


Abstract
The Genomes OnLine Database (GOLD; http://www.genomesonline.org) is a comprehensive online resource to catalog and monitor genetic studies worldwide. GOLD provides up-to-date status on complete and ongoing sequencing projects along with a broad array of curated metadata. Here we report version 5 (v.5) of the database. The newly designed database schema and web user interface supports several new features including the implementation of a four level (meta)genome project classification system and a simplified intuitive web interface to access reports and launch search tools. The database currently hosts information for about 19 200 studies, 56 000 Biosamples, 56 000 sequencing projects and 39 400 analysis projects. More than just a catalog of worldwide genome projects, GOLD is a manually curated, quality-controlled metadata warehouse. The problems encountered in integrating disparate and varying quality data into GOLD are briefly highlighted. GOLD fully supports and follows the Genomic Standards Consortium (GSC) Minimum Information standards.
INTRODUCTION
The Genomes OnLine Database (GOLD) is a data management system for cataloging and continuous monitoring of sequencing projects worldwide. GOLD collects, curates and disseminates metadata associated with those projects. GOLD is currently in its fifth version (1–6). With rapidly decreasing costs for sequencing, the number of sequencing projects and the amount of sequence data generated are increasing at an exponential rate. As these data are submitted to various public resources like GenBank (7) and EMBL (8) or analysis platforms like Integrated Microbial Genomes (IMG) (9) and MG-RAST (10), it becomes increasingly important to document the associated metadata in order to facilitate comparative analysis and hypothesis generation. The Genomic Standards Consortium (GSC) mandates the Minimum Information about any (x) Sequence (MIxS) specifications to be used when making sequence data available in public repositories (11,12). GOLD is fully compliant with the GSC's MIxS standards in capturing metadata and provides a platform to query projects based on various metadata features.
GOLD supports the IMG family of data management systems (9,13–15) as a gatekeeper of projects and metadata and requires that projects are annotated with at least minimal metadata. In fact, an entry in GOLD and compliance with required metadata is a prerequisite to submit a project to the IMG systems for annotation. The main steps in the process include project registration in GOLD, project submission to IMG for annotation and finally publication of results in the GSC's journal, Standards in Genomic Sciences (http://www.standardsingenomics.com/), or other journals of your choice. Since GOLD complies with MIxS, all available required metadata is already in place to publish in SIGS.
In the past, when sequencing was still expensive and only a limited number of high-interest organism genomes were sequenced, maintaining the associated information in a catalog format was sufficient. With lower sequencing costs, many more genomes are now being sequenced as part of a single study. Initiatives such as the Human Microbiome Project (HMP) (16) and Genomic Encyclopedia of Bacteria and Archaea (GEBA) (17,18) are a couple of examples where several thousands of genomes were sequenced as part of a single initiative. The emergence of high-throughput sequencing technologies and the development of analysis tools for studying metagenomes has facilitated the rapid growth in metagenome studies as well. It is also becoming more common to use multiple sequencing approaches on the same sample(s), such as the Functional Encyclopedia of Bacteria and Archaea (FEBA) (19). In such cases, it is important to collect common metadata pertaining to these samples and organize all of the samples under one or more relevant studies.
The increasing variety of sequencing and analysis projects needs to be linked and tracked in a seamlessly integrated system. One of the major limitations of the previous versions of the database has been the assumption of a one-to-one relationship between related components. For example, the previous versions could not correlate multiple sequencing projects to a single sample. In the event an isolate genome and metagenome were derived from a single sample, a separate record for each sequence would need to be created. Similarly the previous versions could not capture the multiple sequencing projects of a combined assembly nor was it possible to connect multiple analyses to a single sequencing project. Another limitation was that all genome projects were designated as isolates, an incorrect assignment for a genome assembled from a metagenome. These issues necessitated a new mechanism to organize various components of sequencing studies.
