

Abstract
The ubiquitin-proteasome system (UPS) is the main ATP-dependent protein degradation pathway in the cytosol and nucleus of eukaryotic cells. At its centre is the 26S proteasome, which degrades regulatory proteins and mis-folded or damaged proteins. In a major breakthrough, several groups have determined high-resolution structures of the entire 26S proteasome particle in different nucleotide conditions and with and without substrate using cryo-electron microscopy combined with other techniques. These structures bring some surprising insights into the functional mechanism of the proteasome and will provide invaluable guidance for genetic and biochemical studies of this key regulatory system.
The concentrations of proteins in the cell are set by their rates of synthesis and degradation. Since the discovery of a eukaryotic intracellular protein degradation pathway in the 1970s, it has become increasingly clear that degradation is regulated intricately even if this regulation is not as well understood as that of gene expression1–4. Most degradation is controlled by the ubiquitin proteasome system (UPS) in the nucleus and cytosol of eukaryotic cells5. At its centre is a proteolytic machine, the proteasome, which is almost as large as the ribosome with its relative molecular weight of 2.5 MDa. The proteasome must be able to degrade any protein in the cell but do so with exquisite specificity. Indeed, the proteasome can even extract individual subunits out of complexes, leaving the rest of the complex intact6, 7. Specificity is achieved in part by a tagging system. Proteins are typically targeted to the proteasome by chains of at least four copies of the 8.5 kDa protein ubiquitin. The first ubiquitin is attached to a lysine residue in a target protein, the second ubiquitin is attached to a specific lysine in the first ubiquitin, and so on. Ubiquitin tags are also used to control other cellular events such as membrane trafficking and chromosome condensation; indeed ubiquitin was first discovered as a chromatin modifier attached to histone tails8–11. However, in the case of non-proteolytic functions for ubiquitin, the tags consist of a single ubiquitin or chains in which the ubiquitin moieties are linked to each other through different lysine residues12, 13. In vitro degradation experiments indicate that ubiquitin tags alone are not enough to target folded proteins for proteolysis, and proteasome substrates must also contain an unstructured region at which the proteasome can initiate degradation14.
Once degradation begins, the proteasome runs along the polypeptide chain of the substrate, hydrolysing ATP to ADP and cutting the substrate protein sequentially into small peptides, while the ubiquitin tag is cleaved off and recycled15, 16 (Figure 1). This allows the proteasome to remodel protein complexes by degrading only the subunit at which it first initiates degradation6, 17–19. It also ensures that the activity of the target protein is removed in its entirety. Many regulatory proteins are designed from modules and leaving one of these undegraded could create an unwanted activity. Nonetheless there are a handful of cases in which the proteasome sculpts proteins by degrading them partially, for example, to remove an inhibitory domain or an activating domain in response to a signal20–26. Thus, the proteasome is a powerful protease, the destructive potential of which is finely tuned.
Figure 1. Steps of proteasomal degradation.
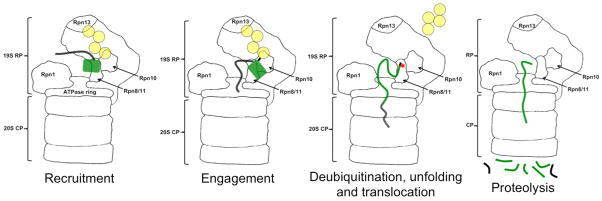
The proteasome recognizes ubiquitin tags in substrates through its receptors, here Rpn10 and Rpn13, and then initiates degradation at an unstructured region in the substrate. As the ATPase motors pull the substrate into the degradation channel, the ubiquitin chain is cleaved off, the substrate unfolds and is finally cleaved into peptides.
In this Review, we highlight the recent advances that have been made in the structural understanding of the proteasome, in terms of how it is arranged as a network of subunit interactions and the resulting implications for substrate recognition and its mechanism of action as an ATP-dependent machine.
A complex of two halves
The proteolytic core particle
The proteasome is a complex of ~33 different proteins arranged into an elongated particle composed of a central core with cap structures at one or both ends. At the centre is the ~700 kDa 28 subunit core particle, which contains the proteolytic active sites. By the early 2000s, atomic-resolution crystal structures of the 20S core particle from a few different species, including from yeast and mammals had been solved27–29. These structures showed that the subunits of the core particle are arranged into four seven-subunit rings: two rings consisting of related α subunits and two consisting of related β subunits. The rings are stacked on top of each other to form a compact cylinder with two β subunit rings in the centre that are flanked at each end by a ring of α subunits. The particle is hollow and contains a set of large chambers running along its central axis that are accessible only through a narrow pore at each end of the core particle27–30. The proteolytic sites of the proteasome are located on three of the β subunits in the central cavity. They show relatively weak preferences for specific target amino acids but together they can cleave almost any sequence31. These proteolytic sites pose no danger for cellular proteins because the pores at the entrance to the core particle are so narrow that folded proteins cannot pass through the pores without being unfolded27, 28, 30. Thus, a major determinant of the proteasome’s specificity lies in its quaternary structure: the proteolytic activity is sequestered away and access to it is controlled sterically. This strategy of controlling catalytic activity by restricting access to reactive sites is similar to the approach used by subcellular compartments such as mitochondria, except that the proteasome forms the compartment itself from a protein shell32, 33.
Controlled access through regulators
The pores at the entrance to the degradation channel on the core particle are gated. They are generally closed in the free core particle and can open once activators or caps dock onto the core particle30, 34–36. There are three or perhaps four different types of activators37. The 11S (PA28 and REG in mammals) and Blm10/PA200 caps stimulate protein degradation without ATP hydrolysis and without recognizing ubiquitin5. Presumably, they have relatively specialized functions and allow largely unstructured proteins or peptides to pass through the degradation channel. The 19S regulatory particle recognizes ubiquitin tags and hydrolyses ATP to unfold its substrates and translocate them into the core particle. The majority of proteasomes in the cell are thought to contain one or two 19S regulatory particles in combination with a 20S core particle and this form is referred to as the 26S proteasome5. However, hybrid proteasomes that are comprised of a 19S regulatory particle on one end of the core particle and a different activator at the opposite end also exist but it is not yet understood how the different caps work together5, 38–41. Recently, it was discovered that yet another form of the proteasome may exist in cells in which the AAA protein Cdc48/p97/VCP serves as yet an additional ATPase cap42, 43.
The 11S cap was the first to be understood structurally through a high-resolution crystal structure of a yeast core particle bound to an 11S cap from Trypanosome34, 44. The 11S cap consists of a ring of seven identical subunits, which dock onto the core particle through their C-termini. Activation loops from each of the 11S cap subunits inserts into cavities that exist between α subunits of the core particle and induces rearrangements in the α subunits to open the central pore34, 45. We know from biochemical experiments that the 19S regulatory particle docks onto the core particle by a similar mechanism46–48.
The 19S regulatory particle
The 19S regulatory particle is a ~900 kDa complex of at least 19 subunits and two catalytic activities: ATP-hydrolysis and a specialized proteolytic cleavage that removes ubiquitin chains from substrate proteins. Biochemical experiments organized the regulatory particle into two sub-complexes, the base and the lid49. The base consists of six ATPases (Rpt1-6; we use yeast S. cerevisiae names here unless otherwise noted): two large organizing subunits, Rpn1 and Rpn2; and two established ubiquitin receptors, Rpn10 and Rpn13. The lid consists of a deubiquitinating enzyme Rpn11, which is a MPN/JAMM domain metal protease, its homologous but non-catalytic binding partner Rpn8, as well as seven scaffolding subunits (Rpn 3, 5, 6, 7, 9,12 and 15 (also called Sem1))5. The scaffolding subunits, except for Sem1, contain protein-protein interaction motifs called PCI (Proteasome–CSN–eIF3) domains5.
In addition to the stoichiometric proteasome subunits, a large number of proteins associate with the 19S regulatory particle to modulate degradation. Some of these, such as the UbL-UBA proteins Rad23, Dsk2 and Ddi1, serve as alternative ubiquitin receptors; some, such as Ubp6 in yeast and Uch37 and Usp14 in mammals, trim ubiquitin chains; and others, such as yeast Hul5, extend the ubiquitin chains of substrates on the proteasome50–59.
