

Abstract
Epilepsy is one of the most prevalent chronic neurological disorders, afflicting about 3.5–6.5 per 1000 children and 10.8 per 1000 elderly people. With intensive effort made during the last two decades, numerous genes and mutations have been published to be associated with the disease. An organized resource integrating and annotating the ever-increasing genetic data will be imperative to acquire a global view of the cutting-edge in epilepsy research. Herein, we developed EpilepsyGene (http://61.152.91.49/EpilepsyGene). It contains cumulative to date 499 genes and 3931 variants associated with 331 clinical phenotypes collected from 818 publications. Furthermore, in-depth data mining was performed to gain insights into the understanding of the data, including functional annotation, gene prioritization, functional analysis of prioritized genes and overlap analysis focusing on the comorbidity. An intuitive web interface to search and browse the diversified genetic data was also developed to facilitate access to the data of interest. In general, EpilepsyGene is designed to be a central genetic database to provide the research community substantial convenience to uncover the genetic basis of epilepsy.
INTRODUCTION
Epilepsy is a group of neurological disorders characterized by recurrent epileptic seizures (1). As one of the most prevalent chronic neurological disorders, it affects about 3.5–6.5 per 1000 children (2) and 10.8 per 1000 elderly people (3). With ages of onset varying from infancy to adulthood, epilepsy encompasses a broad range of clinical phenotypes, such as infantile spasms, childhood absence epilepsy and juvenile myoclonic epilepsy. Idiopathic epilepsy, representing up to 47% of all epilepsies, is considered to have a genetic basis with a monogenic or polygenic mode of inheritance (4). Meanwhile, individuals with epilepsy are consistently reported to show clinical features of other disorders, or vice versa. In particular, autism spectrum disorder (ASD) and attention-deficit/hyperactivity disorder (ADHD) are the most common comorbid conditions associated with epilepsy (2). Besides, the prevalence of epilepsy in patients with autism and mental retardation (MR) is up to 40% (5,6), and individuals with epilepsy are at an increased risk of developing schizophrenia (SCZ) like psychosis (7). Therefore, to unveil the genetic architecture of epilepsy, it is of vital importance to investigate the phenotypic and genetic complexity of epilepsy and its comorbidity with ASD/MR/ADHD/SCZ.
In the past two decades, with intensive effort made to explore genetic susceptibility of epilepsy, numerous genes and mutations have been discovered to be associated with the disease. Over the last 2 years, particularly, rapid progress in its gene discovery has been accelerated by the application of massively parallel sequencing technologies (8,9). An organized resource integrating and annotating the ever-increasing genetic data will be imperative for researchers to acquire a global view of the cutting-edge in epilepsy research. However, genetic database that integrates and analyzes the scattered genetic data on epilepsy is still in its infancy when compared with other disease-specific databases, such as AutismKB (10) and ADHDgene (11). Therefore, it is urgently required to conduct thorough collection, systematic integration and detailed annotation of existing genes and mutations underlying epilepsy.
The currently available genetic databases for epilepsy are: GenEpi (http://epilepsy.hardwicklab.org/), CarpeDB (http://www.carpedb.ua.edu/), epiGAD (12), The Lafora Gene Mutation Database (13) and MeGene (http://www.epigene.org/mutation/). However, they are far from a comprehensive genetic database: either lacking complete genetic information, or restricted on specific diseases or researches. In this study, we present EpilepsyGene, a comprehensive genetic database aimed to fulfill the growing needs of data integration and mining from all available resources. It integrates and annotates 499 genes, 3931 variants and 331 clinical phenotypes collected from 818 eligible publications. An intuitive web interface with versatile searching and browsing functionalities was also developed to help researchers access the data of interest conveniently and perform further data analysis. In general, EpilepsyGene is designed to be a central genetic database to provide research communities substantial convenience to uncover the phenotypic and genetic complexity of epilepsy and its comorbidities with other disorders.