NEW TO THIS RELEASE
Version 5 of the database is founded on a fundamentally redesigned schema to accommodate a four level project classification system (Figure 1). The new classification system is comprised of Studies, Biosamples, Sequencing Projects (SPs) and Analysis Projects (APs). Studies constitute the highest level of classification in the system, containing Biosamples, SPs and APs that are part of a single initiative. GOLD's Biosamples represent the physical isolate or environmental material from which genetic material is extracted for sequencing. GOLD's Biosamples have no relation to NCBI BioSamples. GOLD's SPs represent sequencing protocols such as whole genome sequencing (WGS), transcriptomes, metagenomes, metatranscriptomes, methylation sequencing, etc. applied to Biosamples. APs are the analytical processes applied to the SPs. Multiple different assemblies or annotations of the same SPs would result in multiple different APs with varying metadata that need to be captured. These four components are described in more detail below.
Figure 1.
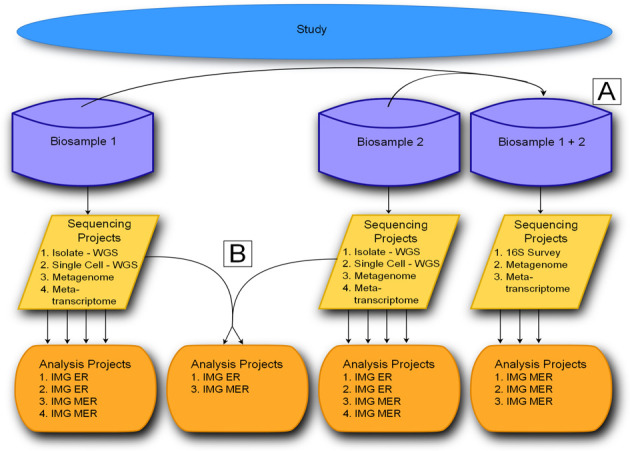
The four level project classification system implemented in v.5 to describe Studies, Biosamples, Sequencing Projects and Analysis Projects. Studies group one or more related Biosamples. Biosamples describe an individual sample of genetic material. Sequencing projects are the sequencing deliverables from the Biosamples. Analysis projects are the data processing methods applied to sequencing projects. (A) Biosamples may be merged prior to sequencing projects (e.g., 16S amplicon data combined prior to sequencing). (B) Sequencing Projects may be merged prior to analysis (e.g., multiple single-cell genomes combined for assembly).
In addition to the four levels described above, one more entity has been introduced in the new schema to provide metadata information for the individual organisms. In the previous versions of the database, each sequencing project of an isolate organism included both the metadata for the sequencing information and the organism in a single record. Increasingly, the genome of a single organism is being sequenced more than once, by different groups, making it inefficient to associate the same organism metadata individually with every different project. GOLD v.5 defines and curates the organism records with core taxonomy, environmental, and other metadata independently of their associated SPs. As a result, this entity can be used by all SPs without the need for curating and propagating redundant metadata. By doing so, v.5 now enables the identification of all the organisms with different but synonymous names.
Historically, the focus of the database was to provide a comprehensive coverage to all prokaryotic genomes and metagenomes. We are in the process of systematically integrating eukaryotic SPs into GOLD. Projects are introduced in the database from three main streams: (i) projects sequenced at the JGI are automatically added following a number of quality control checks; (ii) projects submitted to the database from individual researchers around the world; and (iii) projects available at the NCBI's BioProject portal.
The previous versions of the database provided a read-only project reporting system. This served user needs for accessing project information and searching for projects based on specific metadata. However, the user interface for project creation and curation was provided through a separate system called IMG-GOLD. The new version has enabled the seamless integration of these two formerly separated functions into a single resource.
Isolate genomes via their associated Biosamples are now classified using the same five-tier hierarchical classification system previously developed and implemented for metagenomes (20). Over 10 000 public isolate genomes have been classified accordingly. Over 9000 isolate genomes have also been curated to add strain habitat classifications. This field refers to the specific habitat of the strain according to the strain isolation information, as opposed to the previous general habitat in the database which corresponds to the species. The controlled vocabulary of the strain habitat has been mapped to the hierarchical ecosystem classification. For example, there are 161 genomes for organisms with the classification path Host-associated (ecosystem) -> mammals (ecosystem category)-> digestive system (ecosystem type) -> foregut (ecosystem subtype)-> rumen (specific ecosystem). The strain habitats within this group include ‘sheep rumen’, ‘cattle rumen’ and ‘goat rumen’ with 37, 53 and 1 genomes, respectively. Thus, there is manual curation of organism Biosamples with specific habitat terms.
