

Abstract
The synthesis of α-aminosilanes by a highly enantio- and regioselective copper-catalyzed hydroamination of vinylsilanes is reported. The system employs Cu-DTBM-SEG-PHOS as the catalyst, diethoxymethylsilane as the stoichiometric reductant, and O-benzoylhydroxylamines as the electrophilic nitrogen source. This hydroamination reaction is compatible with differentially substituted vinylsilanes, thus providing access to amino acid mimics and other valuable chiral organosilicon compounds.
Keywords: asymmetric synthesis, copper, hydroamination, silicon
Organosilicon compounds have recently gained prominence within the field of medicinal chemistry. Silicon is often used as an isostere for carbon due to its similar valency and tetrahedral bonding pattern.[1] In addition, because of its larger covalent radius, electropositive/lipophilic nature, and low intrinsic toxicity, silicon is complementary to carbon and is valuable in pharmaceutical research.[2] Among the many subclasses of organosilicon compounds, chiral α-aminosilanes in particular have demonstrated significant bioactivities.[2, 3] Several potent inhibitors of proteolytic enzymes possess the α-aminosilane motif, and α-aminosilanes have been incorporated into peptide isosteres (Scheme 1).[2–4] Thus, the development of robust methods for the construction of chiral α-aminosilanes is an important area of research.
Scheme 1.

Examples of silicon-containing peptidomimetics and amino acids. Boc = tert-butoxycarbonyl.
Although there has been progress in the synthesis of racemic α-aminosilanes,[5, 6] enantioselective approaches remain limited. Previous methods include asymmetric deprotonation followed by reverse aza-Brook rearrangement [Scheme 2, Eq. (1)].[7] Other approaches have utilized the chiral auxiliary bearing aldimines developed by Davis or Ellman in conjunction with silyllithium reagents [Eq. (2)].[3, 8] Recently, Oestreich and co-workers reported an elegant process catalyzed by a chiral N-heterocyclic carbene/copper complex using Suginome's Ph(Me)2SiBpin reagent [Eq. (3)].[8i] However, these methods have limitations with respect to the scope of the amine, and, with the exception of the report by Oestreich and co-workers, require the use of a stoichiometric chiral auxiliary or reagent.
Scheme 2.
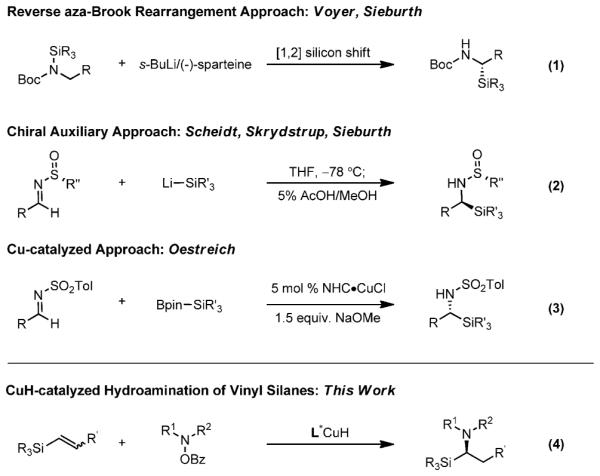
Previous approaches towards the synthesis of chiral α-aminosilanes and the development of our strategy. Bpin = pinacolborane, Bz = benzoyl, Tol = tolyl.
Recently, we, as well as Hirano, Miura, and co-workers, reported the CuH-catalyzed asymmetric Markovnikov hydroamination of styrenes.[9] We felt that this method, when applied to vinylsilane substrates, would allow the generation of a broad range of chiral α-aminosilanes [Eq. (4)]. As vinylsilanes are readily prepared and bench-stable, they are attractive as starting materials for the synthesis of α-aminosilanes.[10] The intermolecular hydroamination of vinylsilanes would likely, based on literature precedent, proceed regioselectively to give chiral α-aminosilanes (III), via the α-silylalkylcopper intermediate II (Scheme 3),[11] on reaction with the O-benzoylhydroxylamine electrophile 2.[9]
Scheme 3.
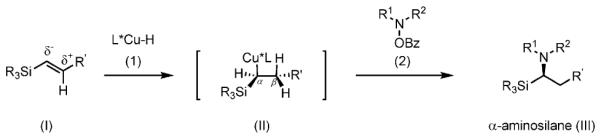
Regioselectivity in the hydroamination of β-vinylsilanes.