DATA COLLECTION AND ANALYSIS
Data collection
To obtain a complete list of genes and mutations relevant to epilepsy, comprehensive searches were performed for epilepsy-related genetic studies. Initially, we retrospectively searched the PubMed database (http://www.ncbi.nih.gov/pubmed) with the following query terms: ‘epilepsy [Title/Abstract] OR specific phenotype such as West syndrome [Title/Abstract] AND gene [Title/Abstract] OR genetic [Title/Abstract] AND mutation [Title/Abstract] OR variant [Title/Abstract] OR variation [Title/Abstract]’. Additionally, EpilepsyGene also includes genetic variants selected with discretion from existing databases, including MITOMAP (14), The Lafora Gene Mutation Database (13), epiGAD (12), GenEpi (http://epilepsy.hardwicklab.org/) and MeGene (http://www.epigene.org/mutation/).
Overall, more than 1000 publications dating from 1995 to 2014 were obtained. The abstracts of these articles were manually screened, and those with negative results or performing only functional analysis of known variants were excluded. In all, 818 studies were recruited for further information extraction. Genetic data such as nucleotide change, gene symbol and clinical phenotype, were extracted through in-depth reading the full text of each publication and double-checked manually. Besides, clinical information relevant to the variant was also collected, including ethnicity, gender (male or female), age-of-onset and inheritance (de novo, maternal, paternal, etc.). Since copy number variations (CNVs) correspond to relatively large regions of the genome covering both associated and irrelevant genes, the genes and phenotypes relevant to the CNVs were then not included. Consequently, the EpilepsyGene database contains 2658 SNVs, 694 InDels, 499 genes, 331 phenotypes, 579 CNVs and the corresponding detailed clinical information from 818 eligible publications.
Functional annotation
To present a detailed report for each variant, ANNOVAR (15) was applied to annotate genetic variants with all available resources. It produced not only general information such as gene regions, effects and band, but also broad annotations from another 25 resources such as 1000 Genomes Project (16), Exome Sequencing Project (17) and dbSNP (18). In addition, mutation spectrum (19) addressing the mutation distribution was provided to present an overview of all mutations at gene level. All the mutations were mapped onto the locus of the corresponding gene, with different colors denoting different mutation types (MTs) or effects. Mutations reported more than once were separated from those initially identified.
To provide an informative gene card for each epileptic gene, gene annotation was produced including basic gene information, related Gene Ontology (GO) (20) terms and pathways, and brain expression details. Firstly, gene annotation file ‘Homo_sapiens.gene_info.gz’ was downloaded from NCBI (http://www.ncbi.nlm.nih.gov/) to obtain basic information of all epileptic genes. Secondly, WebGestalt (21) was used to enrich GO (20) terms, WIKI pathways (22), KEGG pathways (23) and Pathway Commons (24) associated with the genes. Thirdly, brain expression levels of each gene spanning 12 periods and 16 brain regions were obtained from RNA-seq (22) to present the expression pattern of the gene. Finally, gene details and relevant phenotypes in Mouse Genome Informatics (MGI) (25) and Online Mendelian Inheritance in Man (OMIM, http://www.omim.org/) (26) were also included in the gene card.
Gene prioritization
To identify high-confidence genes by the relevance to epilepsy, gene prioritization was conducted by adopting the annotations of 10 functional prediction tools (i.e. SIFT (27), phyloP (28), SiPhy (29), LRT (30), MutationTaster (31), MutationAssessor (32), FATHMM (33), GERP++ (34), PolyPhen2_HDIV (35) and PolyPhen2_HVAR (35)). A score of 1 was assigned to the variant, which was predicted as ‘Deleterious’, ‘Disease-causing’, ‘Tolerated’ or ‘Conserved’, and 10 was assigned to the variant whose MT is ‘frame shift’, ‘splicing’, ‘stop gain’ or ‘stop loss’. The score of the variant is then calculated by:
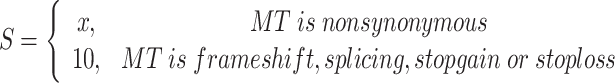 |
where x is the number of tools which predict the variant as ‘Deleterious’, ‘Disease-causing’, ‘Tolerated’ or ‘Conserved’. The final score of the gene is the sum of the score of all variants in the gene:
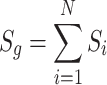 |
where N denotes the number of mutations in the gene, and Si represents the score of the variant. Ultimately, 154 high-confidence genes were obtained on the condition that the total score of the gene (Sg) was no less than 10.
