of the cluster membership or annotation (21).
We report here a major update of the COGs that included assignment to the pre-existing COGs members from 711 genomes that represent the diversity of bacteria and archaea and re-evaluation of the COG annotation that resulted in a name change for more than half of all COGs. Although many of these changes are merely stylistic, aimed at bringing all COG names to a common format, some reflect experimental validation of predictions, whereas others involve functional annotation of a previously uncharacterized COG or reassignment of a COG to a new functional category. The revised version of the COGs is expected to become an important tool for microbial genomics.
CHANGES IN THE COG DATABASE
Compared to the previous versions of the COG database, the current release provides substantially expanded genome coverage and updated annotation of the COGs. However, the new release offers less stand-alone functionality than the previous ones and relies instead on NCBI databases and tools (22).
The organisms are sorted according to the NCBI Taxonomy database (23), and the organism names are directly linked to the respective entries in that database. The only exception is that mycoplasmas are still listed in class Mollicutes within the phylum Firmicutes, not as a separate phylum Tenericutes, as proposed in the recent taxonomic update (24) but questioned by some phylogenetic studies (25–27). The organism names in each COG are abbreviated to a six-letter code that consists of three first letters of the organism's genus name and three letters (or two letters and a number) from the species name (Figure 1). Each organism code is linked to the respective entry in the NCBI Taxonomy database (23).
Figure 1.
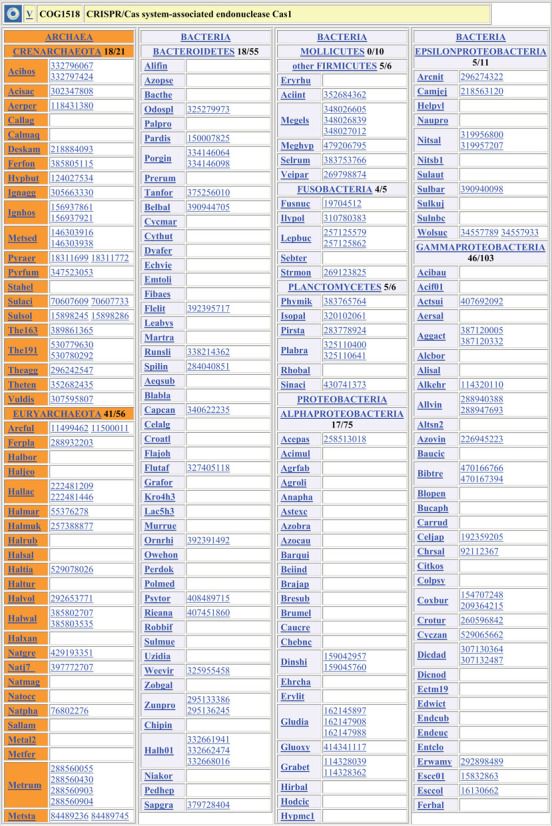
A protein family (COG) page in the COG database. For each phylum (or for Firmicutes and Proteobacteria, for each class), the numbers indicate the number of organisms that have a COG member and the total number of organisms from that phylum (class) in the COG database. Each COG member is represented by its gene index (gi) number in the NCBI Protein database (22) and is linked to the respective entry in RefSeq database (28). The phyla (classes) with no COG members are collapsed. The phylum, class and organism names are linked to the respective entries in the NCBI Taxonomy database (23).
COG format
Each gene entry in a COG is now denoted by its gene index (gi) number in the NCBI protein database and is linked to the respective entry in the NCBI's RefSeq database (28) which provides links to the nucleotide sequence of the encoding gene in GenBank (29), its chromosomal location in Entrez Gene (30), protein domain organization in the CDD (3), known or predicted protein structure, if available, in the NCBI's Molecular Modeling Database (31), pertinent references in PubMed and PubMed Central and a variety of other tools (22). Accordingly, the new version of the COGs does not include sequences of the 1.96 million genes included in the current release and does not show their alignments or phylogenetic trees.
Organisms covered
The new release concentrates on prokaryotes (bacteria and archaea) and no longer includes genes from two yeasts and a microsporidian that have been present in the previous versions. The COG assignments of protein-coding genes from these organisms are still available on the NCBI FTP site in the ftp://ftp.ncbi.nih.gov/pub/COG/COG/whog file. In addition to removing these three eukaryotes, the genome list has been trimmed by removing duplicate entries for Escherichia coli O157:H7 (strain EDL933), Helicobacter pylori (strain J99), Mycobacterium tuberculosis (strain CDC1551) and Neisseria meningitidis (strain Z2491). The remaining 58 organisms from the previous release (including two strains of E. coli, K-12 and O157:H7) were retained and supplemented with 653 organisms including 70 archaea and 583 bacteria. These organisms are classified into three archaeal phyla (Crenarchaeota, Euryarchaeota and Thaumarchaeota) and 14 bacterial phyla (Acidobacteria, Actibobacteria, Bacteroidetes, Chlamydia, Chlorobi, Chloroflexi, Cyanobacteria, Deinococcus-Thermus, Firmicutes, Fusobacteria, Proteobacteria, Spirochaetes, Synergistetes and Thermotogae). Several organisms that do not belong to these phyla have been included in ‘Other archaea’ and ‘Other bacteria’ groupings. The two largest phyla, Firmicutes and Proteobacteria, are further divided into classes.