We began our investigation by examining the hydroamination of (E)-vinylsilane 1a using conditions previously developed for the hydroamination of styrene (Table 1).[9] The reaction furnished α-aminosilane 3a regioselectively in quantitative yield with > 99 % ee after 8 h (entry 1). Switching the solvent to cyclohexane, diethyl ether, or toluene (entries 2–4) had no effect, but no conversion was seen in dichloromethane (entry 5). We also examined other chiral ligands that were previously shown to be effective in reactions catalyzed by a copper(I) hydride complex (entries 6–8).[9b, 17] However, the use of (R)-DTBM-SEGPHOS was found to give the highest reactivity and selectivity.
Table 1.
Reaction optimization.[a]

| ||||
|---|---|---|---|---|
| Entry | Solvent | L | Yield 3a [%][b] | ee [%][c] |
| 1 | THF | L1 | >99 | >99 |
| 2 | cyclohexane | L1 | >99 | >99 |
| 3 | Et2O | L1 | >99 | >99 |
| 4 | toluene | L1 | >99 | >99 |
| 5 | CH2CI2 | L1 | 0[[e],[f],[g]] | – |
| 6 | THF | L2 | 37[f] | 95 |
| 7 | THF | L3 | 92[[d]] | 98 |
| 8 | THF | L4 | 0[[e],[f],[h]] | – |
Reaction conditions: 1a (0.5 mmol), 2a (0.6 mmol), Cu(OAc)2 (0.02 mmol), ligand (0.022 mmol), solvent (1 mL).
Yields of isolated products.
Determined by HPLC analysis on a chiral stationary phase.
16 h.
Yield determined by GC (dodecane as internal standard).
36 h.
77% of 1a remained.
96% of la remained.
We next investigated the influence of the nature of the silyl group and olefin geometry on the reactivity and enantioselectivity (Scheme 4). The reaction was compatible with vinylsilanes containing triethylsilyl (3 a), trimethylsilyl (3 b), dimethylphenylsilyl (3 c), and methyldiphenylsilyl groups (3 d).[18] In all cases, the reactions proceeded regioselectively to give α-aminosilane products. Interestingly, we found: 1) both E and Z isomers provided the same enantiomeric product, and 2) E substrates invariably reacted faster and with a higher level of enantioselectivity than the corresponding Z substrates.
Scheme 4.
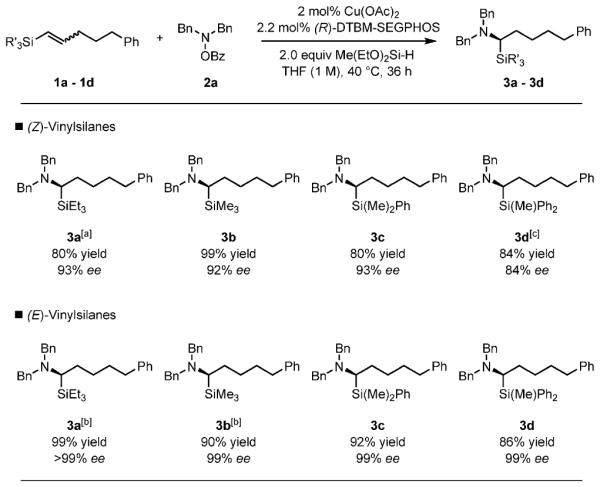
Influence of the silyl group and olefin geometry on yield and enantioselectivity. Reaction conditions: 1a–1d (1 mmol), 2a (1.2 mmol), Cu(OAc)2 (0.02 mmol), (R)-DTBM-SEGPHOS (0.022 mmol), THF (1 mL), 40 8C, 36 h. Yields are of isolated products (average of two runs). [a] Cu(OAc)2 (0.04 mmol), (R)-DTBM-SEGPHOS (0.044 mmol). [b] 8 h. [c] Cu(OAc)2 (0.04 mmol), (R)-DTBM-SEGPHOS (0.044 mmol), THF (0.5 mL, 2 m). Bn = benzyl.