A newly designed web interface provides access to data through various pre-selected reports, project distribution graphs, statistics and an intuitive search interface that allows a user to search based on an array of metadata fields. The new implementation also provides access for public users to search for APs submitted to the IMG systems.
GOLD DATABASE ORGANIZATION AND DATA OVERVIEW
The four-level classification system
The current release organizes genome, metagenome and other sequencing projects into a system of four levels which are described below.
GOLD Study
A study represents the highest-level organization. Studies include one or more Biosamples and their associated SPs and APs that have been grouped to investigate a related research topic of interest. For example, the HMP (16), GEBA (17,18) and KMG (21) studies represent typical cases where researchers set out to explore a specific topic by sequencing thousands of samples. Studies like GEBA-MDM (22) and FEBA (19) applied several different sequencing strategies (e.g. isolate genomes, single-cell genomes, metagenomes, transcriptomes, etc.) as part of a single study. Studies may be composed of one to hundreds of Biosamples from a wide range of ecological settings (Figure 2). Each Biosample may also yield several different SPs, each of which may yield multiple APs (Figure 3 and Table 1). Study IDs are referred to as ‘Gs’ IDs in the new system. A GOLD study is analogous to the NCBI's umbrella BioProject, and may contain one or more NCBI BioSamples.
Figure 2.
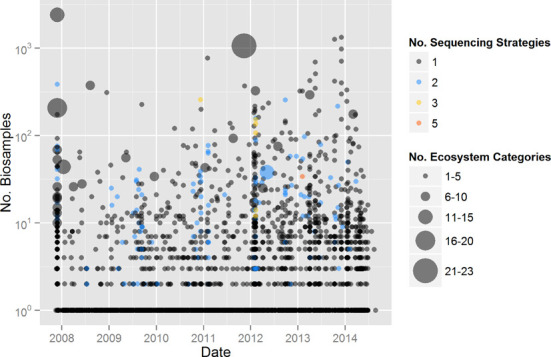
Study Biosamples, ecosystem categories and sequencing strategies. Each point is a GOLD study. The size of the point represents the number of ecosystem categories within a Study. The position on the y-axis denotes the number of Biosamples within a Study. The color of each point indicates the number of unique sequencing strategies used within a Study.
Figure 3.
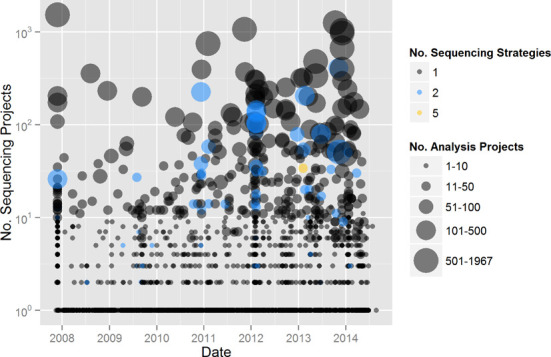
Sequencing and analysis projects per Study over time. Color denotes the number of sequencing strategies used within a Study. The size of the point indicates the number of analysis projects within a Study.
Table 1. GOLD sequencing strategy combinations used within a Study.