New structural understanding
The first high-resolution structure of proteasome complexes with ATPase caps came from the simpler archaeal proteasome particles60, 61. Structures of the 26S proteasome in its entirety came from electron microscopy studies, often combined with mass spectrometry and biochemical methods such as protein-protein crosslinking49, 62, 63 and by 2010 the first sub-nanometre resolution structure of the complete 26S proteasome emerged64. This structure started to place individual subunits within the particle but the locations of many of them, including the substrate receptors, remained ambiguous. Over the past two years, a series of papers from several groups have revealed the structure of the 19S regulatory particle and the whole proteasome from both yeast and human65–70. The initial set of reports presented proteasome structures that were prepared in the presence of saturating ATP levels 65–68 whereas the more recent papers show proteasome in the presence of substrate69 or with a slowly hydrolysable ATP analogue (ATPγS)70. These studies combined high-resolution EM structures of the particle with crystal structures and homology models of isolated subunits, and a wealth of biochemical, proteomic and genetic analyses to determine the structure of the 26S proteasome71, 72. Together, the approaches place all of the known stoichiometric subunits within the particle and in many cases orient their crystal structures well enough to interpret information at atomic resolution in meaningful ways65–68, 73. The substrate-free proteasome structures from the different organisms agree with one another in large part, which highlights that the proteasome structure is largely conserved from yeast to mammals65–68. The substrate- and ATPγS-bound proteasome structures show some intriguing differences compared with the ATP substrate-free structures and it seems that the two sets of structures represent different states of the proteasome, perhaps a resting state for the ATP substrate-free structure and activated states for the substrate-bound and ATPγS structures69, 70.
A fresh view of regulation
The initial models of the proteasome showed the base of the regulatory particle docked onto the core particle through a ring of ATPase subunits. This base was adjacent to a seemingly more loosely attached lid63. The first surprise from the new structures is that they show the regulatory particle as a cohesive structure with a network of interactions within the cap and with the core particle to form an integrated 26S proteasome65–68.
The six ATPases of the base, Rpt1-6, form a ring that docks onto the core particle just as observed in the earlier structures. Each ATPase subunit is composed of extended α helices near the N terminus, followed by an oligonucleotide/oligosaccharide-binding (OB) domain, and then AAA+ domains before the C terminus60, 74. The AAA+ domains and the OB domains each form rings with central channels65–68. The AAA+ domains contain the elements required to bind and hydrolyse ATP and to dock onto the core particle75. The C-terminal tails of three of the AAA+ domains insert into cavities at the surface of the core particle to open the pore to the degradation channel; this gates the passage to the proteolytic sites, reminiscent of the 11S cap structure with the core particle46–48. On the opposite site of the AAA+ domains, the OB domain ring resides next to the AAA+ ring but does not pack tightly against it65–68. Finally, the N-terminal α-helices form coiled-coils with neighbouring ATPase subunits such that three pairs of α-helical coils protrude from the base towards the outer ends of the proteasome cylinder. Thus, the ATPase subunits form an assembly that reaches over 100 Å from the core particle through about two thirds of the regulatory particle and participate in a network of interactions that may allow communication between substrate receptors and the ATPase motor. The organizing subunit Rpn1 binds to one outer side of the ATPase ring near the core particle and Rpn2 stacks on the top of the ATPases at the distal end of the proteasome. The ubiquitin receptors Rpn10 and 13 are located at the periphery of the proteasome particle and near its distal end, where they seem appropriately positioned to capture substrates65–68. Rpn13 binds to Rpn2 and is almost as far away from the centre of the proteasome as possible. Rpn10 binds near Rpn2 to the MPN/JAMM domain protein Rpn11, about a quarter turn away from Rpn13 and slightly closer to the ATPase ring65–68.
The lid binds to the side of the base and forms a wide network of interactions. Two of its subunits, Rpn5 and Rpn6, extend all the way to the core particle and contact it directly65–68 (Figure 2). These contacts form the hinge for a slight rotation of the regulatory particle when substrate binds69. The PCI domains in the scaffolding subunits (Rpn 3, 5, 6, 7, 9, and 12) form a U-shaped structure from which arms of α-helical repeats radiate out. This allows them to connect to the organizing subunit Rpn2, the deubiquitinating subunit Rpn11, the ATPase ring and α subunits of the core particle65, 66, 76. The closed bottom of the U-shaped structure faces towards the core and the opening points towards the distal end65–68. The deubiquitinating subunit Rpn11 and its binding partner Rpn8 are near the open end of this U structure and almost connect the two arms. Rpn11 is also in contact with Rpn2 at the top of the structure. Thus, the lid seems to help attach the regulatory particle onto the core and to stabilize the entire assembly76. This interpretation of the structure is supported by genetic data that show that lid subunits are required for proper proteasome assembly76–78. The network of contacts formed by the lid may also integrate and modulate the enzymatic functions of the different proteasome subunits and provide an interface for allosteric regulation65–68. Indeed, biochemical studies have shown that the binding of ubiquitinated substrates to the proteasome can enhance ATP and peptide hydrolysis rates, suggesting that the proteasome motor may operate in different ‘gears’, as has been demonstrated for related bacterial proteolytic machines79–84.
Figure 2. A scaffold of PCI domain containing subunits integrates RP subunits and the core particle.

The structure of the proteasome in the presence of ATP but without substrate shows the six PCI domain containing subunits (shown in yellow) form a U-shaped structure that interacts with many subunits in the RP and a ring of α-subunits in the core particle (shown in dark grey). These interactions probably stabilize the RP and its interaction with the proteolytic core. They may also provide a mechanism for cooperative interaction of the different biochemical activities of the various subunits. The six ATPase subunits are shown in blue, the ubiquitin receptors Rpn10 and Rpn13 are shown in red, the organizing subunit Rpn2 in orange, and the metallo protease subunits Rpn8/11 heterodimer is in light purple. The figure was generated from the EM structure (EMD-1992) from Lander et al., 2012 and atomic coordinates from Beck et al., 2012 pdb file (PDB code: 4B4T)65, 68. All models were generated and visualized with the UCSF Chimera package154. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). The crystal structures (pdb ID 4B4T) were fitted into the experimental electron microscopy map (EMDB 1992) with the algorithms provided by Chimera and corrected manually. The Segger package was used for the segmentation of the electron density map155.
A puzzling aspect of the ATP substrate-free proteasome structures is that they lack a clear, continuous channel through which substrate can reach the proteolytic sites. The OB ring of the ATPase subunits, the AAA+ domain ring and the adjacent ring of core particle α subunits each form central channels, but the rings are tilted and shifted relative to one another so that the channels do not align65, 68. The AAA+ domain ring and the entrance to the 20S core particle α ring are offset by as much as 10 Å65. However, in the ATPγS and substrate-bound proteasome structures, subunits in the regulatory particle shift and rotate such that all of the rings become coaxially aligned to form a continuous substrate channel69, 70 (Figure 3a). The central pore in the AAA+ subunits widens considerably as compared to the ATP substrate-free structures and this is consistent with biochemical results that measure increased degradation rates of peptides in the presence of ATPγS instead of ATP69, 70, 85 (Figure 3b). The changes in the substrate-bound and ATPγS proteasome structures seem to be largest in the ATPase subunits and cause the ATPase domain interfaces to become more uniform. They also affect a large number of interactions with neighbouring subunits and are not limited to the ATPase subunits.
Figure 3. Structural rearrangements upon substrate and nucleotide analogue binding.


Cutaway views of the central channels of the proteasome in the substrate-free, substrate-bound and ATPγS conditions. 3a: A continuous translocation channel forms upon ATPγS or substrate binding: Top is the substrate-free condition, where the OB domain ring (dark blue), the AAA+ domain ring (light blue) and the entrance to the core particle (grey) are all slightly out of alignment. Rpn11 (green) sits to the side of the OB domain ring. However, upon substrate binding or ATPγS conditions, the rings shift and tilt such that the central pore of each of the rings is aligned coaxially. Thus, a continuous central channel forms in the bottom two structures. Rpn11 moves directly over the OB-ring in both the substrate-bound and ATPγS conditions, aligning its catalytic active site with the translocation channel and possibly preventing substrate escape. 3b: Central pore of AAA+ domain ring expands considerably after substrate binding: Cutaway views looking directly into the translocation channel from the top of the proteasome show that the central pore of the AAA+ ring expands upon substrate binding to allow for substrate entry. AAA+ domain ring is shown in light blue.