Thus, we chose to examine the scope of the hydroamination of (E)-vinylsilanes. This method accommodates a broad range of functional groups (Scheme 5). Vinylsilanes containing a nitrile (5 a), an alkyl chloride (5 b), an ester (5 c), a sulfonamide (5 d), a tert-butyldimethylsilyl ether (5 e), and an allylic ether moiety (5 f) were readily handled. No competitive elimination of alkoxide was observed with an ether (5 f).[19] Additionally, we applied our method to the synthesis of α-amino acid mimics by hydroamination of a secondary benzylamine (5 i), a β,β-disubstituted vinylsilane (5 j), and a β-isopropyl-substituted vinylsilane (5 k), to provide mimics of lysine, valine, and leucine, respectively.
Scheme 5.

Hydroamination of (E)-vinylsilanes. Reaction conditions: 4 a–4k (1 mmol), 2a (1.2 mmol), Cu(OAc)2 (0.02 mmol), (R)-DTBM-SEGPHOS (0.022 mmol), THF (1 mL), 40 8C, 36 h. Yields are of isolated products (average of two runs). [a] 16 h reaction time. [b] Cu-(OAc)2 (0.04 mmol), (R)-DTBM-SEGPHOS (0.044 mmol), THF (1 mL, 1 m). [c] From (Z)-4g. TBS = tert-butyldimethylsilyl, Ts = p-toluenesulfonyl.
The compatibility of this reaction with a variety of O-benzoylhydroxylamine electrophiles was then examined (Scheme 6). Acyclic dialkyl (6 a), cyclic dialkyl (6 b), and alkylbenzylamine-based electrophiles (6 c) were all suitable partners, delivering the hydroaminated products with high yields and enantioselectivities. In addition, a heterocycle-containing electrophile was tolerated (6 d). Hydroamination to install a bis(p-methoxybenzyl)amino group was also successful (6 e).
Scheme 6.
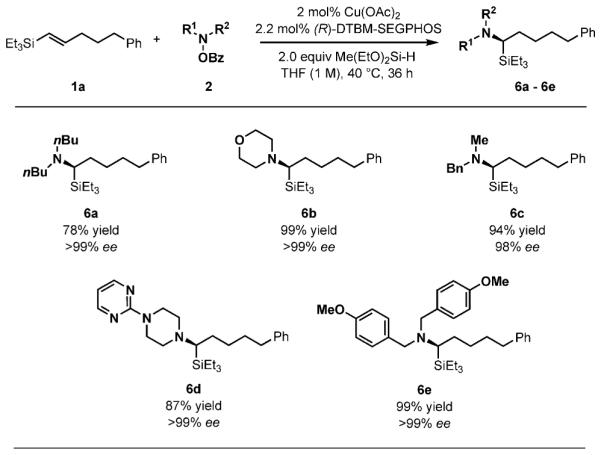
Scope of O-benzoylhydroxylamine electrophiles. Reaction conditions: 1 (1 mmol), 2a (1.2 mmol), Cu(OAc)2 (0.02 mmol), (R)-DTBM-SEGPHOS (0.022 mmol), THF (1 mL), 40 8C, 36 h. Yields are of isolated products (average of two runs).
As previously mentioned (Scheme 4), the E and Z substrates provide the same enantiomer of the product. However, E substrates react faster and afford a higher level of enantioselectivity than the corresponding Z substrates. We thus wondered whether, in the case of (Z)-alkenes, most of the hydroamination product was formed by isomerization of the slower reacting Z isomer, followed by transformation of the nascent E isomer. We investigated this possibility through the use of a deuterated silane reagent. In this case, L*Cu-D would form and the olefin would insert into it through a synaddition process (Scheme 7). For the more reactive E substrate, subsequent reaction with the O-benzoylhydroxylamine would generate the R,R product. For the Z substrate, if no isomerization occurs, the reaction should generate the R,S product. However, if the Z substrate undergoes Cucatalyzed isomerization to the E alkene prior to reaction with O-benzoylhydroxylamine, the major product would contain approximately two deuterium atoms.
Scheme 7.
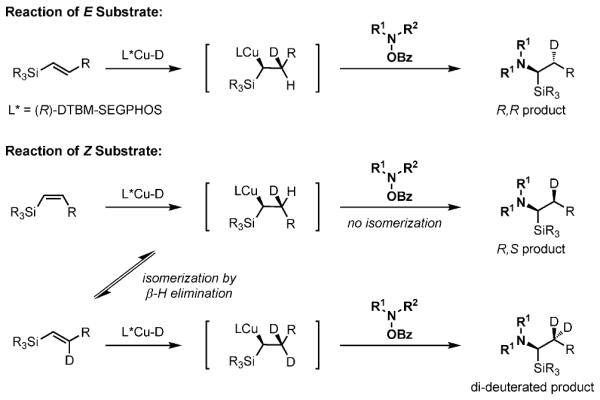
Possible reaction pathways.