| Sequencing strategy combinations | No. Studies | No. Sequencing Projects |
|---|---|---|
| WGS | 18 211 | 46 905 |
| Metagenome | 403 | 3315 |
| Transcriptome, WGS | 76 | 1989 |
| Metagenome, metatranscriptome | 39 | 927 |
| Metagenome, metatranscriptome, targeted gene survey | 6 | 682 |
| Transcriptome | 402 | 596 |
| Metagenome, WGS | 1 | 217 |
| Metatranscriptome | 9 | 54 |
| Metagenome, targeted gene survey | 4 | 53 |
| smRNA, transcription start site, transcriptome, transposon mutagenesis sequencing, WGS | 1 | 34 |
| Metatranscriptome, targeted gene survey | 1 | 21 |
| Methylation | 4 | 15 |
| smRNA, transcriptome | 1 | 14 |
| Plasmid | 2 | 2 |
| smRNA | 1 | 1 |
| Transposon mutagenesis sequencing | 1 | 1 |
GOLD Biosample
Biosamples provide a description of the individual environmental sample, from which the organism or genetic material (DNA or RNA) was isolated for downstream SPs. There are two types of Biosamples, organisms and biomes (environmental samples). Historically, samples were either isolated organisms for WGS or environmental samples for metagenomics. However, it is becoming increasingly common to apply multiple sequencing techniques to a single sample and thus initiating several different SPs from the same starting material. For example, from a single biosample, DNA/RNA can be extracted for a metagenome and a metatranscriptome SP, as well as cells isolated for single-cell genome projects (Figure 2) (19). The need to manage and organize this type of complexity has led to the creation of GOLD Biosamples, which are quite distinct from NCBI's Biosamples. While GOLD Biosamples are organized above the sequencing projects in order to provide linkage of multiple sequencing projects originating from the same physical sample, NCBI's Biosamples are associated with individual sequencing projects, providing metadata only for that sequencing project. NCBI's Biosamples are also used in lieu of BioProjects to represent individual sequencing projects as in the case of multi-isolate projects. GOLD Biosample IDs are represented as ‘Gb’ IDs.
GOLD Sequencing Project
A number of technological advances have enabled an increasing diversity of SP types (Figure 3 and Table 1). SPs represent individual sequencing deliverables such as metagenomes, metatranscriptomes, 16S sequences, single-cell genome sequences, isolate transcriptomes or isolate whole genome sequences. As mentioned above, material from one Biosample can be the basis for more than one SP. GOLD SP's are often connected to a single NCBI BioProject, which could lead to the misconception that there is a one-to-one analogy between them. NCBI's BioProjects represent a mixture of project types that include the umbrella or multi-isolate types that are more analogous to the GOLD's Studies. This lack of standardization in NCBI BioProjects is one of the data management challenges the new GOLD classifications aim to address. GOLD Sequencing Project IDs are represented as ‘Gp’ IDs. Each sequencing project can contain one or more APs.
GOLD Analysis Project
APs represent individual data processing methodologies or approaches that are undertaken for a given SP. As the diversity of data processing and analysis (e.g. assembly, structural and functional annotation) methods has increased, so has the diversity of APs (Figure 3). More specifically, the data generated from a single SP may be processed through multiple different approaches, as researchers have been exploring various different assembly methods or the same assembly with different annotation parameters. As shown in Figure 1, a researcher may also generate a combined assembly from multiple SPs and submit the data for annotation as one AP. This is more common in the case of single-cell genome projects where sparse sequence data from two related single cells can result in a better assembly and thereby more coverage of the genome of the organism being studied. One of the major limitations of the previous systems was the inability to represent these complex APs with their parent SPs. The current release fills this unmet need in representing different APs. AP IDs are represented as ‘Ga’ IDs, and there are currently seven different types:
Default AP. This represents the standard assembly and annotation process applied for any sequencing project.
Default-screened AP and default-unscreened AP. These are applicable only for single-cell genome projects where contamination is a major issue due to extraneous DNA or due to errors during cell sorting/isolation events. Accordingly, there is a need to distinguish between APs that have gone through a decontamination round (screened) and those that have not (unscreened).
Combined assembly AP. These APs use data from multiple SPs that are combined into a single assembly, which is then submitted for annotation. For example whole genome shotgun sequencing may be applied to a set of single-cell genomes from the same Biosample and the data from each single-cell genome can be used to generate a combined assembly for a better genome reconstruction. Alternatively, metagenomic sequences from multiple different Biosamples may be combined into a single assembly. Tracking these many relationships between Biosamples, SPs and APs within a study is a key feature of new GOLD.
Genome from metagenome AP. These APs represent individual genomes extracted from metagenomics data. Advances in metagenomic assembly and binning (http://ggkbase.berkeley.edu/; (23)) have enabled the reconstruction of partial or entire genomes directly from metagenomic sequencing project.