The figure was generated from the EM maps from Lander et al., 2012 (EMD-1992), Matyskiela et al 2013 (EMD-5669) and Sledz et al 2013 (EMD-2348)65, 69, 70. All models were generated and visualized with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). The EM map from Sledz et al 2013 (EMD-2348) was segmented using the Segger package154, 155.
Degradation by concerted effort
Recognizing ubiquitin chains
The degradation signal or degron that targets proteins for destruction by the proteasome consists of a polyubiquitin chain as the proteasome-binding tag and an unstructured region as the proteasome initiation site. Biochemical studies suggest that the polyubiquitin chain has to be at least four ubiquitin moieties long86 to bind tightly to the proteasome and it can be much longer in vivo87. The proteasome in turn contains at least two intrinsic ubiquitin receptors (Rpn10 and Rpn13) and works with several additional detachable ubiquitin receptors50, 88, 89. These receptors recognize ubiquitin through small domains: a Pru (pleckstrin-like receptor for ubiquitin) domain in Rpn13, one (yeast) or two (mammalian) UIM (ubiquitin-interacting motif) domains in Rpn10, and one or two UBA (ubiquitin associated) domains in the detachable ubiquitin receptors. Each of these domains recognizes only one or perhaps two ubiquitin moieties and so it is not immediately obvious how the proteasome recognizes longer ubiquitin chains88, 90, 91. The substrate-free proteasome structures show that Rpn13 and Rpn10 are about 90 Å apart from each other65–68 (Figure 4). Polyubiquitin chains linked through Lysine 48 form relatively compact but flexible structures. A chain of four ubiquitin moieties can take up a structure that is long enough to bind to Rpn10 and Rpn13 receptors simultaneously65, 66, 92–94. By requiring two receptors to bind substrates simultaneously, the proteasome would be able to discriminate between the monoubiquitin tags used as protein interaction signals and longer polyubiquitin chains used in degrons66, 95. However, abolishing ubiquitin binding by Rpn10 or Rpn13 affects yeast growth only minimally so that substrate binding by both intrinsic ubiquitin receptors simultaneously is at least not a strict requirement for proteasome degradation88,92.
Figure 4. Canonical ubiquitin receptors and their locations on the proteasome.

The locations of the two well-characterized ubiquitin receptors on the proteasome, Rpn10 (blue) and Rpn13 (cyan), together with the organizing subunit Rpn1 (dark orange) and the deubiquitinase Rpn11 (purple) are highlighted. The remaining proteasome subunits are shown in grey. The locations of Rpn10 and Rpn13 might allow a polyubiquitin chain to bind both receptors simultaneously or they serve as an adaptive platform to feed structurally diverse substrates into the translocation motor (left). Rpn1 provides a docking site for UbL-UBA proteins such as Rad23 that deliver substrates to the proteasome. The figure was generated using the EM structure (EMD-1992) from Lander et al 2012 and atomic coordinates from Beck 2012 pdb file (PDB code: 4B4T)65, 68 using Chimera and Segger as in Figure 2154, 155.
Engaging substrates
The proteasome structure does not resolve details of the substrate itself and we do not know exactly how the proteasome binds the unstructured region of the degron69. Using archaeal and bacterial ATP-dependent proteases as a guide, the receptor is likely to be formed by series of loops in the central pore of the AAA+ domains near the bottom of the ATPase ring that function as paddles that move the substrates through the degradation channel (we discuss the ATPase motor in more detail below). The ubiquitin receptors have to place the substrate such that the unstructured regions can reach the AAA+ domain paddles and to be engaged by the translocation motor19. The ubiquitin-binding site of Rpn10 is near the N-terminal tips of two of the ATPase subunits but is still ~90 Å from the motor loops, while Rpn13 is even further way. The unstructured regions themselves may bridge some of this distance but they have to be at least long enough to reach from the narrow entrance to the substrate channel formed by the OB domain ring to the motor loops. In vitro experiments show that the unstructured regions must be a certain minimum length and at an appropriate place to allow the proteasome to degrade proteins effectively14,96–101. Thus, the length and position of unstructured regions relative to the ubiquitin tag may contribute to substrate selection.
Adaptive design
So far, we have discussed how the proteasome may avoid the unintended degradation of proteins not targeted for destruction. But the proteasome also faces the opposite challenge: it must be able to degrade all the different regulatory proteins and damaged or misfolded proteins that are targeted for destruction during the cellular stress response. Part of the solution to this challenge may be that ubiquitin tags can be attached to several lysine residues in proteins and the ubiquitin chains themselves vary in length and geometry. By binding to different polyubiquitin chains within a substrate or to different ubiquitin moieties within a long polyubiquitin chain, the proteasome would be able to find a productive binding mode to feed their unstructured regions into the translocation channel. The presence of several ubiquitin receptors on the proteasome may allow the proteasome to choose the appropriate receptors to position structurally diverse substrates optimally for degradation. In addition, the large organizing subunit Rpn1 serves as the docking site for the detachable ubiquitin receptors. These receptors bind the proteasome through UbL (ubiquitin-like) domains and bind substrates through UBA (ubiquitin associated) domains50, 102, 103 and UbL and UBA domains are connected through flexible linkers100, 104. The flexible design of the UbL-UBA receptors may provide even more plasticity to orient substrates to place their initiation region into the degradation channel. Finally, it is possible that the UbL-UBA proteins by themselves or in cooperation with Rpn10 and Rpn13 bind to several ubiquitin chains on a substrate simultaneously105.
Genetic experiments indicate that the known ubiquitin receptors on the proteasome play partially redundant roles and that any one receptor is sufficient for yeast survival, at least under normal culture conditions88,92. Indeed, even abolishing Rpn10’s and Rpn13’s ability to bind ubiquitin at the same time as deleting the three known UbL-UBA proteins is not lethal in yeast. This suggests that other proteasome subunits could contribute to substrate binding or perhaps even that the initiation region in degrons might be sufficient to target some proteins for degradation under certain circumstances106. Thus, Rpn1 and its UbL-UBA receptors, together with Rpn10, Rpn13 and the pore in the ATPase ring, build a versatile and partially redundant interaction platform that can engage a wide range of substrates through several recognition surfaces.
Removing ubiquitin chains
The proteasome does not degrade the ubiquitin tag on a substrate. Instead, a protease activity of Rpn11 cleaves the iso-peptide bond between the substrate and the first ubiquitin to release the entire polyubiquitin chain90, 91. Rpn11 resembles traditional metalloproteases but, in the context of the proteasome, ubiquitin cleavage by Rpn11 is dependent on ATP hydrolysis. The simplest explanation for this is that ubiquitin cleavage is coupled to the ATP-dependent process of substrate translocation into the degradation channel5. The proteasome structures place Rpn11 close to the entrance to the degradation channel but, in the ATP substrate-free structures, the proteolytic site of Rpn11 faces away from the direct path of Rpn10 to the channel entrance65–68. In the substrate-bound structure, Rpn11 is shifted and rotated so that its catalytic groove aligns with the path to the central channel through the protease69. Thus, translocation of the substrate into the degradation channel would automatically position the attachment point of the polyubiquitin chain at the active site of the deubiquitinating enzyme.
Two other deubiquitinating enzymes, Ubp6 and Uch37/Usp14, associate with the proteasome and they trim polyubiquitin chains progressively from the distal end of the chains107. Their action is counteracted by ubiquitin-protein ligases such as Hul5/KIAA10, which extend ubiquitin chains on the proteasome58. These different enzymes seem to contribute another layer in substrate selection, though the mechanism is not entirely understood. Furthermore, knowing their exact positioning in the proteasome structure remains a challenge, perhaps due to heterogeneity amongst the proteasome particles or their conformational flexibility65, 71.