Deuterium-labeling experiments (Scheme 8) revealed that the hydroamination of (E)-4g was completely diastereo-selective with about 99 % deuterium incorporation[20] to give the monodeuterated R,R isomer 5 g’ [Eq. (5)]. The reaction was also diastereoselective for (Z)-4 g, and gave the mono-deuterated R,S isomer 5 g' with about 96 % deuterium incorporation [Eq. (6)].[20] The stereochemical result and the presence of the monodeuterated product from the Z substrate indicates that most of the product does not form by isomerization of the substrate to the E isomer.
Scheme 8.

Deuterium-labeling experiments.
Lastly, to demonstrate the scalability of this transformation, we carried out the hydroamination of 1a with Bn2N-OBz (2 a) on a 10 mmol scale (Scheme 9). Full conversion of the vinylsilane was achieved with a catalyst loading of only 1.5 mol %. The yield and the enantioselectivity was the same as for the 1 mmol scale reaction (Table 1).
Scheme 9.

Large-scale hydroamination reaction of (E)-triethylsilylvinylsilane.
In summary, we have developed an enantioselective Cu-catalyzed hydroamination of vinylsilanes. The reaction proceeds in a regioselective manner to provide enantioenriched α-aminosilanes in high yield with outstanding levels of enantioselectivity. The method is applicable to a variety of substrates, and provides rapid access to a family of valuable chiral organosilicon building blocks and bioactive molecules.
Supplementary Material
Footnotes
Research reported in this publication was supported by the National Institutes of Health under award number GM58160. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We are grateful to Dr. Yiming Wang and Dr. Daniel T. Cohen for insightful discussions, and Phillip J. Milner and Dr. Michael Pirnot for help with the preparation of this manuscript. We thank Dr. Christopher Welch and Dr. Erik Regalado (Merck), and Christina Kraml (Lotus Separations) for SFC analysis.
References
- [1].Apeloig Y. In: The chemistry of organic silicon compounds. Patai S, Rappoport Z, editors. Vol. 1. Wiley; New York: 1989. pp. 57–225. [Google Scholar]
- [2].a Kim J, Hewitt G, Carroll P, Sieburth SM. J. Org. Chem. 2005;70:5781–5789. doi: 10.1021/jo048121v. [DOI] [PubMed] [Google Scholar]; b Franz AK, Wilson SO. J. Med. Chem. 2012;55:388–405. doi: 10.1021/jm3010114. [DOI] [PubMed] [Google Scholar]; c Meanwell NA. J. Med. Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- [3].a Nielsen L, Lindsay KB, Faber J, Nielsen NC, Skrydstrup T. J. Org. Chem. 2007;72:10035–10044. doi: 10.1021/jo701907d. [DOI] [PubMed] [Google Scholar]; b Nielsen L, Skrydstrup T. J. Am. Chem. Soc. 2008;130:13145–13151. doi: 10.1021/ja804720p. [DOI] [PubMed] [Google Scholar]
- [4].a Chen CA, Sieburth SM, Glekas A, Hewitt GW, Trainor GL, Erickson-Viitanen S, Garber SS, Cordova B, Jeffry S, Klabe RM. Chem. Biol. 2001;8:1161–1166. doi: 10.1016/s1074-5521(01)00079-5. [DOI] [PubMed] [Google Scholar]; b Kim J, Sieburth SM. Bioorg. Med. Chem. Lett. 2004;14:2853–2856. doi: 10.1016/j.bmcl.2004.03.042. [DOI] [PubMed] [Google Scholar]
