

Summary
DNA repair deficiencies are common among cancer cells and represent a potential vulnerability that might be exploited by targeting compensatory repair pathways. However, the identification of synthetically lethal combinations of DNA repair defects, while of significant clinical relevance, has been somewhat anecdotal. While numerous models have been proposed to explain synergy among DNA repair mutations, we have only a limited understanding of why a given mutation should render cells sensitive another. In this issue of Cancer Discovery, Reinhardt and colleagues define a general connection between mutations in genes involved in homologous recombination and sensitivity to inhibitors of non-homologous end joining. In doing so, they provide a mechanism to demarcate a set of seemingly diverse tumors that may be highly responsive to established DNA repair-targeted therapeutics.
A central objective of cancer genome sequencing efforts has been to pair specific genomic alterations with actionable therapeutic strategies. This objective stems from the idea that mutations that promote cancer development also represent vulnerabilities that can be exploited given the appropriate therapy. Noteworthy examples of this phenomenon exist in the realm of oncogene addiction, in which the expression of mutant driver oncogenes renders tumor cells exquisitely sensitive to targeted oncogene inhibition. This approach has led to dramatic, albeit sometimes transient, responses in numerous treatment-refractory tumors. However, this type of mutation-targeted therapy represents the exception as opposed to the rule for the treatment of most cancers.
Perhaps the most conceptually advanced paradigms for pairing tumor mutations with targeted therapies stem from studies examining alterations resulting in genomic instability or abrogation of the DNA damage response (1). These studies have yielded two basic strategies to target tumor cell vulnerabilities. The first strategy is to disable a cancer cell’s ability to undergo cell cycle arrest in response to DNA-damaging chemotherapy. Cells with persistent damage undergo a process termed “mitotic catastrophe” if unable to undergo G1/S or G2/M cell cycle arrest. Given that tumor cells frequently carry mutations that abrogate cell cycle checkpoints, targeted therapies that block remaining cell cycle arrest pathways would be expected to have pronounced tumor-specific effects. This process has been described in studies examining combined p53 and ATM (2), p53 and MK2 (3) and p53 and Chk1 deficiency (4, 5). In each case, p53-deficiency disables the G1/S checkpoint, while therapies targeting mediators of the G2/M checkpoint promote mitotic catastrophe in the presence of genotoxic damage.
A second, analogous, approach is to profoundly disrupt a cancer cell’s ability to repair DNA damage. Genomic instability is a common feature of tumor cells and is thought to represent a basic mechanism to acquire the multiple genetic alterations required during the neoplastic process. Cells showing genomic instability retain some capacity to repair DNA damage, as a complete absence of repair mechanisms would lead to a dramatic loss in genomic integrity – particularly in the presence of DNA-damaging therapy. Thus, tumor cells with partial defects in DNA repair are potentially highly sensitized to therapeutics targeting residual DNA repair capabilities. The most dramatic example of this process involves the treatment of breast and ovarian cancers with Brca1/2 deficiency (6, 7). Inactivation of Brca1/2 results in severe defects in the ability of cells to repair DNA damage by homologous recombination (HR). Small molecule inhibition of Poly ADP Ribose Polymerase (PARP) proteins, involved in a diverse set of cellular processes including DNA repair, specifically promotes the death of Brca1/2 deficient cells. This process is thought to arise through the disruption of parallel DNA repair pathways and is further exacerbated in the presence of DNA damage. An additional example of this kind of combined DNA repair defect occurs when targeting both ATM and DNA-PK (2). However, despite an increasing number of “target pairs” that show synthetic lethality, we still lack a systematic and coherent understanding of which specific DNA repair defects render tumor cells particularly sensitive to targeting specific DNA repair proteins. In this issue of Cancer Discovery, Reinhardt and colleagues provide needed resolution to this issue (8). By probing diverse cancer cells lines for sensitivity to a DNA-PK inhibitor, they cement a connection between defects in homologous recombination (HR) and drugs that target non-homologous end joining (NHEJ). In doing so, they provide a clear rationale for the treatment of a diverse set of tumors bearing similar DNA repair deficiencies.