Action of the ATPase motor
The proteasome is processive108 and releases partially degraded proteins only rarely20, 22, 23, 109–112. Thus, the motor that drives the proteasome along the polypeptide chain of a substrate has to be able to act on many different sequences while unfolding any domains it encounters. How this happens is an intriguing question of molecular recognition and protein mechanics. At the heart of the motor is a ring of six distinct ATPase subunits, Rpt1-6. The ATPases belong to the AAA+ family, which is a subset of the much larger class of P-type NTPases113, 114. AAA+ proteins have functions ranging from protein degradation to nucleic acid helix unwinding and membrane protein disassembly but despite this diversity, common sequence and structural features characterize the family. The ATPase site lies between a large AAA+ domain, which corresponds to the nucleotide-binding domain of all P-type ATPases, and a small AAA+ domain that is specific to AAA+ proteins75. The ATP binding, hydrolysis and ADP + Pi release cycle is accompanied by re-arrangements of the AAA+ domains relative to each other and these changes drive the different reactions catalysed by AAA+ proteins, and often allow the protein to perform mechanical work. In many AAA+ proteins, the ATPase subunits are arranged in ring-like structures such as closed rings, open rings or spirals and these proteins give some insight into how the ATPase motor of the proteasome might function75, 115, 116.
Bacteria and archaea have ATP-dependent proteases that are functionally analogous to the eukaryotic proteasome. They share a common architecture of cylindrical particles that are built from rings of subunits stacked on top of each other. Their ATPase caps also consist of six AAA+ subunits, although the subunits are identical116, 117. The bacterial and archaeal degradation systems do not use a ubiquitin tagging system for substrate selection and the protease caps thus lack the proteins involved in ubiquitin recognition and processing. The biochemical mechanism of the ATPase motor of AAA+ proteases has been studied in the greatest detail in the bacterial and archaeal systems, particularly for E. coli ClpXP, ClpAP, HslUV as well as archaeal Pan-20S proteasome. The central pore formed by their ATPase subunits is lined with loops that protrude from the large AAA+ domains116. These loops interact with substrate proteins and undergo conformational changes driven by the ATP hydrolysis cycle118, 119. One of the loops contains an aromatic-hydrophobic-glycine motif and is thought to function as a ‘paddle’ that drives the polypeptide chain of the substrate through the channel into the proteolytic core particle64, 118–122. Indeed, ClpXP can generate pulling forces in the range of 20 to 40 pN on its substrate123, 124.
Structures of the bacterial and archaeal proteases indicate how conformational changes in the ATP subunits might move the loops to perform mechanical work. For example, in ClpX, the large AAA+ domain of one ATPase subunit and the small AAA+ domain of the neighbouring ATPase subunit form rigid intersubunit modules or rigid bodies125, 126. The ATPase cycle results in conformational changes at the interface of the AAA+ domains within one subunit, which are propagated throughout the ring by the rigid units formed by the AAA+ domains of neighbouring subunits125, 126. Crystal structures of ClpX and other related bacterial protease ATPase caps show that the rings can be asymmetric so that some subunits are in conformations that are not bound to nucleotide125, 127, 128. The structures are consistent with biochemical studies of many AAA+ proteases, including the eukaryotic proteasome, which show that only a maximum of four nucleotides are bound within the ring at one time85, 129–131. These observations make it unlikely that the conformational changes that drive the mechanical unraveling of substrates happen in parallel in all subunits as they appear to be in two different conformations at any one time125. Whether the ATPase subunits act in a coordinated fashion to hydrolyze ATP and thereby propel substrates through the translocation channel remains a key question and may differ between ATP-dependent proteases from the three domains of life. Coordinated ATP hydrolysis activity has been demonstrated for the ATP-dependent protease, FtsH, and much of the evidence for the proteasome suggests that at least some degree of ATP hydrolysis coordination around the ring exists85, 132. In contrast, for the bacterial protease, ClpX, the conformational changes do not need to occur sequentially around the ring and the motor works well even when individual subunits are inactivated133. Finally, dramatic conformational rearrangements of the ring structure also do not appear to be a general requirement for the degradation cycle for AAA+ proteases, as constraining the topology of ClpX ring with covalent crosslinks such that the ring remains closed during the entire ATPase-degradation cycle affects degradation only minimally126.
Rearrangements of the ATPase subunits
The eukaryotic proteasome is more processive than the bacterial and archaeal proteases and, unlike the analogous machines in non-eukaryotes, its ATPase motor operates as a hetero-hexamer134, 135. The different ATPase subunits have specific, non-redundant roles in the gating of the translocation channel and even in translocation46, 79, 136–139. All six ATPase subunits contain the aromatic-hydrophobic-glycine motif found in the bacterial protease translocation paddles but the arrangement of the ATPase subunits in the proteasome is noticeably different to what has generally been observed for the bacterial proteases and this arrangement changes with the nucleotide bound and the presence or absence of substrate.
In the substrate-free ATP-bound proteasome structure, the AAA+ domains are arranged along a spiral staircase such that there is a substantial offset between the lowest and highest subunit65, 68. Each ATPase occupies a distinct position within the spiral and the arrangement is consistent with results from disulphide crosslinking studies140. In the substrate free ATP-bound proteasome, Rpt6 forms the bridge between the bottom and top ends of the spiral to close the ATPase ring. The ATPγS proteasome structure keeps the AAA+ domains in a spiraled configuration but the spiral is shifted by half a turn so that Rpt5 becomes the bridging subunit70. The bridging subunit takes up a different conformation from the other ATPase subunits and appears to be in a conformation that is unable to bind ATP. In the substrate-bound structure of the proteasome, the ATPase subunits rearrange into a level plane, reminiscent of the more uniform arrangement of ATPase subunits in the bacterial systems. This alignment allows the large AAA+ domains to form rigid body units with the small AAA+ domains of neighbouring ATPase as seen in ClpX 69, 125. However, each individual ATPase subunit is tilted differently relative to the plane of the ring, which places the aromatic-hydrophobic-glycine motif loops in a spiral69 (Figure 5).
Figure 5. The ATPase arrangements in different conditions.

A cartoon representation of the arrangement of the six ATPases of the proteasome and the aromatic-hydrophobic-glycine loop (black loop from each ATPase structure) for each of the conditions in which proteasome structures were determined. Each ATPase is represented with its large and small domain and in a different color. The spiraled arrangement of the AAA+ domains is obtained in both the substrate-free and ATPγS conditions, though the subunits rearrange in the ATPγS condition, so that now Rpt5 “bridges” the spiral. In the substrate-bound condition, the ATPase domains rearrange to a level plane, but the aromatic-hydrophobic-glycine loops are still arranged in a spiral due to the different tiltings of the ATPase domains. The figure is adapted from Beckwith et al. 2013139.
It is not entirely clear what role the ATPase subunit arrangements obtained in the different structures have during the proteasome degradation cycle or for the overall mechanism of substrate translocation and the structure papers discuss several possible mechanisms65, 67–70. The spiral conformation of the substrate-free ATP proteasome structure may represent a low energy or standby state of the machine65, 67, 69, 70. Substrate binding would then switch the structure into an active flat ring arrangement that drives translocation more effectively65, 67, 69. The switching of the ring conformation to the translocation active state might be induced once the ATPases engage the initiation region of a substrate69. Indeed, biochemical studies show that binding of ubiquitinated substrates to the proteasome enhances ATP and peptide hydrolysis, thus activating the enzymatic activities of the proteasome several fold79–82, 138. The flat ATPase ring could then function similarly to the ClpX mechanism, where the ATPase cycle drives conformational changes of rigid inter-subunit bodies and, as a result, their aromatic-hydrophobic-glycine motif loops125. The arrangement of the loops in a spiral may allow them to interact with a longer stretch of the substrate through local motions of the pore loops, or the ATPase subunits might use larger motions as the subunits move through the different tilt angles to thread substrates to the core particle. This does not mean that the ATPase subunits contribute equally to translocation and, in fact, biochemical experiments show that mutations in the aromatic-hydrophobic-glycine motif loops in the different ATPase subunits affect protein degradation and ATPase activity in distinct ways79, 137–139, 141.
An alternative model is that the arrangements of the AAA+ domains observed in the different conditions represent distinct intermediates in the ATPase cycle and that the active proteasome cycles through spiral conformations as it moves substrates through the substrate channel. For example, the aromatic-hydrophobic-glycine motif loops shift positions when the ATPγS and substrate free ATP structures are compared and the movement is different for each subunit. Some loops shift primarily vertically (along the long axis of the cylindrical particle) and these changes may be responsible for substrate translocation, whereas other loops move largely horizontally (in the plane of the ATPase ring) and these loops may be responsible for holding the substrate in place as the machine resets during the ATP hydrolysis cycle70. Both types of mechanisms are thought to occur in other examples of large ATP-dependent protein machines, and comparison of these with the proteasome may help to understand the mechanics of the proteasome-substrate interaction during translocation.