- [5].a Padwa A, Eisenbarth P, Venkatramanan MK, Wong GSK. J. Org. Chem. 1987;52:2427–2432. For examples of non-transition-metal-catalyzed approaches. [Google Scholar]; b Okazaki S, Sato Y. Synthesis. 1990:36–38. [Google Scholar]; c Noll JE, Speier JL, Daubert BF. J. Am. Chem. Soc. 1991;113:3867–3871. [Google Scholar]; d Eisch JJ, Galle JE. J. Org. Chem. 1976;41:2615–2621. [Google Scholar]; e Sato Y, Toyo'ok T, Aoyama T, Shirai H. J. Org. Chem. 1976;41:3559–3564. [Google Scholar]; f Benkeser RA, Li GS, Mozdzen EC. J. Organomet. Chem. 1979;178:21–28. [Google Scholar]; g Tsuge O, Kanemasa S, Matusda K. Chem. Lett. 1983:1131–1134. [Google Scholar]; h Picard JP, Grelier S, Constantieux T, Dunogues J, Aizpurua JM, Palomo C, Petraud M, Barbe B, Lunazzi L, Leger JM. Organometallics. 1993;12:1378–1385. [Google Scholar]; i Katritzky AR, Hong Q, Yang Z. Organometallics. 1995;14:734–737. [Google Scholar]; j Honda T, Satoh S, Mori M. Organometallics. 1995;14:1548–1550. [Google Scholar]; k Sieburth SM, Somers JJ, O'Hare HK. Tetrahedron. 1996;52:5669–5682. [Google Scholar]; l Fleming I, Mack SR, Clark BP. Chem. Commun. 1998:6199–6202. [Google Scholar]
- [6].Vyas DJ, Frohlich R, Oestreich M. Org. Lett. 2011;13:2094–2097. doi: 10.1021/ol200509c. For an example of a transition-metal-catalyzed approach. [DOI] [PubMed] [Google Scholar]
- [7].a Barberis C, Voyer N. Tetrahedron Lett. 1998;39:6807–6810. [Google Scholar]; b Sieburth SM, O'Hare HK, Xu J, Chen Y, Liu G. Org. Lett. 2003;5:1859–1861. doi: 10.1021/ol034397y. [DOI] [PubMed] [Google Scholar]; c Sieburth SM, Liu G. Org. Lett. 2003;5:4677–4769. doi: 10.1021/ol0358713. [DOI] [PubMed] [Google Scholar]
- [8].a Ballweg DM, Miller RC, Gray DL, Scheidt KA. Org. Lett. 2005;7:1403–1406. doi: 10.1021/ol050244u. [DOI] [PubMed] [Google Scholar]; b Hernandez D, Lindsay KB, Nielsen L, Mittag T, Bjerglund K, Friis S, Mose R, Skrydstrup T. J. Org. Chem. 2010;75:3283–3293. doi: 10.1021/jo100301n. [DOI] [PubMed] [Google Scholar]; c Hernandez D, Nielsen L, Lindsay KB, Lopez-Garcia MA, Bjerglund K, Skrydstrup T. Org. Lett. 2010;12:3528–3531. doi: 10.1021/ol101379t. [DOI] [PubMed] [Google Scholar]; d Hernandez D, Mose R, Skrydstrup T. Org. Lett. 2010;12:732–735. doi: 10.1021/ol102968g. [DOI] [PubMed] [Google Scholar]; e Bo Y, Singh S, Duong HQ, Cao C, Sieburth SM. Org. Lett. 2011;13:1787–1789. doi: 10.1021/ol2002978. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Hensel A, Nagura K, Delvos LB, Oestreich M. Angew. Chem. Int. Ed. Angew. Chem. 2014;2014;53126:4964–4967. 5064–5067. doi: 10.1002/anie.201402086. [DOI] [PubMed] [Google Scholar]
- [9].a Zhu S, Niljianskul N, Buchwald SL. J. Am. Chem. Soc. 2013;135:15746–15749. doi: 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Miki Y, Hirano K, Satoh T, Miura M. Angew. Chem. Int. Ed. Angew. Chem. 2013;2013;52125:10830–10834. 11030–11034. doi: 10.1002/anie.201304365. [DOI] [PubMed] [Google Scholar]
- [10].a Hamze A, Provot O, Brion J-D, Alami M. J. Organomet. Chem. 2008;693:2789–2797. [Google Scholar]; b Lu B, Falck JR. J. Org. Chem. 2010;75:1701–1705. doi: 10.1021/jo902678p. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Trost BM, Ball TZ. J. Am. Chem. Soc. 2005;127:17644–17655. doi: 10.1021/ja0528580. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ojima I, Li Z, Zhu J. Recent advances in the hydrosilylation and related reactions. Patai’s Chemistry of Functional Groups; Wiley, New York: 2009. pp. 30–46. [Google Scholar]