As a result of removing the eukaryotic species, 178 COGs that contained exclusively eukaryotic proteins and 64 COGs that included only one or two prokaryotic genes were removed from the COG database, leaving a total of 4631 COGs. The removed COGs can still be found at the NCBI FTP site mentioned above.
COG pipeline
Sequences of the proteins from 4873 COGs of the 2003 COG version (15) were aligned using the MUSCLE program (32); these multiple alignments were used to derive PSI-BLAST (33) position-specific scoring matrices (PSSMs). PSI-BLAST searches with COG-derived PSSMs were used to assign annotated proteins from 711 genomes to COGs.
Except for these essential modifications, changes to the COGs were kept to the minimum. The list of functional categories was expanded to 26, with the last remaining letter ‘X’ used to denote phage-derived proteins, transposases and other mobilome components. Many of these proteins have been previously assigned to the category L ‘DNA replication, recombination and repair’ which was hardly an appropriate placing for them. It should be noted that this new category includes many proteins whose functions are uncharacterized or poorly characterized.
COG STATISTICS
The current update of COGs does not include any newly created COGs. The removal of 242 COGs with predominantly yeast proteins left 4631 COGs in the system. The great majority of these, 4215 COGs, include less than 1000 genes. However, there are five COGs that include more 10 000 proteins each: COG0457 ‘Tetratricopeptide (TPR) repeat’, COG0583 ‘DNA-binding transcriptional regulator, LysR family’, COG0745 ‘DNA-binding response regulator, OmpR family, contains REC and winged-helix (wHTH) domain’, COG1028 ‘NAD(P)-dependent dehydrogenase, short-chain alcohol dehydrogenase family’, and COG1309 ‘DNA-binding transcriptional regulator, AcrR family’. Fifty-five more COGs contain between 3000 and 9000 genes, making them difficult to handle and display online.
The COGs are classified into 26 functional categories, with the largest numbers of COGs, 507 and 959, respectively, still assigned to the categories R ‘General function prediction only’ and S ‘Function unknown’. Analysis of the total genome coverage of various bacterial and archaeal phyla shows that even the limited set of 4631 COGs includes between 60 and 86% of the respective proteomes (Figure 2). The fraction of the total proteome with specific functional annotation (excluding R- and S-COG categories) varies from 51–53% in Thaumarchaeota, Cyanobacteria and Planctomycetes to 72–76% in Aquificae, Thermotogae and Synergistetes. These numbers also show that, despite substantial progress in understanding the core proteome contents of prokaryotic genomes, a sizable fraction of proteins encoded in any given genome remains without functional annotation.
Figure 2.
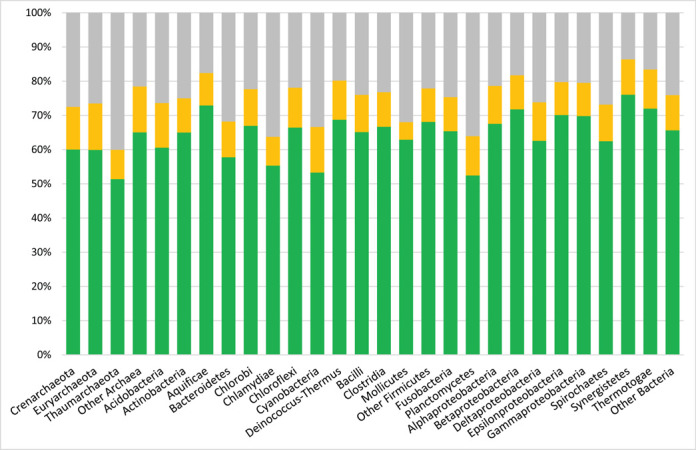
COG coverage of various bacterial phyla. The columns represent the average fraction of proteins from the organisms in the given phylum that are not included in COGs (gray), assigned to the R or S categories in COGs (yellow) or assigned to other COG functional categories (green). For Firmicutes and Proteobacteria, coverage is shown at the class level.
IMPROVED COG ANNOTATION
In the previous releases, COG names had been assigned with the goal of providing the most complete description of the range of functions (demonstrated or predicted) within the respective protein family (COG). Although these COG names were not always suitable for functional annotation of individual proteins, in practice, this has been their most common use. With that in mind, the current version has undergone a variety of changes, both substantive and merely stylistic, to simplify that task.
The existing COG annotations were verified by comparing them to the annotations of the COG members in UniProt and RefSeq databases (28,34), protein domain names in the Pfam, InterPro and CDD databases (3,7,12), and, for COGs that contained representatives of the respective model organisms, functional assignments from EcoGene, CyanoBase and Pseudomonas Genome Database (35–37). For COGs that (previously) had yeast members, the annotations from Saccharomyces Genome Database (SGD) (38) have been checked as well. Finally, the ‘y’ gene names, which typically indicate the absence of a known function, have been searched against PubMed and PubMed Central (22). In addition, the annotations for COGs that are specific for archaea have been reconciled with those in the arCOG database (19,20).