Mechanistic insights from helicases
AAA+ helicases and Rec-A type (which is another subgroup of P-type NTPases) helicases are two families of ATP-dependent machines and the diverse mechanisms by which they translocate their nucleic acid substrates inform the discussion of the proteasome mechanism. The helicases contain rings of ATPase subunits that adopt a variety of conformational states to interact with their substrates65, 67, 142–145. The Drosophila melanogaster helicase MCM (a AAA+ ATPase) and the bacterial Rho transcription termination factor helicase (a Rec-A type ATPase) switch between an inactive conformation characterized by a helical ATPase ring and an active conformation characterized by a flat ATPase ring. The conformation change is triggered by accessory factor binding in the MCM helicase109, 111 and by substrate binding in the Rho transcription termination factor143. Rho termination factor interacts with its RNA substrate through hairpin loops that project from each ATPase subunit into a central pore. In the active flat ring structure, the loops are arranged in a spiral and they are thought to cycle through the different positions of the staircase as the subunits hydrolyse ATP145, 146. A similar arrangement is seen in the Bovine papilloma virus replicative helicase E1 (also a AAA+ protein)145, 146. Thus, these helicases function by a mechanism that is similar to one that envisages the ATP, substrate-free structure of the proteasome representing a resting state for the machine.
In another Rec-A type helicase, DnaB, the ATPase subunits are arranged in a spiral but here the spiral structure represents the active state of the enzyme. A crystal structure shows DnaB in a spiral staircase structure with its substrate bound and the entire ATPase domain is thought to cycle through the different positions of the staircase as part of the translocation mechanism147. Thus, DnaB provides a precedent for a proteasome mechanism in which the ATPase ring undergoes larger structural changes and subunits cycle through the arrangements seen in the different proteasome structures.
Reconstructions of cryo-EM structures represent the most abundant conformations of the proteasome in the given conditions. In contrast to x-ray structures, there are no crystal constraints that restrain individual proteasome particles into particular conformations148. Thus, the cryo-EM reconstructions provide some insight into how variable or dynamic the proteasome conformation might be in a given condition148. The positions of the ATPase subunits are fairly well defined even in the substrate-bound and ATPγS structures and this makes it unlikely that the ATPase subunits are undergoing large conformational changes in the majority of proteasome particles72. As Matyskiela et al. discuss, the structures could represent dwell states of the machine between bursts of translocation activity65, 69, 70. An example of this type of mechanism has been described for the DNA packaging motor of bacteriophage ϕ2969, 149–151. The motor spends most of its time in a dwell phase, even in the presence of substrate, and only a small fraction of time in an active translocating phase, which is characterized by rapid structural changes69, 151. Similarly, the ATPγS and substrate-bound proteasome structures might represent different aspects of a dwell phase in which the machine is in a translocation-competent but not actively translocating state69, 70. Translocation of the substrate would occur in burst phases during rapid ATP hydrolysis around the ring and could even be characterized by the movement of entire AAA+ domains, similarly to the large conformational changes proposed for the DnaB helicase or to local motions in a static ring arrangement. Indeed, the ATPγS and substrate-bound proteasome structures are thought to have more structural variability compared with the ATP substrate-free structure, suggesting that the proteasome conformation under these conditions may be more dynamic and could populate a range of other structural states, however in lower abundance69, 70. Once degradation of the substrate is complete, ATP hydrolysis in the bridging subunit could reset the ring to a resting state, which would be represented by the substrate-free ATP structure.
Thus, the different proteasome structures provide intriguing models for how the proteasome might function but they do not yet yield a definitive picture of how translocation occurs. Future biochemical and structural work should define this mechanism.
Conclusions
Over the past two years, several exciting new proteasome structures have redefined our understanding of this large protein machine that lies at the centre of cellular regulation. They show the proteasome as an integrated structure that allows communication between the different enzymatic activities of the machine. The proteasome seems to be malleable and adaptive, a possible requirement of a changing cellular environment, in which precise control is required for protein turnover. The structures in the presence or absence of substrate, or in different nucleotide conditions, illustrate that the conformational changes, particularly in the motor domains of the proteasome, are even more marked than expected. At the same time, the new structures support many aspects of the current view of proteasome action and will be instrumental in generating new hypotheses for how the machine functions during the degradation cycle.
The new structures are also likely to influence drug development. The proteasome is directly involved in many diseases, particularly in cancers such as multiple myeloma, and thus is an important pharmacological target. All of the current proteasome drugs are inhibitors that target the proteolytic sites31, 37. The new structures of the proteasome could allow the design of an entirely new class of drugs that modulate conformational changes in the regulatory particle or target one of the enzymatic activities in the regulatory particle107. By stabilizing specific conformations or states of the proteasome, these drugs might activate or inhibit the proteasome. Proteasome activators could also conceivably be useful tools for clearing toxic protein aggregates that accumulate in many neurodegenerative diseases152, 153. Thus, it is difficult to overestimate the impact that the new structures will have on our understanding of how cellular protein concentrations are regulated and it seems likely that protein destruction will turn out to be as finely orchestrated as its synthesis.
References
- 1.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 2.Varshavsky A. The ubiquitin system. Trends Biochem Sci. 1997;22:383–7. doi: 10.1016/s0968-0004(97)01122-5. [DOI] [PubMed] [Google Scholar]
- 3.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–47. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 4.Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–39. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 5.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verma R, McDonald H, Yates JR, 3rd, Deshaies RJ. Selective degradation of ubiquitinated Sic1 by purified 26S proteasome yields active S phase cyclin-Cdk. Mol Cell. 2001;8:439–48. doi: 10.1016/s1097-2765(01)00308-2. [DOI] [PubMed] [Google Scholar]
- 7.Nishiyama A, et al. A nonproteolytic function of the proteasome is required for the dissociation of Cdc2 and cyclin B at the end of M phase. Genes Dev. 2000;14:2344–57. doi: 10.1101/gad.823200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hicke L, Riezman H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell. 1996;84:277–87. doi: 10.1016/s0092-8674(00)80982-4. [DOI] [PubMed] [Google Scholar]
- 9.Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 10.Bonner WM, Stedman JD. Histone 1 is proximal to histone 2A and to A24. Proc Natl Acad Sci U S A. 1979;76:2190–4. doi: 10.1073/pnas.76.5.2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jentsch S. The ubiquitin-conjugation system. Annu Rev Genet. 1992;26:179–207. doi: 10.1146/annurev.ge.26.120192.001143. [DOI] [PubMed] [Google Scholar]
- 12.Pickart CM. Back to the future with ubiquitin. Cell. 2004;116:181–90. doi: 10.1016/s0092-8674(03)01074-2. [DOI] [PubMed] [Google Scholar]
- 13.Trempe JF. Reading the ubiquitin postal code. Curr Opin Struct Biol. 2011;21:792–801. doi: 10.1016/j.sbi.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 14.Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol. 2004;11:830–7. doi: 10.1038/nsmb814. [DOI] [PubMed] [Google Scholar]
- 15.Nussbaum AK, et al. Cleavage motifs of the yeast 20S proteasome beta subunits deduced from digests of enolase 1. Proc Natl Acad Sci U S A. 1998;95:12504–9. doi: 10.1073/pnas.95.21.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kisselev AF, Akopian TN, Woo KM, Goldberg AL. The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation. J Biol Chem. 1999;274:3363–71. doi: 10.1074/jbc.274.6.3363. [DOI] [PubMed] [Google Scholar]