- [11]. For vinylsilanes, the partial positive charge is more pronounced at the b-position (to the silicon) due to the b-silicon effect (see Refs. [12] and [13]). The partial negative charge that develops at the a-position is stabilized by the silyl group (see Refs. [14] and [15]). Furthermore, intermediate II (Scheme 3) is thermodynamically stabilized by hyperconjugation of the s-orbital of the Cu-alkyl bond with a s*-orbital on the silicon–carbon bond (see Ref. [16])
- [12].Wierschke SG, Chandrasekhar J, Jorgensen WL. J. Am. Chem. Soc. 1985;107:1496–1500. [Google Scholar]
- [13].Lambert JB, Liu C, Kouliev T. J. Phys. Org. Chem. 2002;15:667–671. [Google Scholar]
- [14].Brinkman EA, Berger S, Brauman JI. J. Am. Chem. Soc. 1994;116:8304–8310. [Google Scholar]
- [15].a Ito H, Kosaka Y, Nonoyama K, Sasaki Y, Sawamura M. Angew. Chem. Int. Ed. Angew. Chem. 2008;2008;47120:7424–7427. 7534–7537. doi: 10.1002/anie.200802342. Similar regioselectivity has been observed in the Cu-catalyzed borylation of vinylsilanes. [DOI] [PubMed] [Google Scholar]; b Ito H, Toyoda T, Sawamura M. J. Am. Chem. Soc. 2010;132:5990–5992. doi: 10.1021/ja101793a. [DOI] [PubMed] [Google Scholar]; c Kubota K, Yamamoto E, Ito H. Adv. Synth. Catal. 2013;355:3527–3531. [Google Scholar]
- [16].a Brook MA. Silicon in Organic, Organometallic, and Polymer Chemistry. Wiley, New York: 2000. [Google Scholar]; b Zweifel G, Backlund SJI. J. Am. Chem. Soc. 1977;99:3184–3185. similar regioselectivity has also been observed in the hydroboration of alkynylsilanes to generate a,a-boryl(silyl)alkenes. [Google Scholar]
- [17].Noh D, Chea H, Ju J, Yun J. Angew. Chem. Int. Ed. Angew. Chem. 2009;2009;48121:6062–6064. 6178–6180. doi: 10.1002/anie.200902015. [DOI] [PubMed] [Google Scholar]
- [18]. The hydroamination reaction is slow for vinylsilanes with large silyl groups such as TIPS and TBDPS. After 36 h, no conversion was observed for (Z)-triisopropyl(5-phenylpent-1-en-1-yl)silane and only 16 % conversion was observed for (Z)-tert-butyldiphenyl(5-phenylpent-1-en-1-yl)silane. [Google Scholar]
- [19].a Kacprzynski M, Hoveyda AH. J. Am. Chem. Soc. 2004;126:10676–10681. doi: 10.1021/ja0478779. Competitive elimination of an allylic leaving group was previously observed in related Cu-catalyzed processes. [DOI] [PubMed] [Google Scholar]; b Nakatani A, Hirano K, Satoh T, Miura M. Org. Lett. 2012;14:2586–2589. doi: 10.1021/ol300886k. [DOI] [PubMed] [Google Scholar]; c Harada A, Makida Y, Sato T, Ohmiya H, Sawamura M. J. Am. Chem. Soc. 2014 doi: 10.1021/ja5084333. DOI: 10.1021/ja508433. [DOI] [PubMed] [Google Scholar]; d Hojoh K, Shido Y, Ohmiya H, Sawamura M. Angew. Chem. Int. Ed. Angew. Chem. 2014;2014;53126:4954–4958. 5054–5058. doi: 10.1002/anie.201402386. [DOI] [PubMed] [Google Scholar]; e Makida Y, Takayama Y, Ohmiya H, Sawamura M. Angew. Chem. Int. Ed. 2013;52:5350–5354. doi: 10.1002/anie.201300785. [DOI] [PubMed] [Google Scholar]; f Shido Y, Yoshida M, Tanabe M, Ohmiya H, Iwai T, Sawamura M. Adv. Synth. Catal. 2012;354:3440–3444. [Google Scholar]; g Nagao K, Yokobori U, Makida Y, Ohmiya H, Sawamura M. J. Am. Chem. Soc. 2012;134:8982–8987. doi: 10.1021/ja302520h. [DOI] [PubMed] [Google Scholar]; h Nagao K, Ohmiya H, Sawamura M. Synthesis. 2012:1535–1541. [Google Scholar]
- [20]. See the Supporting Information section II, part C for a detailed analysis. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.