Reassembly AP. This represents the APs created when an already processed genome is subjected to different assembly methods to generate a new assembly.
Reannotation AP. This represents the AP created for annotating a genome that has been annotated before.
Metatranscriptome mapping AP. These APs represent the mapping of the metatranscriptomic data on the metagenomic sequences in order to connect functional processes to genes.
GOLD BY NUMBERS
Studies
As of September 2014, there are 19 242 studies in GOLD. These include 472 metagenomic studies (i.e. have at least one metagenome sequencing project) and 18 770 non-metagenomic studies. Studies have been generally growing in size and complexity and are increasingly composed of Biosamples from more diverse environments (Figure 2). There are also an increasing number of sequencing strategies applied to each Biosample (Figure 2 and Table 1) as well as a growing number of APs used within a study (Figure 3).
Biosamples
There are currently 56 403 Biosamples in the database which are classified as host-associated (11 755 samples), engineered (1563 samples), environmental (6619 samples) and unclassified (36 466 samples). Organism Biosamples represent more than 150 GOLD phylogenetic classifications. Biome Biosamples represent more than 200 unique GOLD ecosystem classifications.
Sequencing Projects
There are currently 56 458 sequencing projects reported in the database. These include 47 932 WGS projects distributed across 36 824 bacteria, 5822 eukaryal and 851 archaeal projects. There are also 4351 metagenomic SPs, distributed across 1567 host-associated, 239 engineered and 2545 environmental projects. In addition to the genomic and metagenomic SP, the database provides information on 1200 transcriptomic and 797 metatranscriptomic SPs. While there are only 34 targeted gene survey SPs, all of these are part of studies that include metagenomic data and most include metatranscriptomic data (Table 1). The database also provides information on 13 transposon mutagenesis SPs. As this technique is becoming more high-throughput more projects of this type can be expected (19). A similar growth is expected for the methylation SPs, only 15 of which are currently available in the database.
Analysis Projects
Thirty-eight thousand five hundred seventy-three APs are currently reported of which 36 755 are default APs. For single-cell SPs there are 856 default-screened and 1082 default-unscreened APs. There are also 107 transcriptome mapping and 80 metatranscriptome mapping APs. Finally, 30 combined assembly APs from 310 SPs in 11 studies are available in GOLD. All of the sequencing projects used for combined assembly were also used for ‘default’ APs.
ACCESSING GOLD
GOLD provides free access to all publicly available data, project status reports and other statistical information. Data can be accessed by various pre-computed reports or by querying the database using search functions. Menu tabs to allow users to choose Search, Distribution Graphs, Biogeographical Metadata and Statistics options to access data are also available from the front page. A list of all public projects in the database is also available for download.
Distribution Graphs
Automatically generated pie charts that describe the different types of projects in the database are now available. These include data organized by SP type, sequencing status, phylogenetic table, phylogenetic tree and Biosample classification in separate tabs.
Biogeographical Metadata
The geographic distribution of Biosamples can be visualized via the Google Map and Google Earth options. These can also be used to select Biosamples based on their geographic location. The Google Map feature aggregates Biosamples by geographic location into circles noting the number of Biosamples in a group when viewing larger spatial extents. These groupings are ungrouped as the map view is focused using the zoom feature. The map view can be focused on the location of a biosample when it is selected from a list next to the map. The Google Earth feature provides a similar tool but with a 3-dimensional global perspective.
Statistics
The GOLD statistics page provides several pre-computed user friendly, easy to interpret graphs, bar charts and pie charts about various sequencing projects. Refer to the Supplementary material for more details about various pre-computed charts.
SEARCHING THE GOLD DATABASE
The Search function can be used to query the database based on various search criteria that encompass all four levels of the project classification system or based on various metadata features. A drop-down menu allows the choice of three search options, Quick Search, Advanced Search and Metadata Search.
Quick Search
Quick Search allows a user to search through the most frequently used fields/identifiers across the four levels in the database (Studies, Biosamples, Sequencing Projects and APs).