Functional analysis based on the prioritized genes
To schematize the functional relevance of epilepsy-related genes, further data mining was performed based on the high-confidence genes, including co-expression analysis, phenotype and GO (20) enrichment. Firstly, to cluster the high-confidence genes on the basis of their expressions during the different developmental periods of human brain, we applied the WGCNA program (weighted gene co-expression network analysis) (36) and identified four co-expression modules with varying sizes labeled with different colors (i.e. blue, green, brown and gray). The human brain data from different periods in life were acquired from BrainSpan by RNA-seq (http://www.brainspan.org/) (22). This dataset includes gene expression profiles of 16 cortical and subcortical structures throughout the full life of human brain development (from 8 post-conception weeks to 40 years). Each module shows special pattern of expression profile and biological progress. Secondly, to identify epileptic phenotypes associated with the four gene sets, the enrichment of phenotypes was performed using the hypergeometric test (37), with significance threshold set as 1E-02 and minimum number of genes for an enriched phenotype three. Moreover, the four gene sets were used to enrich GO (20) terms separately with WebGestalt (21) to gain insights into the biological implications of the differentially expressed genes.
Overlap with ASD, MR, ADHD and SCZ
For the purpose of exploring the comorbidity of other disorders with epilepsy, overlap analysis was performed based on the shared genes, intersectional epileptic phenotypes and enriched pathways. Relative genes of each disorder were firstly collected from three existing databases (AutismKB (10), ADHDgene (11) and SZGR (38)). Due to the absence of genetic databases specific for MR, HGMD (39) was chosen as the resource to collect MR-related genes. The collected genes underlying the four disorders were then compared with genes in EpilepsyGene. As a result, 65 genes in EpilepsyGene were found to be shared by MR (MR-EP genes), 18 genes by ADHD (ADHD-EP genes), 67 genes by SCZ (SCZ-EP genes) and 146 genes by ASD (ASD-EP genes). To identify the specific phenotypes associated with respective overlapping genes, epileptic phenotypes associated with the common genes were retrieved and classified. Furthermore, pathway enrichment analysis for the common genes was undertaken separately by WebGestalt (21), in order to reveal the biological implications indicated by the overlapped genes.
Gene–disease network
For understanding the phenotypic and genetic complexity in epilepsy, gene–disease network, developed by SVG, was constructed to demonstrate the overlapping features between two characters. Three types of nodes were used to respectively denote gene, disease and common disease/gene. Gene and disease nodes are connected through edges if the corresponding gene–disease association exists in the EpilepsyGene database. Common disease/gene nodes will be connected to two gene (disease) nodes if the genes (diseases) share a disease (gene). Total number of mutations associated with the phenotype or the number of mutations in a specific gene related to the phenotype was displayed. Overall, it aims to address the following three issues: (i) ‘gene–gene network’ to represent the common associated phenotypes between two genes, (ii) ‘disease–disease network’ to demonstrate the shared genes associated with both phenotypes and (iii) ‘gene–disease network’ for users to know whether associations exist between the selected gene and phenotype.
DATABASE INTERFACE
A user-friendly web interface of EpilepsyGene was developed, supported with versatile and facilitated browsing and searching functionalities. All the data were stored and managed in MySQL relational database. The users may access genetic data or extended analysis results freely through the web interface (http://61.152.91.49/EpilepsyGene).