Stylistically, COGs annotations were adjusted to satisfy a simple convention: the COG name represents the protein function (to the degree it is known or predicted), followed by its constitutive domains (if these domains are sufficiently widespread and are likely to produce hits with non-orthologous proteins), followed by a family (or superfamily) assignment, where appropriate. Examples of such COG annotations are listed in Table 1 and Supplementary Table S1. We expect that these new, uniform annotations will make COG-based annotation of new genomes more accurate and straightforward.
Table 1. Examples of newly annotated COGs in the ‘Translation…’ (J) category.
| COG no. | 2003 funca | Gene | New COG name |
|---|---|---|---|
| COG0144 | J | sun | 16S rRNA C967 or C1407 C5-methylase RsmB/F |
| COG0275 | M | yabC | 16S rRNA C1402 N4-methylase RsmH |
| COG0313 | R | yraL | 16S rRNA C1402 (ribose-2′-O)-methylase RsmI |
| COG0357 | M | gidB | 16S rRNA G527 N7-methylase RsmI/GidB |
| COG0742 | L | yhhF | 16S rRNA G966 N2-methylase RsmD |
| COG1385 | S | yggJ | 16S rRNA U1498 N3-methylase RsmE |
| COG0116 | L | ycbY1 | 23S rRNA G2445 N2-methylase RlmL |
| COG1092 | R | yccW | 23S rRNA G2069 N7-methylase RlmK or C1962 C5-methylase RlmI |
| COG1576 | S | ybeA | 23S rRNA pseudouridine1915 N3-methylase RlmH |
| COG3129 | R | ybiN | 23S rRNA A1618 N6-methylase RlmF |
| COG2961 | R | yhiR | 23S rRNA A2030 N6-methylase RlmJ |
| COG2933 | R | ygdE | 23S rRNA C2498 (ribose-2′-O)-methylase RlmM |
| COG0820 | R | yfgB | 23S rRNA A2503 and tRNA A37 C2-methylase RlmN |
| COG2603 | R | ybbB | tRNA 2-selenouridine synthase SelU, contains rhodanese domain |
| COG0802 | R | yjeE | tRNA A37 threonylcarbamoyladenosine modification protein TsaE |
| COG1179 | H | ygdL | tRNA A37 threonylcarbamoyladenosine dehydratase TcdA |
| COG1214 | O | yeaZ | tRNA A37 threonylcarbamoyladenosine modification protein TsaB |
| COG0009 | J | yrdC | tRNA A37 threonylcarbamoyl synthetase subunit TsaC/SUA5/YrdC |
| COG0533 | O | ygjD | tRNA A37 threonylcarbamoyltransferase TsaD |
| COG0220 | R | yggH | tRNA G46 methylase TrmB |
| COG4121 | S | yfcK | tRNA U34 5-methylaminomethyl-2-thiouridine-forming methyltransferase MnmC |
| COG0445 | D | gidA | tRNA U34 5-carboxymethylaminomethyl modifying enzyme MnmG/GidA |
| COG0486 | R | thdF | tRNA U34 5-carboxymethylaminomethyl modifying GTPase MnmE/TrmE |
| COG0585 | S | ygbO | tRNA(Glu) U13 pseudouridine synthase TruD |
| COG0037 | D | mesJ | tRNA(Ile)-lysidine synthase TilS/MesJ |
| COG1444 | R | ypfI | tRNA(Met) C34 acetyltransferase TmcA |
| COG4123 | R | yfiC | tRNA1(Val) A37 N6-methylase TrmN |
| COG0590 | F | yfhC | tRNA(Arg) A34 adenosine deaminase TadA/CumB |
| COG1720 | S | yaeB | tRNA(Thr-GGU) A37 N-methylase TsaA |
| COG0799 | S | ybeB | Ribosomal silencing factor RsfS, regulates association of 30S and 50S subunits (Iojap protein) |
| COG1690 | S | rtcB | RNA-splicing ligase RtcB, repairs tRNA damage |
| COG0684 | H | menG | Regulator of RNase E activity RraA |
| COG3076 | S | yjgD | Regulator of RNase E activity RraB |
| COG1944 | S | ycaO | Ribosomal protein S12 methylthiotransferase accessory factor YcaO |
| COG2001 | S | yabB | MraZ, inhibitor of RsmH methyltransferase activity |
| COG2850 | S | ycfD | Ribosomal protein L16 Arg81 hydroxylase, contains JmjC domain |
| COG3101 | S | yfcM | Elongation factor P hydroxylase (EF-P beta-lysylation pathway) |
| COG4575 | S | elaB | Membrane-anchored ribosome-binding protein, inhibits growth in stationary phase, ElaB/YqjD/DUF883 family |
| COG4680 | S | ygjN | mRNA-degrading endonuclease (mRNA interferase) HigB, toxin component of the HigAB toxin–antitoxin module |
| COG3041 | S | yafQ | mRNA-degrading endonuclease (mRNA interferase) YafQ, toxin component of the YafQ–DinJ toxin–antitoxin module |
| COG2606 | S | ybaK | Cys-tRNA(Pro) deacylase, prolyl-tRNA editing enzyme EbsC |
aFunctional category previously assigned to the COG: J, translation, ribosomal structure and biogenesis; D, cell cycle control, cell division, chromosome partitioning; F, nucleotide transport and metabolism; H, coenzyme transport and metabolism; L, cell wall/membrane/envelope biogenesis; O, post-translational modification, protein turnover, chaperones; R, general function prediction only; S, function unknown.