As a precursor to this study, Reinhardt and colleagues identified ATM deficiency as a tumor alteration that sensitized tumor cells to DNA-PK chemical or genetic targeting (9). While it is clear that DNA-PK plays a central role in double-strand break repair by homologous recombination, ATM is putatively involved in a diverse set of cellular processes including DNA damage signaling and DNA repair. Thus, the basic mechanism of synergy for these two alterations remained somewhat speculative. In the extant study, they used a set of 94 well-characterized cell lines to explore sensitivity to DNA-PK inhibition in a more unbiased manner. The mutational spectra in each cell line allowed them to stratify relative sensitivity to DNA-PK inhibition based on defined genetic alterations. Strikingly, alterations that correlated with the greatest sensitivity to DNA-PK inhibition represented mutations in genes implicated in HR. These included mutations in BRCA2, RAD50, CHEK2, PAXIP and FANCD2, as well as, at lower frequency, mutations in BRCA1 and ATM. The alterations most frequently associated with DNA-PK sensitivity were inactivating mutations in MSH3 – a gene with established roles in mismatch repair (MMR) and HR. MSH3 mutations are found in as many as 7% of all colon cancers (10), making this connection between MSH3 mutation and sensitivity to a targeted therapeutic a finding of considerable clinical relevance. Indeed, a great strength of their approach is that by screening cells lines for sensitivity to a targeted therapeutic, the identification of robust sensitizing mutations leads immediately to a actionable pairing of an existing drug with a cancer-relevant lesion.
While the DNA-PK/MSH3 interdependency represents a clinically important concept, perhaps even more relevant is the global connection between defects in HR and sensitivity to inhibition of NHEJ. To more extensively explore this phenomenon, this authors examined whether their sensitive cell lines actually exhibited HR defects. Using the appearance of Rad51 foci as a marker for HR following DNA damage, they showed an absence of these foci in cells sensitive to DNA-PK inhibition. Moreover HR-defective cells treated with a DNA-PK inhibitor showed a persistence of DNA breaks following genotoxic damage that is characteristic of a global inability to repair DNA damage. Finally, to establish the reciprocal nature of this HR/NHEJ dependency, they examined whether additional NHEJ defects render cells sensitive to MSH3 deficiency. Here, they used RNAi to silence Ku70 and Ku80, two proteins essential for NHEJ, in cancer cell lines with MSH3 mutations. Here, again, they observed synergy between defects in NHEJ and HR.
These data strongly suggest that one can move beyond anecdotal pairs of synthetically lethal drug/genotype combinations and begin to think about how a diversity of defects in the same pathway may yield common vulnerabilities. Thus, tumors might be binned into categories of repair defects rather than more complex mutational spectra. However, multiple significant challenges remain in moving this approach towards clinical implementation. First, our estimation of the relevance of a gene to a specific repair process does not necessarily correlate with the degree to which its inactivation might sensitize cells to a second repair defect. For example, in this study Brca1 and ATM mutations were only weakly correlated with sensitivity to DNA-PK inhibition. Moreover, some, but not other, ATM mutations sensitized cells to NHEJ deficiency. Thus, we need to more completely understand the specific nature of the DNA repair defect to predict the effect of a given mutation has on a repair process. Second, tumors bear a diverse set of alterations, and it may be difficult to infer the effect of any single alteration on global repair processes. In this study, the authors describe a cell line that was heterozygous for a BRCA1 mutation but was, unexpectedly, sensitive to DNA-PK inhibition. Further analysis of this cell line revealed a MSH3 mutation that likely underlies drug sensitivity. Thus, a clear understanding of how mutations act in concert to affect repair processes may be necessary to predict the response to drugs targeting parallel repair pathways.