Data statistics
To provide an overview of the genetic and clinical information in EpilepsyGene, all the data were demonstrated schematically, including the composition of MT and effect, gender distribution, age-of-onset and inheritance patterns. A four-set Venn diagram was firstly generated to present the intersections over subsets of the de novo, recurrent, rare and entire CNVs. Secondly, scatter diagrams were provided to present the distribution of age-of-onset in each phenotype. It should be noted that only those phenotypes with which more than 10 mutations are associated and the relevant ages of onset are not all ‘NA’ were selected. The same approach was applied on the distribution of gender or inheritance in each phenotype as well. Thirdly, bar plots were charted out to summarize distribution of mutations on each chromosome. Lastly, composition of the MTs and effects were also presented through a 3-D pie chart or a bar plot. All the statistical results are freely available in the web page of the EpilepsyGene database.
Data browse
To support facilitating access to genetic data in EpilepsyGene, four browsing approaches have been provided: ‘Browse by gene’, ‘Browse by mutation’, ‘Browse by phenotype’ and ‘Browse by chromosome’. ‘Browse by gene’ lists all genes related to epilepsy, and genes overlapped by epilepsy and four other disorders are also available in this module. ‘Browse by mutation’ displays variants wisely according to the MTs (SNV, InDel or CNV), and separates de novo mutations from inherited ones, rare CNVs from recurrent ones. ‘Browse by phenotype’ classifies the majority of the epileptic phenotypes into eight sets, and each set contains phenotypes that can be linked to a detailed page accommodating all genes and mutations associated with the phenotype. ‘Browse by chromosome’ mapped all mutations onto 24 chromosomes, and each rectangle either colored in red (SNVs) or in blue (InDels) on the chromosomes can be linked to a detailed gene card containing gene annotations, related phenotypes and mutation spectrum.
EpilepsyGene provides a specified gene card for each gene. Take the gene PCDH19 for example, a gene card specific for PCDH19 can be obtained through ‘Browse by gene’, and the card is documented with ‘gene annotations’, ‘mutations in the PCDH19’, ‘mutation spectrum of PCDH19’ and ‘related phenotypes’ (Figure 1). Gene annotation covers basic information, related GO (20) terms and pathways, information in MGI (25) and OMIM (26), gene expression level in 12 developmental periods and 16 brain regions, and the corresponding expression pattern in the brain. Mutations in PCDH19 are displayed through a detailed table, and a hyperlink is provided for each mutation to be linked to a detailed mutation report. Mutation spectrum visualizes the distribution of mutations, and may indicate that mutations in PCDH19 mainly distribute on the first coding exon. Phenotypes associated with the gene are schematized, with each circle (gene or phenotype) linked to a detailed page.
Figure 1.

An example of data access in EpilepsyGene. Gene card mainly consists of three parts: gene annotation, mutation spectrum of the gene and related phenotypes. Gene annotation includes basic information, related GO and pathways, information in OMIM and MGI, and brain expression level in different periods and regions. Mutation spectrum depicts the mutations of the gene schematically, which can be linked to a detailed report of the variant. Phenotypes associated with the gene are presented by gene–disease network.
Data search
EpilepsyGene supports multiple search approaches in a user-friendly environment, including keyword search, advanced search and BLAST search. First, a keyword search in the home page was provided for searching by gene symbol or phenotype. Second, advanced search was incorporated to query variants, phenotypes or publications by filtering diversified options (i.e. gene symbol, gene region, MT, inheritance or locus). Additionally, EpilepsyGene also provides BLAST search to query against the nucleotide or protein sequences of all epilepsy-related genes in EpilepsyGene.
DISCUSSION AND PERSPECTIVES
As a comprehensive genetic resource for epilepsy, EpilpsyGene integrates and annotates cumulative to date 499 epilepsy-related genes and 3931 mutations through in-depth reading 818 publications. The genes overlapped with ASD/MR/SCZ/ADHD are separated accordingly. The integrated data will provide a global view of the cutting-edge of genetic research in epilepsy. Meanwhile, gene–disease network makes it possible to demonstrate correlations between two genes or phenotypes, and thus facilitates the inquiry into phenotypic and genetic complexity of epilepsy. Supported with versatile searching and browsing tools, the EpilepsyGene database could act as not only an integrated genetic resource for epilepsy, but also the first disease database addressing the comorbidity of epilepsy.