Advanced Search
Advanced Search provides options to query metadata fields in each level of the new classification system. Results are provided as a list according to the search criteria, with fields used displayed in separate columns. Search result can be redefined by removing any search term by clicking ‘remove’ next to the search term in the column header. Search results may also be refined directly in the results table by modifying the search term to any field by clicking the ‘+’ under the column header. There is also a ‘Select Fields’ button on the left, which allows the user to add additional fields.
Metadata Search
Metadata Search is designed to query the database using various metadata identifiers. These include the classification by the domains of the project organism, Archaea, Bacteria, Eukarya or all. The various search tabs contain graphical and tabular representation of the numbers of projects or organisms. This approach serves to obtain an overall picture of projects and samples according to chosen criteria which produces a sortable table and also plots these lists in a pie-chart for easy reference.
CREATING AND EDITING PROJECTS IN GOLD
Registered users can submit new projects or edit their existing entries.
Editing
Existing projects can be updated using a new inline-editing user interface. For editing existing entries, a user needs to login and select the entry of their interest. When clicking on a field, an edit box is launched with existing values in it. One can update the value and save. The inline-edit feature seamlessly integrates the edit functionality with user interface without the need for launching a separate edit form.
Creating new projects
Registered users can create new SPs using the new project entry interface. Creating a new SP also requires defining all related database entities like Study, Biosample and Organism when applicable for isolate genome projects. As shown in the Supplementary material, the new project entry landing page provides the following options: (i) create a new SP, (ii) create a new AP; (iii) review your Studies, Biosamples and Sequencing Projects.
The new SP creation interface will walk a user through a series of steps to define new projects or select existing projects. For example, launching ‘Create a new Sequencing Project’ will first ask if this is metagenome (biome) or isolate (organism) project. This information is used to launch appropriate forms and guide users through the process. Next, a user will be asked to enter a Study for the SP. If this is a returning user adding additional SPs to an existing study, the user will be able to choose the existing study. Otherwise the user will be asked to define a new one. Once the Study is created a Biosample must be defined. Again the user may define a new Biosample or select an existing Biosample. If the SP is for an isolate organism, the user must select an existing organism from the database or define a new organism. After the Study, Biosample and/or organism are created, the user will be able to define a new SP. All the required fields are marked with an asterisk and tool tips are provided with appropriate examples to guide a user in defining new projects. Help pages are available to provide explanation on specific database terminology. If an SP is defined a user can select ‘Create a new Analysis Project for submission to IMG’ to define an AP. A single SP can have multiple APs to represent different assemblies and/or gene calling methodologies applied. Study, Biosample and Organism entries created but not yet associated with a sequencing project are saved as drafts. Users can access these from the ‘My Data’ table as well as select these from the pull-down list as part of new SP creation interface.
DATA IMPORT AND CURATION CHALLENGES
GOLD continuously monitors sequencing projects around the world both through direct submissions from users and through data imports from major public resources, such as NCBI (7). A series of cross checks have been implemented to ensure high data quality, manually verify data conflicts and curate metadata during and after import into the database. Due to the nature of data organization and data quality enforcement standards at different public resources, it is challenging and curation intensive to keep the import processes working. For a list of examples see the Supplementary material.
The aim of listing these issues is 2-fold: (i) to express the difficulties that any integrated public database resource like GOLD is facing in representing disparate information and (ii) to highlight the need for more manual data curation and quality control checks at major public resources like NCBI. If the data are corrected at the source, it saves time and effort for several groups around the world. For example, correctly representing the sequencing center names and geographic coordinates at the source would eliminate the need for all other databases who import data from NCBI to come up with their own procedures for finding and resolving these issues. NCBI systems serve as a large democratizing force providing unrestricted access for users around the world to submit their data and freely share with the rest of the world. With such a broad mandate and unhindered access, it is difficult to enforce strict standards, but at least some of the above listed issues can be mitigated with more manual curation and quality control processes in place. These challenges are not unique to this database, but to all who rely on public database resources. Thus, there is a strong case for the stakeholders and funding agencies to support data curation efforts at public resources (23).
FUTURE DIRECTIONS
Future developments will focus on data integration, expanding metadata fields and providing sophisticated search options across the metadata fields at different classification levels.