One solution to these challenges would be to attempt to stratify tumors based on certain functional processes as opposed to specific mutations. Given the idea that we might target global repair defects like “HR-deficiency”, it would be of tremendous value to develop assays that could interrogate ongoing repair processes in human cancers. Most pathological analysis of tumors relies on examining a static entity, yet the identification of specific DNA repair defects requires the observation of DNA integrity over time. In some cases, it requires the introduction of an exogenous substrate into tumor cells to examine a repair process. While such approaches are certainly challenging, they would allow one to define a tumor in terms of an ongoing repair defect as opposed to intuiting a repair defect based on a set of characteristic mutations.
Figure 1.
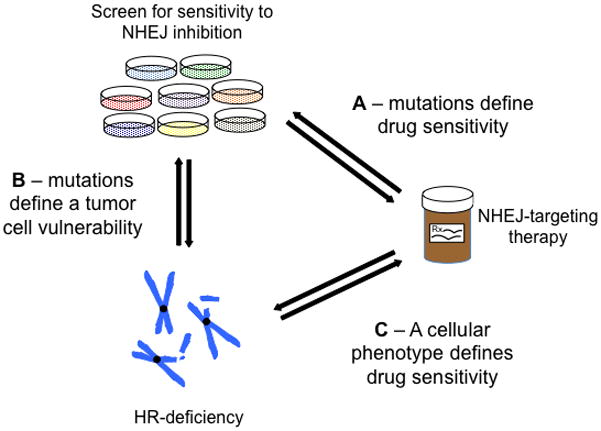
Mutually informative processes govern the use DNA repair inhibitors in repair-deficient cell lines. A) Screening for cell lines sensitive to NHEJ inhibition identifies mutations that define a set of DNA-PK-dependent tumors. B) Sensitizing mutations implicate HR-deficiency as the major defect underlying DNA-PK dependence. This suggests that other HR-mutations not found in this study may similarly sensitize tumors to DNA-PK inhibition. C) Tumor cells can similarly be defined as sensitive to a DNA-PK inhibitor or having defects in HR. This suggests that tumor cells with an HR defect – even in the absence of an HR-related mutation – would be sensitive to DNA-PK inhibition.
Footnotes
The author declares no conflicts of interest related to this manuscript.
References
- 1.Reinhardt HC, Jiang H, Hemann MT, Yaffe MB. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle. 2009;8:3112–9. doi: 10.4161/cc.8.19.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, et al. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;23:1895–909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11:175–89. doi: 10.1016/j.ccr.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mukhopadhyay UK, Senderowicz AM, Ferbeyre G. RNA silencing of checkpoint regulators sensitizes p53-defective prostate cancer cells to chemotherapy while sparing normal cells. Cancer Res. 2005;65:2872–81. doi: 10.1158/0008-5472.CAN-04-2502. [DOI] [PubMed] [Google Scholar]
- 5.Origanti S, Cai SR, Munir AZ, White LS, Piwnica-Worms H. Synthetic lethality of Chk1 inhibition combined with p53 and/or p21 loss during a DNA damage response in normal and tumor cells. Oncogene. 2013;32:577–88. doi: 10.1038/onc.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 7.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 8.Dietlein F, Thelen L, Jokic M, Jachimowicz RD, Ivan L, Knittel G, et al. A functional cancer genomics screen identifies a druggable synthetic lethal interaction between MSH3 and PRKDC. Cancer Discov. 2014 doi: 10.1158/2159-8290.CD-13-0907. [DOI] [PubMed] [Google Scholar]
- 9.Riabinska A, Daheim M, Herter-Sprie GS, Winkler J, Fritz C, Hallek M, et al. Therapeutic targeting of a robust non-oncogene addiction to PRKDC in ATM-defective tumors. Sci Transl Med. 2013;5:189ra78. doi: 10.1126/scitranslmed.3005814. [DOI] [PubMed] [Google Scholar]
- 10.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]