Naming functionally diverse COGs
Although sequence conservation among the proteins within a COG typically implies functional similarity, there are cases when members of the same COG perform dramatically different (biological) functions. New functions typically evolve in a particular lineage and often involve change (or even loss) of the respective enzymatic activity (39). In such cases, we list both (or all) known function, separating them with either a slash or with a conjunction ‘and’ or ‘or’. One such example is COG0252 in which the bacterial members possess L-asparaginase activity, whereas the archaeal members function as subunits of the four-protein complex involved in the synthesis of glutaminyl-tRNA (40). Accordingly, this COG is named ‘L-asparaginase/archaeal Glu-tRNAGln amidotransferase subunit D’. Similarly, in COG0816, the RNase H-fold protein YqgF is predicted to function as a Holliday junction resolvase in firmicutes and mycoplasmas, but is also involved in anti-termination at Rho-dependent terminators (41,42); all this information is reflected in the COG name. Other examples include COG0608 ‘Single-stranded DNA-specific exonuclease RecJ/archaeal DNA replication initiation protein CDC45’ (43), COG2132 ‘Multicopper oxidase with three-cupredoxin domains, includes cell division protein FtsP and spore coat protein CotA’, COG0455 ‘MinD-like ATPase involved in chromosome partitioning or flagellar assembly’, COG2141 ‘Flavin-dependent oxidoreductase, luciferase family, includes alkanesulfonate monooxygenase SsuD and F420:5,10-methylene tetrahydromethanopterin reductase’ and several other COGs with similarly long and complex names.
Annotation of functionally uncharacterized COGs
Highlighting widespread protein families (COGs) for which the biological functions remain unknown is vital to the progress of microbial genome annotation and more broadly genome-based microbiology (44–47). In the COG database, COGs of unknown function are assigned to the S category and are named ‘Uncharacterized protein’ with additional characterization based on either predicted membrane localization or widespread distribution. For consistency, the ‘Uncharacterized conserved protein’ designation was reserved for those COGs that include at least 100 proteins from at least two different phyla (a more detailed analysis of protein conservation in bacterial and archaeal COGs is currently in preparation). For COGs that include proteins from one of the two best-studied model organisms, E. coli and/or Bacillus subtilis, these names were supplemented by the respective ‘Y’ designations of the respective genes. Furthermore, such COGs were cross-referenced with the two other resources that list uncharacterized protein families, namely Uncharacterized Protein Families (UPFs; http://www.uniprot.org/docs/upflist) in UniProt and Domains of Unknown Function (DUFs) in Pfam (7,34), and respective family designations have been added to many COG names. As a result, typical names for S-COGs include ‘Uncharacterized conserved protein YbjQ, UPF0145 family’, ‘Uncharacterized conserved protein YggU, DUF167 family’, ‘Uncharacterized membrane protein YbhN, UPF0104 family’, ‘Uncharacterized protein YjgD, DUF1641 family’ and so on. This category also includes several named COGs, for which the absence of known specific biological function is indicated in parentheses, for example, COG1915 ‘Pheromone shutdown protein TraB, contains GTxH motif (function unknown)’. The complete COG list, available on the http://www.ncbi.nlm.nih.gov/COG/ site, provides the number of organisms and proteins included in each COG, allowing one to search for widespread uncharacterized genes.
LESSONS FROM COG REANNOTATION
Because most COG names have not been updated since the last COG release in 2003, the present COG reannotation project offered a unique opportunity to obtain an estimate of the accuracy of the original COG assignments and evaluate the progress in microbial genome annotation over the past 12 years.
Whenever COG names were changed, this change was scored as either: (i) essentially stylistic, or (ii) validation of the computationally predicted function, or (iii) substantial improvement in functional annotation, or (iv) correction of a previous erroneous annotation. The last category was found to represent less than 0.5% of the total COG names. The reasons for erroneous assignments included misleading experimental reports, failure to recognize distinct protein families, assignment of the function to a wrong domain in a multidomain protein, as well as human error (Supplementary Table S1). A common error, for example, involved routinely annotating proteins that carried predicted Fe-S clusters as ‘Fe-S oxidoreductases’; several families of such proteins have been subsequently shown to belong to the radical S-Adenosyl Methionine (SAM) superfamily, where the Fe-S clusters catalyze a variety of reactions but are not involved in redox processes (48,49).
Apart from the small number of mis-assignments, now corrected, tentative functional annotations of many COGs previously placed in the R category ‘General function prediction only’ have been verified, either by direct experiments or through the use of high-throughput methods. In about 200 cases, predicted methyltransferases, oxidoreductases, ATPases, GTPases, DNA- or RNA-binding proteins or membrane permeases could be assigned a (more) specific function in line with the previous annotation. Examples include various rRNA methylases (Table 1), cell division proteins, proteins involved in the biogenesis of the cell envelope and several other functional groups (50–64; Table 2).