- 17.Johnson ES, Gonda DK, Varshavsky A. cis-trans recognition and subunit-specific degradation of short-lived proteins. Nature. 1990;346:287–91. doi: 10.1038/346287a0. [DOI] [PubMed] [Google Scholar]
- 18.Hochstrasser M, Varshavsky A. In vivo degradation of a transcriptional regulator: the yeast alpha 2 repressor. Cell. 1990;61:697–708. doi: 10.1016/0092-8674(90)90481-s. [DOI] [PubMed] [Google Scholar]
- 19.Prakash S, Inobe T, Hatch AJ, Matouschek A. Substrate selection by the proteasome during degradation of protein complexes. Nat Chem Biol. 2009;5:29–36. doi: 10.1038/nchembio.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aza-Blanc P, Ramirez-Weber FA, Laget MP, Schwartz C, Kornberg TB. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell. 1997;89:1043–53. doi: 10.1016/s0092-8674(00)80292-5. [DOI] [PubMed] [Google Scholar]
- 21.Amir RE, Haecker H, Karin M, Ciechanover A. Mechanism of processing of the NF-kappa B2 p100 precursor: identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(beta-TrCP) ubiquitin ligase. Oncogene. 2004;23:2540–7. doi: 10.1038/sj.onc.1207366. [DOI] [PubMed] [Google Scholar]
- 22.Lin L, Ghosh S. A glycine-rich region in NF-kappaB p105 functions as a processing signal for the generation of the p50 subunit. Mol Cell Biol. 1996;16:2248–54. doi: 10.1128/mcb.16.5.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson MD, et al. Proteasome-mediated processing of def1, a critical step in the cellular response to transcription stress. Cell. 2013;154:983–95. doi: 10.1016/j.cell.2013.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schrader EK, Harstad KG, Holmgren RA, Matouschek A. A three-part signal governs differential processing of Gli1 and Gli3 proteins by the proteasome. J Biol Chem. 2011;286:39051–8. doi: 10.1074/jbc.M111.274993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan Y, Wang B. A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J Biol Chem. 2007;282:10846–52. doi: 10.1074/jbc.M608599200. [DOI] [PubMed] [Google Scholar]
- 26.Chen CH, et al. Nuclear trafficking of Cubitus interruptus in the transcriptional regulation of Hedgehog target gene expression. Cell. 1999;98:305–16. doi: 10.1016/s0092-8674(00)81960-1. [DOI] [PubMed] [Google Scholar]
- 27.Lowe J, et al. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995;268:533–9. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 28.Groll M, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463–71. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 29.Unno M, et al. The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure. 2002;10:609–18. doi: 10.1016/s0969-2126(02)00748-7. [DOI] [PubMed] [Google Scholar]
- 30.Groll M, et al. A gated channel into the proteasome core particle. Nat Struct Biol. 2000;7:1062–7. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- 31.Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 32.Lupas A, Flanagan JM, Tamura T, Baumeister W. Self-compartmentalizing proteases. Trends Biochem Sci. 1997;22:399–404. doi: 10.1016/s0968-0004(97)01117-1. [DOI] [PubMed] [Google Scholar]
- 33.Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–80. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 34.Whitby FG, et al. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature. 2000;408:115–20. doi: 10.1038/35040607. [DOI] [PubMed] [Google Scholar]
- 35.Stadtmueller BM, Hill CP. Proteasome activators. Mol Cell. 2011;41:8–19. doi: 10.1016/j.molcel.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sadre-Bazzaz K, Whitby FG, Robinson H, Formosa T, Hill CP. Structure of a Blm10 complex reveals common mechanisms for proteasome binding and gate opening. Mol Cell. 2010;37:728–35. doi: 10.1016/j.molcel.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kish-Trier E, Hill CP. Structural biology of the proteasome. Annu Rev Biophys. 2013;42:29–49. doi: 10.1146/annurev-biophys-083012-130417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cascio P, Call M, Petre BM, Walz T, Goldberg AL. Properties of the hybrid form of the 26S proteasome containing both 19S and PA28 complexes. EMBO J. 2002;21:2636–45. doi: 10.1093/emboj/21.11.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanahashi N, et al. Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J Biol Chem. 2000;275:14336–45. doi: 10.1074/jbc.275.19.14336. [DOI] [PubMed] [Google Scholar]
- 40.Shibatani T, et al. Global organization and function of mammalian cytosolic proteasome pools: Implications for PA28 and 19S regulatory complexes. Mol Biol Cell. 2006;17:4962–71. doi: 10.1091/mbc.E06-04-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt M, et al. The HEAT repeat protein Blm10 regulates the yeast proteasome by capping the core particle. Nat Struct Mol Biol. 2005;12:294–303. doi: 10.1038/nsmb914. [DOI] [PubMed] [Google Scholar]
- 42.Barthelme D, Sauer RT. Identification of the Cdc48*20S proteasome as an ancient AAA+ proteolytic machine. Science. 2012;337:843–6. doi: 10.1126/science.1224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barthelme D, Sauer RT. Bipartite determinants mediate an evolutionarily conserved interaction between Cdc48 and the 20S peptidase. Proc Natl Acad Sci U S A. 2013;110:3327–32. doi: 10.1073/pnas.1300408110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forster A, Masters EI, Whitby FG, Robinson H, Hill CP. The 1.9 A structure of a proteasome-11S activator complex and implications for proteasome-PAN/PA700 interactions. Mol Cell. 2005;18:589–99. doi: 10.1016/j.molcel.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Z, et al. Identification of an activation region in the proteasome activator REGalpha. Proc Natl Acad Sci U S A. 1998;95:2807–11. doi: 10.1073/pnas.95.6.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith DM, et al. Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol Cell. 2007;27:731–44. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gillette TG, Kumar B, Thompson D, Slaughter CA, DeMartino GN. Differential roles of the COOH termini of AAA subunits of PA700 (19 S regulator) in asymmetric assembly and activation of the 26 S proteasome. J Biol Chem. 2008;283:31813–22. doi: 10.1074/jbc.M805935200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rabl J, et al. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell. 2008;30:360–8. doi: 10.1016/j.molcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glickman MH, et al. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell. 1998;94:615–23. doi: 10.1016/s0092-8674(00)81603-7. [DOI] [PubMed] [Google Scholar]
- 50.Elsasser S, Chandler-Militello D, Muller B, Hanna J, Finley D. Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J Biol Chem. 2004;279:26817–22. doi: 10.1074/jbc.M404020200. [DOI] [PubMed] [Google Scholar]
- 51.Chen L, Madura K. Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol Cell Biol. 2002;22:4902–13. doi: 10.1128/MCB.22.13.4902-4913.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaplun L, et al. The DNA damage-inducible UbL-UbA protein Ddi1 participates in Mec1-mediated degradation of Ho endonuclease. Mol Cell Biol. 2005;25:5355–62. doi: 10.1128/MCB.25.13.5355-5362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Funakoshi M, Sasaki T, Nishimoto T, Kobayashi H. Budding yeast Dsk2p is a polyubiquitin-binding protein that can interact with the proteasome. Proc Natl Acad Sci U S A. 2002;99:745–50. doi: 10.1073/pnas.012585199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saeki Y, Saitoh A, Toh-e A, Yokosawa H. Ubiquitin-like proteins and Rpn10 play cooperative roles in ubiquitin-dependent proteolysis. Biochem Biophys Res Commun. 2002;293:986–92. doi: 10.1016/S0006-291X(02)00340-6. [DOI] [PubMed] [Google Scholar]
- 55.Hanna J, et al. Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell. 2006;127:99–111. doi: 10.1016/j.cell.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 56.Guterman A, Glickman MH. Complementary roles for Rpn11 and Ubp6 in deubiquitination and proteolysis by the proteasome. J Biol Chem. 2004;279:1729–38. doi: 10.1074/jbc.M307050200. [DOI] [PubMed] [Google Scholar]
- 57.Borodovsky A, et al. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001;20:5187–96. doi: 10.1093/emboj/20.18.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crosas B, et al. Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell. 2006;127:1401–13. doi: 10.1016/j.cell.2006.09.051. [DOI] [PubMed] [Google Scholar]
- 59.Wilkinson CR, et al. Proteins containing the UBA domain are able to bind to multi-ubiquitin chains. Nat Cell Biol. 2001;3:939–43. doi: 10.1038/ncb1001-939. [DOI] [PubMed] [Google Scholar]
- 60.Zhang F, et al. Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009;34:473–84. doi: 10.1016/j.molcel.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang F, et al. Mechanism of substrate unfolding and translocation by the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009;34:485–96. doi: 10.1016/j.molcel.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walz J, et al. 26S proteasome structure revealed by three-dimensional electron microscopy. J Struct Biol. 1998;121:19–29. doi: 10.1006/jsbi.1998.3958. [DOI] [PubMed] [Google Scholar]