On the basis of the data integrated from publications, extensive data mining was performed and some promising results could be generated. For example, among the high-confidence genes, the ‘blue’ genes and the ‘green’ genes, representing two utterly opposite expression patterns in brain (Figure 2A), were unexpectedly found to be enriched in almost similar phenotypes, e.g. West syndrome (WS), Lennox–Gastaut syndrome (LGS), severe myoclonic epilepsy of infancy (SMEI), early-onset epileptic encephalopathy (EOEE) and Ohtahara syndrome (OS) (Figure 2B). By contrast, the ‘blue’ and ‘gray’ genes were enriched in channel activities, and both sets represent similar expression pattern in brain after 18 months after birth. In addition, the ‘green’ genes displayed statistically significant enrichment in ‘ion transport’ (Figure 2C), a major role in ion channels (40), which are of crucial importance in various processes, such as nerve excitation, cell proliferation, sensory transduction, and learning and memory (41,42). This may explain why the corresponding expression pattern (Figure 2A) suggests an increase since 8–9 post-conception weeks. On the other side, the overlap analysis (Figure 2D) revealed that ‘MR-EP’ genes and ‘ASD-EP’ genes have the largest number of common epileptic phenotypes (86 phenotypes), which may account for the high prevalence of epilepsy in individuals with autism and MR. In pathway analysis (Table 1), ‘Neuroactive ligand-receptor interaction’, a pathway concerning neuronal brain function (43), was found to exhibit the highest enrichment scores in three overlapping gene sets (‘ADHD-EP’, ‘ASD-EP’ and ‘SCZ-EP’). Although the pathway did not appear in the top three enriched pathways in ‘MR-EP’, it ranked fifth with high significance (P-value: 4.86E-05). The analysis suggests that perturbations of the genes involved in the pathway may alter the spectrum of neurological functions resulting in epileptic phenotypes and its comorbidities with other disorders. Noteworthily, pathway ‘Alzheimer's disease’ (AD) is overlapped by ‘SCZ-EP’ and ‘MR-EP’ sets, indicating that AD may be a comorbid condition in epilepsy. This supposition has found underpinning from several publications (44–47).
Figure 2.
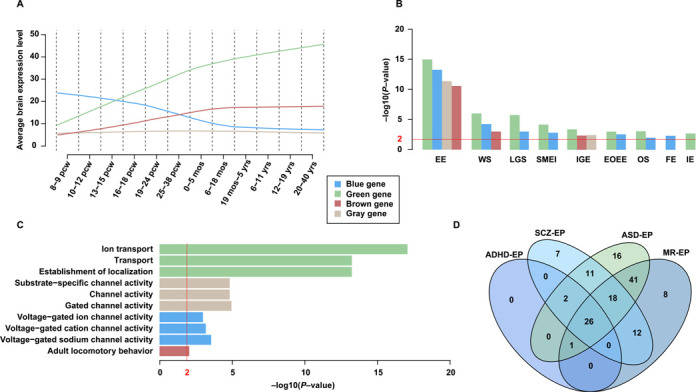
Gene prioritization and overlap analysis. (A) Four co-expression modules derived from WGCNA clustering. The green module includes 57 genes, and expressions of these genes suggest increasing throughout the full life of human brain development. The brown module, including 30 genes, suggests a moderate increasing expression pattern, and tends to keep constant after birth. The blue module contains 43 genes. These genes are highly expressed in embryonic period (8–9 pcw), with a slow decrease during prenatal period and reach stable after birth. The gray module having 24 genes does not demonstrate obvious tendencies (‘pcw’ represents post-conception weeks, ‘mos’ and ‘yrs’ means months and years, respectively). (B) Enriched epileptic phenotypes based on the four modules (EE, epileptic encephalopathy; WS, West syndrome; LGS, Lennox–Gastaut syndrome; SMEI, severe myoclonic epilepsy of infancy; IGE, idiopathic generalized epilepsy; EOEE, early-onset EE; OS, Ohtahara syndrome; FE, focal epilepsy; IE, idiopathic epilepsy). (C) Relative enriched GO terms of each module. (D) A four-set Venn diagram displaying intersectional epileptic phenotypes over subsets of four overlapping genes.