Table 2. Functional category reassignment for poorly characterized COGs.
| Example | ||||
|---|---|---|---|---|
| Changea | Number of COGs | COG no. | Gene | New COG name, reference |
| S to known function | 294 | COG3681 | cdsB (yhaM) | L-cysteine desulfidase (50,51) |
| S to J | 37 | COG1617 | – | tRNA threonylcarbamoyladenosine modification (KEOPS) complex, Cgi121 subunit (52) |
| S to K | 25 | COG5503 | ykzG | DNA-dependent RNA polymerase auxiliary subunit epsilon (53) |
| S to T | 19 | COG1774 | yaaT | Cell fate regulator YaaT, PSP1 superfamily (controls sporulation, competence, biofilm development) (54) |
| S to R | 130 | COG0718 | ebfC (ybaB) | Conserved DNA-binding protein YbaB (function unknown) (55,56) |
| S to X | 32 | COG3645 | yoqD | Phage antirepressor protein YoqD, KilAC domain (57) |
| R to known function | 210 | COG1623 | disA | Diadenylate cyclase (c-di-AMP synthetase), DNA integrity scanning protein DisA (58,59) |
| R to J | 42 | COG0319 | ybeY | ssRNA-specific RNase YbeY, 16S rRNA maturation enzyme (60) |
| R to M | 18 | COG3107 | yraM | Outer membrane lipoprotein LpoA, binds and activates PBP1a (61–69) |
| R to X | 42 | COG3941 | gp42 | Phage tail tape-measure protein, controls tail length |
| R to S | 52 | COG3193 | glcG | Uncharacterized conserved protein GlcG, DUF336 family (64) |
aFunctional category designations are as in Table 1 with the following additions: K, transcription; M, cell wall/membrane/envelope biogenesis; T, signal transduction mechanisms; X, mobilome: phages and transposons.
A particularly notable development was the availability of functional assignments for some conserved proteins from the ‘Function unknown’ category. Several widespread proteins from that category had been shown to participate in translation, including rRNA maturation, tRNA modification and similar processes (Table 1). Although most of these newly recognized functions already have been recorded in UniProt and the Modomics database (34,65), not all of them have been propagated to the entire protein families and used in genome annotation. While many previously poorly characterized or uncharacterized proteins (R- or S-COGs) have been now moved to better-defined functional categories, analysis of the functional predictions in the R category resulted in the reassignment of 54 R-COGs to the ‘Function unknown’ (S) category as the previous general functional predictions were found to be poorly justified (Table 2).
As described previously (66), analysis of the COG phyletic profiles (patterns of presence and absence of proteins from given genomes in a given set of COGs) revealed numerous cases of potentially erroneous genome annotation, where certain genes, including some essential ones, appeared to be missing in certain genomes. As an example, the COG for glutamyl-tRNA synthetase (COG0008), an essential enzyme, is missing representatives from two archaeal species, Thermoproteus tenax and Candidatus Nitrososphaera gargensis. Examination of the respective genome sequences shows that the corresponding Open Reading Frames (ORFs) are present but contain frameshifts and are therefore marked as pseudogenes and omitted from the deduced protein sets. Obviously, these organisms would not be able to grow without glutamyl-tRNA synthetases which in archaea is required for charging both tRNAGlu and tRNAGln (67). As noted previously, protein sets translated from many sequenced genomes lack some short ribosomal proteins (27). Thus, the use of COG phyletic patterns offers a possibility to identify missed genes and improve genome annotation (66,68).
AVAILABILITY
The new version of the COGs is publicly available at http://www.ncbi.nlm.nih.gov/COG/. The 2003 version of the COG database, which includes yeast genes, is available on the NCBI FTP site ftp://ftp.ncbi.nih.gov/pub/COG/COG/. All queries and comments regarding the COG database should be directed to the authors at cogs@ncbi.nlm.nih.gov.
FUTURE DEVELOPMENTS
The current updated release of the COGs did not involve creation of new COGs and 242 COGs have been removed from the database. The accumulation of COGs containing thousands of protein illuminated certain problems in the COG approach that might need to be addressed by dividing these COGs into smaller ones based on phylogenetic analysis. We anticipate adding to the system new COGs, primarily archaeal, cyanobacterial and sporulation-related COGs described in our previous publications (17,19–20,69). To assist the structural genomics efforts, we also plan providing links to the Protein Data Bank (70) and highlighting those R- and S-COGs for which structures remain unavailable.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the U.S. National Institutes of Health at the National Library of Medicine. Funding for open access charge: Intramural funds of the U.S. Department of Health and Human Services [to the National Library of Medicine, National Institutes of Health].
Conflict of interest statement. None declared.