Data integration. We will continue importing public metagenome sequencing projects from NCBI and EBI. We will expand our semi-automatic NCBI isolate genome import process to include multi-isolate NCBI BioProjects, where more than one isolate genome is listed under a single NCBI BioProject with different NCBI BioSamples as opposed to represented by individual NCBI BioProjects.
Expanding metadata fields. The growing complexity of the SPs and the diversity of the GOLD Biosamples collected from specific locations and conditions necessitate GOLD to constantly expand metadata fields. We plan to incorporate all of the MIxS environmental packages and include metadata fields that are not currently available in the database.
Metadata Miner. The advanced search feature in the current release provides an option to search among a multitude of metadata fields within each of the four project classification levels. For data mining and hypothesis generation often it is important to search across different levels using different metadata fields at the same time. For example the search for ‘aerobic bacterial WGS projects that have a project relevance of medical, with human as a host and project status of complete’ in the current implementation would need to be executed in multiple steps at different GOLD classification levels. We plan to implement an integrated Metadata Miner that would facilitate complex searches across all four levels of GOLD. Such an advanced metadata mining tool will make it easy for users to execute searches similar to the above example.
CONCLUSION
The steady increase in the number of sequencing studies carried out around the world coupled with the complexity of the samples, diversity of sequencing strategies and expanding analysis methods necessitates an integrated metadata warehouse like GOLD. As outlined above both through our current release and proposed feature enhancements like Metadata Miner, GOLD is uniquely positioned to organize sequence metadata and provide unhindered access both for hypothesis generation and testing. GOLD's rich metadata coupled with seamless integration with the IMG analysis systems provides users with the ability to look at their data and analyze results from a whole different perspective with associated metadata. This helps in understanding the observations as well as asking questions to find answers hitherto impossible without curated metadata. Toward this goal, GOLD will continue expanding in terms of metadata fields as well as the numbers of projects integrated from various sources around the world.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to all the colleagues who kindly provide information for the more accurate monitoring of the genome projects and their associated metadata. We thank the members of the microbial genomics and metagenomics programs at the JGI for support, useful discussions and exchange of ideas. We would also like to thank Lynn Schriml and Lynette Hirschman for feedback and useful discussion on the EnvO ontology. Statistical analysis and visualizations were generated using R with ggplot2.
FUNDING
This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract no. DE-AC02-05CH11231. Funding for open access charge: US Department of Energy.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kyrpides N.C. Genomes OnLine Database (GOLD 1.0): a monitor of complete and ongoing genome projects world-wide. Bioinformatics. 1999;15:773–774. doi: 10.1093/bioinformatics/15.9.773. [DOI] [PubMed] [Google Scholar]
- 2.Bernal A., Ear U., Kyrpides N. Genomes OnLine Database (GOLD): a monitor of genome projects world-wide. Nucleic Acids Res. 2001;29:126–127. doi: 10.1093/nar/29.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liolios K., Tavernarakis N., Hugenholtz P., Kyrpides N.C. The Genomes On Line Database (GOLD) v.2: a monitor of genome projects worldwide. Nucleic Acids Res. 2006;34:D332–D334. doi: 10.1093/nar/gkj145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liolios K., Mavromatis K., Tavernarakis N., Kyrpides N.C. The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2008;36:D475–D479. doi: 10.1093/nar/gkm884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liolios K., Chen I.-M., Mavromatis K., Tavernarakis N., Hugenholtz P., Markowitz V.M., Kyrpides N.C. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2010;38:D346–D354. doi: 10.1093/nar/gkp848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pagani I., Liolios K., Jansson J., Chen I.-M., Smirnova T., Nosrat B., Markowitz V.M., Kyrpides N.C. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012;40:D571–D579. doi: 10.1093/nar/gkr1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benson D.A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2013;41:D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leinonen R., Akhtar R., Birney E., Bower L., Cerdeno-Tárraga A., Cheng Y., Cleland I., Faruque N., Goodgame N., Gibson R., et al. The European Nucleotide Archive. Nucleic Acids Res. 2011;39:D28–D31. doi: 10.1093/nar/gkq967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Markowitz V.M., Chen I.-M., Palaniappan K., Chu K., Szeto E., Pillay M., Ratner A., Huang J., Woyke T., Huntemann M., et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42:D560–D567. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyer F., Paarmann D., D'Souza M., Olson R., Glass E.M., Kubal M., Paczian T., Rodriguez A., Stevens R., Wilke A., et al. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Field D., Amaral-Zettler L., Cochrane G., Cole J.R., Dawyndt P., Garrity G.M., Gilbert J., Glöckner F.O., Hirschman L., Karsch-Mizrachi I., et al. The Genomic Standards Consortium. PLoS Biol. 2011;9:e1001088. doi: 10.1371/journal.pbio.1001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yilmaz P., Kottmann R., Field D., Knight R., Cole J.R., Amaral-Zettler L., Gilbert J.A., Karsch-Mizrachi I., Johnston A., Cochrane G., et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 2011;29:415–420. doi: 10.1038/nbt.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markowitz V.M., Chen I.-M., Chu K., Szeto E., Palaniappan K., Pillay M., Ratner A., Huang J., Pagani I., Tringe S., et al. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 2014;42:D568–D573. doi: 10.1093/nar/gkt919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Markowitz V.M., Mavromatis K., Ivanova N.N., Chen I.-M., Chu K., Kyrpides N.C. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 15.Markowitz V.M., Chen I.-M., Chu K., Szeto E., Palaniappan K., Jacob B., Ratner A., Liolios K., Pagani I., Huntemann M., et al. IMG/M-HMP: a metagenome comparative analysis system for the Human Microbiome Project. PLoS One. 2012;7:e40151. doi: 10.1371/journal.pone.0040151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson K.E., Weinstock G.M., Highlander S.K., Worley K.C., Creasy H.H., Wortman J.R., Rusch D.B., Mitreva M., Sodergren E., Chinwalla A.T., et al. A catalog of reference genomes from the human microbiome. Science. 2010;328:994–999. doi: 10.1126/science.1183605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu D., Hugenholtz P., Mavromatis K., Pukall R., Dalin E., Ivanova N.N., Kunin V., Goodwin L., Wu M., Tindall B.J., et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462:1056–1060. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kyrpides N.C., Hugenholtz P., Eisen J.A., Woyke T., Göker M., Parker C.T., Amann R., Beck B.J., Chain P.S.G., Chun J., et al. Genomic encyclopedia of bacteria and archaea: sequencing a myriad of type strains. PLoS Biol. 2014;12:e1001920. doi: 10.1371/journal.pbio.1001920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blow M.J., Deutschbauer A.M., Hoover C.A., Lamson J., Pennacchio L.A., Price M.N., Waters J., Wetmore K.M., Bristow J., Arkin A.P. Poster Session Presented at: Genomics of Energy & Environment User Meeting. Walnut Creek, CA: 2013. Functional Encyclopedia of Bacteria and Archaea. [Google Scholar]
- 20.Ivanova N., Tringe S.G., Liolios K., Liu W.-T., Morrison N., Hugenholtz P., Kyrpides N.C. A call for standardized classification of metagenome projects. Environ. Microbiol. 2010;12:1803–1805. doi: 10.1111/j.1462-2920.2010.02270.x. [DOI] [PubMed] [Google Scholar]
- 21.Kyrpides N.C., Woyke T., Eisen J.A., Garrity G., Lilburn T.G., Beck B.J., Whitman W.B., Hugenholtz P., Klenk H.-P. Genomic encyclopedia of type strains, phase I: the one thousand microbial genomes (KMG-I) project. Stand. Genomic Sci. 2014;9:1278–1284. doi: 10.4056/sigs.5068949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rinke C., Schwientek P., Sczyrba A., Ivanova N.N., Anderson I.J., Cheng J.-F., Darling A., Malfatti S., Swan B.K., Gies E.A., et al. Insights into the phylogeny and coding potential of microbial darkmatter—Supplementary Information. Nature. 2013;499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 23.Kyrpides N.C. Fifteen years of microbial genomics: meeting the challenges and fulfilling the dream. Nat. Biotechnol. 2009;27:627–632. doi: 10.1038/nbt.1552. [DOI] [PubMed] [Google Scholar]