Table 1. Top three enriched pathways for the overlapped genes.
| Gene set | Term | KEGG ID | P-value |
|---|---|---|---|
| ADHD-EP | Neuroactive ligand-receptor interaction | hsa04080 | 2.85E-06 |
| ASD-EP | Neuroactive ligand-receptor interaction | hsa04080 | 2.22E-11 |
| Calcium signaling pathway | hsa04020 | 8.10E-11 | |
| Long-term potentiation | hsa04720 | 1.42E-08 | |
| MR-EP | Long-term potentiation | hsa04720 | 3.59E-07 |
| Calcium signaling pathway | hsa04020 | 6.32E-07 | |
| Alzheimer's disease | hsa05010 | 9.72E-06 | |
| SCZ-EP | Neuroactive ligand-receptor interaction | hsa04080 | 2.48E-16 |
| Alzheimer's disease | hsa05010 | 4.90E-07 | |
| Amyotrophic lateral sclerosis | hsa05014 | 2.20E-06 |
Note: All the above pathways were enriched from KEGG pathway by Webgestalt 2.0.
In conclusion, EpilepsyGene was developed to fulfill the growing demand of integrating and mining the genetic and clinical information of epilepsy. In the following years, there continue to be considerable advances in the identification of potential epilepsy susceptibility genes with the increasing application of massively parallel sequencing technologies. As is the case with many other databases, we are following up the frontier of epilepsy studies to integrate and annotate the newly generated data, and use these data to make EpilepsyGene up-to-date and comprehensive. Semi-automatic publication-mining methods will be used in the subsequent updates of the database, including (i) automatically filter out large amount of irrelevant publications by the GAPscreener (48) program; (ii) manually confirm the relevance of publications and collect the latest mutation data and clinical information associated with epilepsy; (iii) automatically perform functional annotation based upon the collected data and (iv) automatically update basic statistics, mutation spectrum and other functionalities. Meanwhile, as online submission is an important source of genetic data, a central site has been made available for the submission of genes and variants associated with epilepsy. Taken together, we hope that EpilepsyGene will be a valuable resource for deciphering the genetic architecture of epilepsy and finally the improvement of clinical diagnosis and treatment.
Acknowledgments
We thank all our colleagues and friends at the Institute of Genomic Medicine, Wenzhou Medical University who helped us test the database and provided the valuable suggestions.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Funding for open access charge: National Natural Science Foundation of China [31171236/C060503]; Nation High Technology Research and Development Program of China [2012AA02A201, 2012AA02A202].
Conflict of interest statement. None declared.