REFERENCES
- 1.Tatusov R.L., Koonin E.V., Lipman D.J. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 2.Marchler-Bauer A., Anderson J.B., Cherukuri P.F., DeWeese-Scott C., Geer L.Y., Gwadz M., He S., Hurwitz D.I., Jackson J.D., Ke Z., et al. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 2005;33:D192–D196. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marchler-Bauer A., Zheng C., Chitsaz F., Derbyshire M.K., Geer L.Y., Geer R.C., Gonzales N.R., Gwadz M., Hurwitz D.I., Lanczycki C.J., et al. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013;41:D348–D352. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Overbeek R., Olson R., Pusch G.D., Olsen G.J., Davis J.J., Disz T., Edwards R.A., Gerdes S., Parrello B., Shukla M., et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST) Nucleic Acids Res. 2014;42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Markowitz V.M., Chen I.M., Palaniappan K., Chu K., Szeto E., Pillay M., Ratner A., Huang J., Woyke T., Huntemann M., et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42:D560–D567. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Letunic I., Doerks T., Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40:D302–D305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finn R.D., Bateman A., Clements J., Coggill P., Eberhardt R.Y., Eddy S.R., Heger A., Hetherington K., Holm L., Mistry J., et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galperin M.Y. Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J. Bacteriol. 2006;188:4169–4182. doi: 10.1128/JB.01887-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galperin M.Y. Diversity of structure and function of response regulator output domains. Curr. Opin. Microbiol. 2010;13:150–159. doi: 10.1016/j.mib.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mi H., Muruganujan A., Thomas P.D. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selengut J.D., Haft D.H., Davidsen T., Ganapathy A., Gwinn-Giglio M., Nelson W.C., Richter A.R., White O. TIGRFAMs and Genome Properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res. 2007;35:D260–D264. doi: 10.1093/nar/gkl1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunter S., Jones P., Mitchell A., Apweiler R., Attwood T.K., Bateman A., Bernard T., Binns D., Bork P., Burge S., et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2012;40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tatusov R.L., Galperin M.Y., Natale D.A., Koonin E.V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tatusov R.L., Natale D.A., Garkavtsev I.V., Tatusova T.A., Shankavaram U.T., Rao B.S., Kiryutin B., Galperin M.Y., Fedorova N.D., Koonin E.V. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tatusov R.L., Fedorova N.D., Jackson J.D., Jacobs A.R., Kiryutin B., Koonin E.V., Krylov D.M., Mazumder R., Mekhedov S.L., Nikolskaya A.N., et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galperin M.Y., Kolker E. New metrics for comparative genomics. Curr. Opin. Biotechnol. 2006;17:440–447. doi: 10.1016/j.copbio.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulkidjanian A.Y., Koonin E.V., Makarova K.S., Mekhedov S.L., Sorokin A., Wolf Y.I., Dufresne A., Partensky F., Burd H., Kaznadzey D., et al. The cyanobacterial genome core and the origin of photosynthesis. Proc. Natl Acad. Sci. U.S.A. 2006;103:13126–13131. doi: 10.1073/pnas.0605709103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makarova K., Slesarev A., Wolf Y., Sorokin A., Mirkin B., Koonin E., Pavlov A., Pavlova N., Karamychev V., Polouchine N., et al. Comparative genomics of the lactic acid bacteria. Proc. Natl Acad. Sci. U.S.A. 2006;103:15611–15616. doi: 10.1073/pnas.0607117103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makarova K.S., Sorokin A.V., Novichkov P.S., Wolf Y.I., Koonin E.V. Clusters of orthologous genes for 41 archaeal genomes and implications for evolutionary genomics of archaea. Biol. Direct. 2007;2:33. doi: 10.1186/1745-6150-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolf Y.I., Makarova K.S., Yutin N., Koonin E.V. Updated clusters of orthologous genes for Archaea: a complex ancestor of the Archaea and the byways of horizontal gene transfer. Biol. Direct. 2012;7:46. doi: 10.1186/1745-6150-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powell S., Forslund K., Szklarczyk D., Trachana K., Roth A., Huerta-Cepas J., Gabaldon T., Rattei T., Creevey C., Kuhn M., et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014;42:D231–D239. doi: 10.1093/nar/gkt1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2014;42:D7–D17. doi: 10.1093/nar/gkt1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Federhen S. The NCBI Taxonomy database. Nucleic Acids Res. 2012;40:D136–D143. doi: 10.1093/nar/gkr1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ludwig W., Schleifer K.-H., Whitman W.B. Revised Road Map to the Phylum Firmicutes. In: Vos P De, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer K-H, Whitman WB., editors. Bergey's Manual of Systematic Bacteriology. 2nd edn. Vol. 3. NY: Springer-Verlag; 2009. pp. 1–8. [Google Scholar]
- 25.Wolf M., Muller T., Dandekar T., Pollack J.D. Phylogeny of Firmicutes with special reference to Mycoplasma (Mollicutes) as inferred from phosphoglycerate kinase amino acid sequence data. Int. J. Syst. Evol. Microbiol. 2004;54:871–875. doi: 10.1099/ijs.0.02868-0. [DOI] [PubMed] [Google Scholar]