- 63.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–68. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 64.Bohn S, et al. Structure of the 26S proteasome from Schizosaccharomyces pombe at subnanometer resolution. Proc Natl Acad Sci U S A. 2010;107:20992–7. doi: 10.1073/pnas.1015530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lander GC, et al. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–91. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lasker K, et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci U S A. 2012;109:1380–7. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.da Fonseca PC, He J, Morris EP. Molecular model of the human 26S proteasome. Mol Cell. 2012;46:54–66. doi: 10.1016/j.molcel.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 68.Beck F, et al. Near-atomic resolution structural model of the yeast 26S proteasome. Proc Natl Acad Sci U S A. 2012;109:14870–5. doi: 10.1073/pnas.1213333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matyskiela ME, Lander GC, Martin A. Conformational switching of the 26S proteasome enables substrate degradation. Nat Struct Mol Biol. 2013;20:781–8. doi: 10.1038/nsmb.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sledz P, et al. Structure of the 26S proteasome with ATP-gammaS bound provides insights into the mechanism of nucleotide-dependent substrate translocation. Proc Natl Acad Sci U S A. 2013;110:7264–9. doi: 10.1073/pnas.1305782110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matyskiela ME, Martin A. Design principles of a universal protein degradation machine. J Mol Biol. 2012;425:199–213. doi: 10.1016/j.jmb.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lander GC, Martin A, Nogales E. The proteasome under the microscope: the regulatory particle in focus. Curr Opin Struct Biol. 2013;23:243–51. doi: 10.1016/j.sbi.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bohn S, et al. Localization of the regulatory particle subunit Sem1 in the 26S proteasome. Biochem Biophys Res Commun. 2013;435:250–4. doi: 10.1016/j.bbrc.2013.04.069. [DOI] [PubMed] [Google Scholar]
- 74.Djuranovic S, et al. Structure and activity of the N-terminal substrate recognition domains in proteasomal ATPases. Mol Cell. 2009;34:580–90. doi: 10.1016/j.molcel.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 75.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–29. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 76.Pathare GR, et al. The proteasomal subunit Rpn6 is a molecular clamp holding the core and regulatory subcomplexes together. Proc Natl Acad Sci U S A. 2012;109:149–54. doi: 10.1073/pnas.1117648108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Santamaria PG, Finley D, Ballesta JP, Remacha M. Rpn6p, a proteasome subunit from Saccharomyces cerevisiae, is essential for the assembly and activity of the 26 S proteasome. J Biol Chem. 2003;278:6687–95. doi: 10.1074/jbc.M209420200. [DOI] [PubMed] [Google Scholar]
- 78.Isono E, Saito N, Kamata N, Saeki Y, Toh EA. Functional analysis of Rpn6p, a lid component of the 26 S proteasome, using temperature-sensitive rpn6 mutants of the yeast Saccharomyces cerevisiae. J Biol Chem. 2005;280:6537–47. doi: 10.1074/jbc.M409364200. [DOI] [PubMed] [Google Scholar]
- 79.Kim YC, Li X, Thompson D, Demartino GN. ATP Binding by Proteasomal ATPases Regulates Cellular Assembly and Substrate-induced Functions of the 26 S Proteasome. J Biol Chem. 2013;288:3334–45. doi: 10.1074/jbc.M112.424788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peth A, Besche HC, Goldberg AL. Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol Cell. 2009;36:794–804. doi: 10.1016/j.molcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li X, Demartino GN. Variably modulated gating of the 26S proteasome by ATP and polyubiquitin. Biochem J. 2009;421:397–404. doi: 10.1042/BJ20090528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bech-Otschir D, et al. Polyubiquitin substrates allosterically activate their own degradation by the 26S proteasome. Nat Struct Mol Biol. 2009;16:219–25. doi: 10.1038/nsmb.1547. [DOI] [PubMed] [Google Scholar]
- 83.Gur E, Sauer RT. Degrons in protein substrates program the speed and operating efficiency of the AAA+ Lon proteolytic machine. Proc Natl Acad Sci U S A. 2009;106:18503–8. doi: 10.1073/pnas.0910392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sen M, et al. The ClpXP Protease Unfolds Substrates Using a Constant Rate of Pulling but Different Gears. Cell. 2013;155:636–46. doi: 10.1016/j.cell.2013.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smith DM, Fraga H, Reis C, Kafri G, Goldberg AL. ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell. 2011;144:526–38. doi: 10.1016/j.cell.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9:679–90. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Husnjak K, et al. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature. 2008;453:481–8. doi: 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. A 26 S protease subunit that binds ubiquitin conjugates. J Biol Chem. 1994;269:7059–61. [PubMed] [Google Scholar]
- 90.Schreiner P, et al. Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature. 2008;453:548–52. doi: 10.1038/nature06924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang Q, Young P, Walters KJ. Structure of S5a bound to monoubiquitin provides a model for polyubiquitin recognition. J Mol Biol. 2005;348:727–39. doi: 10.1016/j.jmb.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 92.Sakata E, et al. Localization of the proteasomal ubiquitin receptors Rpn10 and Rpn13 by electron cryomicroscopy. Proc Natl Acad Sci U S A. 2012;109:1479–84. doi: 10.1073/pnas.1119394109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cook WJ, Jeffrey LC, Carson M, Chen Z, Pickart CM. Structure of a diubiquitin conjugate and a model for interaction with ubiquitin conjugating enzyme (E2) J Biol Chem. 1992;267:16467–71. doi: 10.2210/pdb1aar/pdb. [DOI] [PubMed] [Google Scholar]
- 94.Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. 2009;37:937–53. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- 95.Zhang N, et al. Structure of the s5a:k48-linked diubiquitin complex and its interactions with rpn13. Mol Cell. 2009;35:280–90. doi: 10.1016/j.molcel.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhao M, Zhang NY, Zurawel A, Hansen KC, Liu CW. Degradation of some polyubiquitinated proteins requires an intrinsic proteasomal binding element in the substrates. J Biol Chem. 2010;285:4771–80. doi: 10.1074/jbc.M109.060095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Takeuchi J, Chen H, Coffino P. Proteasome substrate degradation requires association plus extended peptide. EMBO J. 2007;26:123–31. doi: 10.1038/sj.emboj.7601476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Verhoef LG, et al. Minimal length requirement for proteasomal degradation of ubiquitin-dependent substrates. FASEB J. 2009;23:123–33. doi: 10.1096/fj.08-115055. [DOI] [PubMed] [Google Scholar]
- 99.Heinen C, Acs K, Hoogstraten D, Dantuma NP. C-terminal UBA domains protect ubiquitin receptors by preventing initiation of protein degradation. Nat Commun. 2011;2:191. doi: 10.1038/ncomms1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fishbain S, Prakash S, Herrig A, Elsasser S, Matouschek A. Rad23 escapes degradation because it lacks a proteasome initiation region. Nat Commun. 2011;2:192. doi: 10.1038/ncomms1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Inobe T, Fishbain S, Prakash S, Matouschek A. Defining the geometry of the two-component proteasome degron. Nat Chem Biol. 2011;7:161–7. doi: 10.1038/nchembio.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Elsasser S, et al. Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat Cell Biol. 2002;4:725–30. doi: 10.1038/ncb845. [DOI] [PubMed] [Google Scholar]
- 103.Saeki Y, Sone T, Toh-e A, Yokosawa H. Identification of ubiquitin-like protein-binding subunits of the 26S proteasome. Biochem Biophys Res Commun. 2002;296:813–9. doi: 10.1016/s0006-291x(02)02002-8. [DOI] [PubMed] [Google Scholar]
- 104.Walters KJ, Lech PJ, Goh AM, Wang Q, Howley PM. DNA-repair protein hHR23a alters its protein structure upon binding proteasomal subunit S5a. Proc Natl Acad Sci U S A. 2003;100:12694–9. doi: 10.1073/pnas.1634989100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang D, et al. Together, Rpn10 and Dsk2 can serve as a polyubiquitin chain-length sensor. Mol Cell. 2009;36:1018–33. doi: 10.1016/j.molcel.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hoyt MA, Coffino P. Ubiquitin-free routes into the proteasome. Cell Mol Life Sci. 2004;61:1596–600. doi: 10.1007/s00018-004-4133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee BH, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–84. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A. ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell. 2001;7:627–37. doi: 10.1016/s1097-2765(01)00209-x. [DOI] [PubMed] [Google Scholar]