REFERENCES
- 1.Guidelines for epidemiologic studies on epilepsy. Commission on Epidemiology and Prognosis, International League Against Epilepsy. Epilepsia. (1993;34:592–596. doi: 10.1111/j.1528-1157.1993.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 2.Lo-Castro A., Curatolo P. Epilepsy associated with autism and attention deficit hyperactivity disorder: is there a genetic link. Brain Dev. 2014;36:185–193. doi: 10.1016/j.braindev.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 3.Faught E., Richman J., Martin R., Funkhouser E., Foushee R., Kratt P., Kim Y., Clements K., Cohen N., Adoboe D., et al. Incidence and prevalence of epilepsy among older U.S. Medicare beneficiaries. Neurology. 2012;78:448–453. doi: 10.1212/WNL.0b013e3182477edc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicita F., De Liso P., Danti F.R., Papetti L., Ursitti F., Castronovo A., Allemand F., Gennaro E., Zara F., Striano P., et al. The genetics of monogenic idiopathic epilepsies and epileptic encephalopathies. Seizure. 2012;21:3–11. doi: 10.1016/j.seizure.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Tuchman R.F., Rapin I., Shinnar S. Autistic and dysphasic children. II: Epilepsy. Pediatrics. 1991;88:1219–1225. [PubMed] [Google Scholar]
- 6.Tuchman R., Moshe S.L., Rapin I. Convulsing toward the pathophysiology of autism. Brain Dev. 2009;31:95–103. doi: 10.1016/j.braindev.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cascella N.G., Schretlen D.J., Sawa A. Schizophrenia and epilepsy: is there a shared susceptibility. Neurosci. Res. 2009;63:227–235. doi: 10.1016/j.neures.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helbig I., Lowenstein D.H. Genetics of the epilepsies: where are we and where are we going. Curr. Opin. Neurol. 2013;26:179–185. doi: 10.1097/WCO.0b013e32835ee6ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jensen F.E. Epilepsy in 2013: progress across the spectrum of epilepsy research. Nat. Rev. Neurol. 2014;10:63–64. doi: 10.1038/nrneurol.2013.277. [DOI] [PubMed] [Google Scholar]
- 10.Xu L.M., Li J.R., Huang Y., Zhao M., Tang X., Wei L. AutismKB: an evidence-based knowledgebase of autism genetics. Nucleic Acids Res. 2012;40:D1016–D1022. doi: 10.1093/nar/gkr1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L., Chang S., Li Z., Zhang K., Du Y., Ott J., Wang J. ADHDgene: a genetic database for attention deficit hyperactivity disorder. Nucleic Acids Res. 2012;40:D1003–D1009. doi: 10.1093/nar/gkr992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan N.C., Berkovic S.F. The Epilepsy Genetic Association Database (epiGAD): analysis of 165 genetic association studies, 1996–2008. Epilepsia. 2010;51:686–689. doi: 10.1111/j.1528-1167.2009.02423.x. [DOI] [PubMed] [Google Scholar]
- 13.Ianzano L., Zhang J., Chan E.M., Zhao X.C., Lohi H., Scherer S.W., Minassian B.A. Lafora progressive Myoclonus Epilepsy mutation database-EPM2A and NHLRC1 (EMP2B) genes. Hum. Mutat. 2005;26:397. doi: 10.1002/humu.9376. [DOI] [PubMed] [Google Scholar]
- 14.Ruiz-Pesini E., Lott M.T., Procaccio V., Poole J.C., Brandon M.C., Mishmar D., Yi C., Kreuziger J., Baldi P., Wallace D.C. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007;35:D823–D828. doi: 10.1093/nar/gkl927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu W., O'Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Acland A., Agarwala R., Barrett T., Beck J., Benson D.A., Bollin C., Bolton E., Bryant S.H., Canese K., Church D.M. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2014;42 doi: 10.1093/nar/gkt1146. doi:10.1093/nar/gkt1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ran X., Cai W.J., Huang X.F., Liu Q., Lu F., Qu J., Wu J., Jin Z.B. ‘RetinoGenetics’: a comprehensive mutation database for genes related to inherited retinal degeneration. Database. 2014;2014 doi: 10.1093/database/bau047. doi:10.1093/database/bau047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gene Ontology Consortium. Gene Ontology annotations and resources. Nucleic Acids Res. 2013;41:D530–D535. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J., Duncan D., Shi Z., Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77–W83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelder T., van Iersel M.P., Hanspers K., Kutmon M., Conklin B.R., Evelo C.T., Pico A.R. WikiPathways: building research communities on biological pathways. Nucleic Acids Res. 2012;40:D1301–D1307. doi: 10.1093/nar/gkr1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerami E.G., Gross B.E., Demir E., Rodchenkov I., Babur O., Anwar N., Schultz N., Bader G.D., Sander C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39:D685–D690. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blake J.A., Bult C.J., Eppig J.T., Kadin J.A., Richardson J.E. The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res. 2014;42:D810–D817. doi: 10.1093/nar/gkt1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amberger J., Bocchini C., Hamosh A. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM(R)) Hum. Mutat. 2011;32:564–567. doi: 10.1002/humu.21466. [DOI] [PubMed] [Google Scholar]