- 26.Yutin N., Galperin M.Y. A genomic update on clostridial phylogeny: Gram-negative spore formers and other misplaced clostridia. Environ. Microbiol. 2013;15:2631–2641. doi: 10.1111/1462-2920.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yutin N., Puigbo P., Koonin E.V., Wolf Y.I. Phylogenomics of prokaryotic ribosomal proteins. PLoS One. 2012;7:e36972. doi: 10.1371/journal.pone.0036972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tatusova T., Ciufo S., Fedorov B., O'Neill K., Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2014;42:D553–D559. doi: 10.1093/nar/gkt1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benson D.A., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2014;42:D32–D37. doi: 10.1093/nar/gkt1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maglott D., Ostell J., Pruitt K.D., Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011;39:D52–D57. doi: 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madej T., Lanczycki C.J., Zhang D., Thiessen P.A., Geer R.C., Marchler-Bauer A., Bryant S.H. MMDB and VAST+: tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014;42:D297–D303. doi: 10.1093/nar/gkt1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The UniProt Consortium. Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winsor G.L., Lam D.K., Fleming L., Lo R., Whiteside M.D., Yu N.Y., Hancock R.E., Brinkman F.S. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 2011;39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujisawa T., Okamoto S., Katayama T., Nakao M., Yoshimura H., Kajiya-Kanegae H., Yamamoto S., Yano C., Yanaka Y., Maita H., et al. CyanoBase and RhizoBase: databases of manually curated annotations for cyanobacterial and rhizobial genomes. Nucleic Acids Res. 2014;42:D666–D670. doi: 10.1093/nar/gkt1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou J., Rudd K.E. EcoGene 3.0. Nucleic Acids Res. 2013;41:D613–D624. doi: 10.1093/nar/gks1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Costanzo M.C., Engel S.R., Wong E.D., Lloyd P., Karra K., Chan E.T., Weng S., Paskov K.M., Roe G.R., Binkley G., et al. Saccharomyces genome database provides new regulation data. Nucleic Acids Res. 2014;42:D717–D725. doi: 10.1093/nar/gkt1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galperin M.Y., Koonin E.V. Divergence and convergence in enzyme evolution. J. Biol. Chem. 2012;287:21–28. doi: 10.1074/jbc.R111.241976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rampias T., Sheppard K., Soll D. The archaeal transamidosome for RNA-dependent glutamine biosynthesis. Nucleic Acids Res. 2010;38:5774–5783. doi: 10.1093/nar/gkq336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aravind L., Makarova K.S., Koonin E.V. Holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 2000;28:3417–3432. doi: 10.1093/nar/28.18.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iwamoto A., Osawa A., Kawai M., Honda H., Yoshida S., Furuya N., Kato J. Mutations in the essential Escherichia coli gene, yqgF, and their effects on transcription. J. Mol. Microbiol. Biotechnol. 2012;22:17–23. doi: 10.1159/000336517. [DOI] [PubMed] [Google Scholar]
- 43.Makarova K.S., Koonin E.V., Kelman Z. The CMG (CDC45/RecJ, MCM, GINS) complex is a conserved component of the DNA replication system in all archaea and eukaryotes. Biol. Direct. 2012;7:7. doi: 10.1186/1745-6150-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anton B.P., Chang Y.C., Brown P., Choi H.P., Faller L.L., Guleria J., Hu Z., Klitgord N., Levy-Moonshine A., Maksad A., et al. The COMBREX project: design, methodology, and initial results. PLoS Biol. 2013;11:e1001638. doi: 10.1371/journal.pbio.1001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galperin M.Y., Koonin E.V. From complete genome sequence to ‘complete’ understanding. Trends Biotechnol. 2010;28:398–406. doi: 10.1016/j.tibtech.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bateman A., Coggill P., Finn R.D. DUFs: families in search of function. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 2010;66:1148–1152. doi: 10.1107/S1744309110001685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goodacre N.F., Gerloff D.L., Uetz P. Protein domains of unknown function are essential in bacteria. MBio. 2014;5:e00744–e00813. doi: 10.1128/mBio.00744-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shisler K.A., Broderick J.B. Emerging themes in radical SAM chemistry. Curr. Opin. Struct. Biol. 2012;22:701–710. doi: 10.1016/j.sbi.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J., Woldring R.P., Roman-Melendez G.D., McClain A.M., Alzua B.R., Marsh E.N. Recent advances in radical SAM enzymology: new structures and mechanisms. ACS Chem. Biol. 2014;9:1929–1938. doi: 10.1021/cb5004674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tchong S.I., Xu H., White R.H. L-cysteine desulfidase: an [4Fe-4S] enzyme isolated from Methanocaldococcus jannaschii that catalyzes the breakdown of L-cysteine into pyruvate, ammonia, and sulfide. Biochemistry. 2005;44:1659–1670. doi: 10.1021/bi0484769. [DOI] [PubMed] [Google Scholar]