- 109.Tian L, Holmgren RA, Matouschek A. A conserved processing mechanism regulates the activity of transcription factors Cubitus interruptus and NF-kappaB. Nat Struct Mol Biol. 2005;12:1045–53. doi: 10.1038/nsmb1018. [DOI] [PubMed] [Google Scholar]
- 110.Hoppe T, et al. Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell. 2000;102:577–86. doi: 10.1016/s0092-8674(00)00080-5. [DOI] [PubMed] [Google Scholar]
- 111.Piwko W, Jentsch S. Proteasome-mediated protein processing by bidirectional degradation initiated from an internal site. Nat Struct Mol Biol. 2006;13:691–7. doi: 10.1038/nsmb1122. [DOI] [PubMed] [Google Scholar]
- 112.Zhang M, Coffino P. Repeat sequence of Epstein-Barr virus-encoded nuclear antigen 1 protein interrupts proteasome substrate processing. J Biol Chem. 2004;279:8635–41. doi: 10.1074/jbc.M310449200. [DOI] [PubMed] [Google Scholar]
- 113.Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 114.Iyer LM, Leipe DD, Koonin EV, Aravind L. Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol. 2004;146:11–31. doi: 10.1016/j.jsb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 115.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 116.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 117.Smith DM, Benaroudj N, Goldberg A. Proteasomes and their associated ATPases: a destructive combination. J Struct Biol. 2006;156:72–83. doi: 10.1016/j.jsb.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 118.Wang J, et al. Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure. 2001;9:177–84. doi: 10.1016/s0969-2126(01)00570-6. [DOI] [PubMed] [Google Scholar]
- 119.Hinnerwisch J, Fenton WA, Furtak KJ, Farr GW, Horwich AL. Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell. 2005;121:1029–41. doi: 10.1016/j.cell.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 120.Martin A, Baker TA, Sauer RT. Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat Struct Mol Biol. 2008;15:1147–51. doi: 10.1038/nsmb.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang J, et al. Nucleotide-dependent conformational changes in a protease-associated ATPase HsIU. Structure. 2001;9:1107–16. doi: 10.1016/s0969-2126(01)00670-0. [DOI] [PubMed] [Google Scholar]
- 122.Yamada-Inagawa T, Okuno T, Karata K, Yamanaka K, Ogura T. Conserved pore residues in the AAA protease FtsH are important for proteolysis and its coupling to ATP hydrolysis. J Biol Chem. 2003;278:50182–7. doi: 10.1074/jbc.M308327200. [DOI] [PubMed] [Google Scholar]
- 123.Aubin-Tam ME, Olivares AO, Sauer RT, Baker TA, Lang MJ. Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell. 2011;145:257–67. doi: 10.1016/j.cell.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Maillard RA, et al. ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell. 2011;145:459–69. doi: 10.1016/j.cell.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell. 2009;139:744–56. doi: 10.1016/j.cell.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Glynn SE, Nager AR, Baker TA, Sauer RT. Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat Struct Mol Biol. 2012;19:616–22. doi: 10.1038/nsmb.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sousa MC, et al. Crystal and solution structures of an HslUV protease-chaperone complex. Cell. 2000;103:633–43. doi: 10.1016/s0092-8674(00)00166-5. [DOI] [PubMed] [Google Scholar]
- 128.Bochtler M, et al. The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature. 2000;403:800–5. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- 129.Hersch GL, Burton RE, Bolon DN, Baker TA, Sauer RT. Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell. 2005;121:1017–27. doi: 10.1016/j.cell.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 130.Horwitz AA, et al. ATP-induced structural transitions in PAN, the proteasome-regulatory ATPase complex in Archaea. J Biol Chem. 2007;282:22921–9. doi: 10.1074/jbc.M702846200. [DOI] [PubMed] [Google Scholar]
- 131.Yakamavich JA, Baker TA, Sauer RT. Asymmetric nucleotide transactions of the HslUV protease. J Mol Biol. 2008;380:946–57. doi: 10.1016/j.jmb.2008.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Augustin S, et al. An intersubunit signaling network coordinates ATP hydrolysis by m-AAA proteases. Mol Cell. 2009;35:574–85. doi: 10.1016/j.molcel.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Martin A, Baker TA, Sauer RT. Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature. 2005;437:1115–20. doi: 10.1038/nature04031. [DOI] [PubMed] [Google Scholar]
- 134.Herman C, Prakash S, Lu CZ, Matouschek A, Gross CA. Lack of a robust unfoldase activity confers a unique level of substrate specificity to the universal AAA protease FtsH. Mol Cell. 2003;11:659–69. doi: 10.1016/s1097-2765(03)00068-6. [DOI] [PubMed] [Google Scholar]
- 135.Koodathingal P, et al. ATP-dependent proteases differ substantially in their ability to unfold globular proteins. J Biol Chem. 2009;284:18674–84. doi: 10.1074/jbc.M900783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kohler A, et al. The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol Cell. 2001;7:1143–52. doi: 10.1016/s1097-2765(01)00274-x. [DOI] [PubMed] [Google Scholar]
- 137.Rubin DM, Glickman MH, Larsen CN, Dhruvakumar S, Finley D. Active site mutants in the six regulatory particle ATPases reveal multiple roles for ATP in the proteasome. EMBO J. 1998;17:4909–19. doi: 10.1093/emboj/17.17.4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Peth A, Nathan JA, Goldberg AL. The ATP costs and time required to degrade ubiquitinated proteins by the 26 S proteasome. J Biol Chem. 2013;288:29215–22. doi: 10.1074/jbc.M113.482570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Beckwith R, Estrin E, Worden EJ, Martin A. Reconstitution of the 26S proteasome reveals functional asymmetries in its AAA+ unfoldase. Nat Struct Mol Biol. 2013;20:1164–1172. doi: 10.1038/nsmb.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tomko RJ, Jr, Funakoshi M, Schneider K, Wang J, Hochstrasser M. Heterohexameric ring arrangement of the eukaryotic proteasomal ATPases: implications for proteasome structure and assembly. Mol Cell. 2010;38:393–403. doi: 10.1016/j.molcel.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Erales J, Hoyt MA, Troll F, Coffino P. Functional asymmetries of proteasome translocase pore. J Biol Chem. 2012;287:18535–43. doi: 10.1074/jbc.M112.357327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Enemark EJ, Joshua-Tor L. On helicases and other motor proteins. Curr Opin Struct Biol. 2008;18:243–57. doi: 10.1016/j.sbi.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Skordalakes E, Berger JM. Structural insights into RNA-dependent ring closure and ATPase activation by the Rho termination factor. Cell. 2006;127:553–64. doi: 10.1016/j.cell.2006.08.051. [DOI] [PubMed] [Google Scholar]
- 144.Costa A, et al. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat Struct Mol Biol. 2011;18:471–7. doi: 10.1038/nsmb.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Enemark EJ, Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–5. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 146.Thomsen ND, Berger JM. Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell. 2009;139:523–34. doi: 10.1016/j.cell.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Itsathitphaisarn O, Wing RA, Eliason WK, Wang J, Steitz TA. The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell. 2012;151:267–77. doi: 10.1016/j.cell.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Frank J. Single-particle imaging of macromolecules by cryo-electron microscopy. Annu Rev Biophys Biomol Struct. 2002;31:303–19. doi: 10.1146/annurev.biophys.31.082901.134202. [DOI] [PubMed] [Google Scholar]
- 149.Chemla YR, et al. Mechanism of force generation of a viral DNA packaging motor. Cell. 2005;122:683–92. doi: 10.1016/j.cell.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 150.Aathavan K, et al. Substrate interactions and promiscuity in a viral DNA packaging motor. Nature. 2009;461:669–73. doi: 10.1038/nature08443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Moffitt JR, et al. Intersubunit coordination in a homomeric ring ATPase. Nature. 2009;457:446–50. doi: 10.1038/nature07637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kessler BM. Ubiquitin - omics reveals novel networks and associations with human disease. Curr Opin Chem Biol. 2013;17:59–65. doi: 10.1016/j.cbpa.2012.12.024. [DOI] [PubMed] [Google Scholar]
- 153.Tai HC, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008;9:826–38. doi: 10.1038/nrn2499. [DOI] [PubMed] [Google Scholar]
- 154.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 155.Pintilie GD, Zhang J, Goddard TD, Chiu W, Gossard DC. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J Struct Biol. 2010;170:427–38. doi: 10.1016/j.jsb.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]