- 27.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 28.Siepel A., Pollard K.S., Haussler D. New methods for detecting lineage-specific selection. Lect. Notes Comput. Sci. 2006;3909:190–205. [Google Scholar]
- 29.Lindblad-Toh K., Garber M., Zuk O., Lin M.F., Parker B.J., Washietl S., Kheradpour P., Ernst J., Jordan G., Mauceli E., et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chun S., Fay J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwarz J.M., Rodelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 32.Reva B., Antipin Y., Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:E118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shihab H.A., Gough J., Cooper D.N., Stenson P.D., Barker G.L.A., Edwards K.J., Day I.N.M., Gaunt T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput. Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peter Langfelder S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2012;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang B., Kirov S., Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jia P., Sun J., Guo A., Zhao Z. SZGR: a comprehensive schizophrenia gene resource. Mol. Psychiatry. 2010;15:453–462. doi: 10.1038/mp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stenson P.D., Mort M., Ball E.V., Shaw K., Phillips A.D., Cooper D.N. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014;133:1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oosterwijk E., Gillies R. Targeting ion transport in cancer. Philos. Trans. R. Soc. B Biol. Sci. 2014;369:20130107. doi: 10.1098/rstb.2013.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Askland K., Read C., O'Connell C., Moore J.H. Ion channels and schizophrenia: a gene set-based analytic approach to GWAS data for biological hypothesis testing. Hum. Genet. 2012;131:373–391. doi: 10.1007/s00439-011-1082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashcroft F.M. From molecule to malady. Nature. 2006;440:440–447. doi: 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- 43.Bocchio-Chiavetto L., Maffioletti E., Bettinsoli P., Giovannini C., Bignotti S., Tardito D., Corrada D., Milanesi L., Gennarelli M. Blood microRNA changes in depressed patients during antidepressant treatment. Eur. Neuropsychopharmacol. 2013;23:602–611. doi: 10.1016/j.euroneuro.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 44.Borroni B., Pilotto A., Bonvicini C., Archetti S., Alberici A., Lupi A., Gennarelli M., Padovani A. Atypical presentation of a novel Presenilin 1 R377W mutation: sporadic, late-onset Alzheimer disease with epilepsy and frontotemporal atrophy. Neurol. Sci. 2012;33:375–378. doi: 10.1007/s10072-011-0714-1. [DOI] [PubMed] [Google Scholar]
- 45.Rudzinski L.A., Fletcher R.M., Dickson D.W., Crook R., Hutton M.L., Adamson J., Graff-Radford N.R. Early onset Alzheimer's disease with spastic paraparesis, dysarthria and seizures and N135S mutation in PSEN1. Alzheimer Dis. Assoc. Disord. 2008;22:299–307. doi: 10.1097/WAD.0b013e3181732399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Velez-Pardo C., Arellano J.I., Cardona-Gomez P., Jimenez Del Rio M., Lopera F., De Felipe J. CA1 hippocampal neuronal loss in familial Alzheimer's disease presenilin-1 E280A mutation is related to epilepsy. Epilepsia. 2004;45:751–756. doi: 10.1111/j.0013-9580.2004.55403.x. [DOI] [PubMed] [Google Scholar]
- 47.Ezquerra M., Carnero C., Blesa R., Gelpi J., Ballesta F., Oliva R. A presenilin 1 mutation (Ser169Pro) associated with early-onset AD and myoclonic seizures. Neurology. 1999;52:566–566. doi: 10.1212/wnl.52.3.566. [DOI] [PubMed] [Google Scholar]
- 48.Yu W., Clyne M., Dolan S.M., Yesupriya A., Wulf A., Liu T., Khoury M.J., Gwinn M. GAPscreener: an automatic tool for screening human genetic association literature in PubMed using the support vector machine technique. BMC Bioinformatics. 2008;9:205. doi: 10.1186/1471-2105-9-205. [DOI] [PMC free article] [PubMed] [Google Scholar]