- 51.Mendez J., Reimundo P., Perez-Pascual D., Navais R., Gomez E., Guijarro J.A. A novel cdsAB operon is involved in the uptake of L-cysteine and participates in the pathogenesis of Yersinia ruckeri. J. Bacteriol. 2011;193:944–951. doi: 10.1128/JB.01058-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perrochia L., Guetta D., Hecker A., Forterre P., Basta T. Functional assignment of KEOPS/EKC complex subunits in the biosynthesis of the universal t6A tRNA modification. Nucleic Acids Res. 2013;41:9484–9499. doi: 10.1093/nar/gkt720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keller A.N., Yang X., Wiedermannova J., Delumeau O., Krasny L., Lewis P.J. ϵ, a new subunit of RNA polymerase found in Gram-positive bacteria. J. Bacteriol. 2014;196:3622–3632. doi: 10.1128/JB.02020-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carabetta V.J., Tanner A.W., Greco T.M., Defrancesco M., Cristea I.M., Dubnau D. A complex of YlbF, YmcA and YaaT regulates sporulation, competence and biofilm formation by accelerating the phosphorylation of Spo0A. Mol. Microbiol. 2013;88:283–300. doi: 10.1111/mmi.12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cooley A.E., Riley S.P., Kral K., Miller M.C., DeMoll E., Fried M.G., Stevenson B. DNA-binding by Haemophilus influenzae and Escherichia coli YbaB, members of a widely-distributed bacterial protein family. BMC Microbiol. 2009;9:137. doi: 10.1186/1471-2180-9-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jutras B.L., Bowman A., Brissette C.A., Adams C.A., Verma A., Chenail A.M., Stevenson B. EbfC (YbaB) is a new type of bacterial nucleoid-associated protein and a global regulator of gene expression in the Lyme disease spirochete. J. Bacteriol. 2012;194:3395–3406. doi: 10.1128/JB.00252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iyer L.M., Koonin E.V., Aravind L. Extensive domain shuffling in transcription regulators of DNA viruses and implications for the origin of fungal APSES transcription factors. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-3-research0012. RESEARCH0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Witte G., Hartung S., Buttner K., Hopfner K.P. Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol. Cell. 2008;30:167–178. doi: 10.1016/j.molcel.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 59.Oppenheimer-Shaanan Y., Wexselblatt E., Katzhendler J., Yavin E., Ben-Yehuda S. c-di-AMP reports DNA integrity during sporulation in Bacillus subtilis. EMBO Rep. 2011;12:594–601. doi: 10.1038/embor.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grinwald M., Ron E.Z. The Escherichia coli translation-associated heat shock protein YbeY is involved in rRNA transcription antitermination. PLoS One. 2013;8:e62297. doi: 10.1371/journal.pone.0062297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Typas A., Banzhaf M., van den Berg van Saparoea B., Verheul J., Biboy J., Nichols R.J., Zietek M., Beilharz K., Kannenberg K., von Rechenberg M., et al. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell. 2010;143:1097–1109. doi: 10.1016/j.cell.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paradis-Bleau C., Markovski M., Uehara T., Lupoli T.J., Walker S., Kahne D.E., Bernhardt T.G. Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell. 2010;143:1110–1120. doi: 10.1016/j.cell.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jean N.L., Bougault C.M., Lodge A., Derouaux A., Callens G., Egan A.J., Ayala I., Lewis R.J., Vollmer W., Simorre J.P. Elongated structure of the outer-membrane activator of peptidoglycan synthesis LpoA: implications for PBP1A stimulation. Structure. 2014;22:1047–1054. doi: 10.1016/j.str.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pellicer M.T., Badia J., Aguilar J., Baldoma L. glc locus of Escherichia coli: characterization of genes encoding the subunits of glycolate oxidase and the glc regulator protein. J. Bacteriol. 1996;178:2051–2059. doi: 10.1128/jb.178.7.2051-2059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Machnicka M.A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K.M., et al. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Natale D.A., Galperin M.Y., Tatusov R.L., Koonin E.V. Using the COG database to improve gene recognition in complete genomes. Genetica. 2000;108:9–17. doi: 10.1023/a:1004031323748. [DOI] [PubMed] [Google Scholar]
- 67.Nureki O., O'Donoghue P., Watanabe N., Ohmori A., Oshikane H., Araiso Y., Sheppard K., Soll D., Ishitani R. Structure of an archaeal non-discriminating glutamyl-tRNA synthetase: a missing link in the evolution of Gln-tRNAGln formation. Nucleic Acids Res. 2010;38:7286–7297. doi: 10.1093/nar/gkq605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koonin E.V., Galperin M.Y. Sequence–Evolution–Function: Computational Approaches in Comparative Genomics. Boston, MA: Kluwer Academic; 2003. [PubMed] [Google Scholar]
- 69.Galperin M.Y., Mekhedov S.L., Puigbo P., Smirnov S., Wolf Y.I., Rigden D.J. Genomic determinants of sporulation in Bacilli and Clostridia: towards the minimal set of sporulation-specific genes. Environ. Microbiol. 2012;14:2870–2890. doi: 10.1111/j.1462-2920.2012.02841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rose P.W., Bi C., Bluhm W.F., Christie C.H., Dimitropoulos D., Dutta S., Green R.K., Goodsell D.S., Prlic A., Quesada M., et al. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res. 2013;41:D475–D482. doi: 10.1093/nar/gks1200. [DOI] [PMC free article] [PubMed] [Google Scholar]