

Fragile X Syndrome (FXS) is commonly thought to arise from dysfunction of the synapse, the site of communication between neurons. However, loss of the protein that results in FXS occurs early in embryonic development, while synapses are formed relatively late.
Keywords: dendrite development, FMRP, Fragile X Syndrome, in vivo imaging, neurogenesis, Xenopus
Abstract
Fragile X Syndrome (FXS) is the leading known monogenic form of autism and the most common form of inherited intellectual disability. FXS results from silencing the FMR1 gene during embryonic development, leading to loss of Fragile X Mental Retardation Protein (FMRP), an RNA-binding protein that regulates mRNA transport, stability, and translation. FXS is commonly thought of as a disease of synaptic dysfunction; however, FMRP expression is lost early in embryonic development, well before most synaptogenesis occurs. Recent studies suggest that loss of FMRP results in aberrant neurogenesis, but neurogenic defects have been variable. We investigated whether FMRP affects neurogenesis in Xenopus laevis tadpoles that express a homolog of FMR1. We used in vivo time-lapse imaging of neural progenitor cells and their neuronal progeny to evaluate the effect of acute loss or overexpression of FMRP on neurogenesis in the developing optic tectum. We complimented the time-lapse studies with SYTOX labeling to quantify apoptosis and CldU labeling to measure cell proliferation. Animals with increased or decreased levels of FMRP have significantly decreased neuronal proliferation and survival. They also have increased neuronal differentiation, but deficient dendritic arbor elaboration. The presence and severity of these defects was highly sensitive to FMRP levels. These data demonstrate that FMRP plays an important role in neurogenesis and suggest that endogenous FMRP levels are carefully regulated. These studies show promise in using Xenopus as an experimental system to study fundamental deficits in brain development with loss of FMRP and give new insight into the pathophysiology of FXS.
Significance Statement
Fragile X Syndrome (FXS) is commonly thought to arise from dysfunction of the synapse, the site of communication between neurons. However, loss of the protein that results in FXS occurs early in embryonic development, while synapses are formed relatively late. This suggests that deficits may occur earlier in neuronal development. We show that changes in FMRP expression in the brains of intact Xenopus laevis tadpoles have profound effects on neurogenesis, the generation of neurons. Therefore, neuronal function in FXS may be affected by events that have gone awry during embryonic development. These studies show promise in using Xenopus as a model of FXS and give new insight into the pathophysiology of FXS.
Introduction
The developmental neurological disease, Fragile X Syndrome (FXS), is the most common form of inherited intellectual disability and the leading monogenic cause of autism (Bhakar et al., 2012; Santoro et al., 2012; Wijetunge et al., 2013). FXS is typically caused by expansion of a trinucleotide (CGG) repeat in the 5′ untranslated region of the Fragile X Mental Retardation 1 (FMR1) gene (Fu et al., 1991; Verkerk et al., 1991). The full mutation contains CGG repeats in excess of 200 leading to hypermethylation and transcriptional silencing of FMR1, preventing expression of Fragile X Mental Retardation Protein (FMRP) (Oberlé et al., 1991; Verheij et al., 1993). FMRP is an RNA-binding protein that interacts with mRNAs and regulates mRNA transport, stability, and translation (Santoro et al., 2012). FMRP inhibits protein synthesis downstream of group 1 metabotropic glutamate receptor (mGluR) activation (Waung and Huber, 2009). Unchecked protein synthesis at the synapse is thought to play an important role in the disease mechanism. The morphological hallmark of the FXS brain is the prevalence of immature dendritic spines, the predominant site of excitatory synapse formation (Irwin et al., 2000; He and Portera-Cailliau, 2013). Together, these findings have led to the theory that synaptic dysfunction is largely responsible for the clinical phenotypes of FXS (Zoghbi and Bear, 2012).
FMR1 and FMRP are ubiquitously expressed in the developing CNS of many animals, including humans. Expression begins during early embryogenesis and continues into adulthood. FMR1 and FMRP are expressed within proliferating cells in the embryonic brain and later expression is more restricted to neurons (Abitbol et al., 1993; Devys et al., 1993; Hinds et al., 1993; Castrén et al., 2005; Pacey and Doering, 2007; Saffary and Xie, 2011). The expression of FMRP in proliferating cells suggests that loss of FMRP in FXS may affect neurogenesis, which includes cell proliferation, survival, migration, and differentiation of neurons. Brain development requires strict spatial and temporal regulation of these processes, so errors in the regulation of neurogenesis are expected to have profound effects on brain development and function. Recent studies in rodents, Drosophila, and stem cell preparations support a role for FMRP in neurogenesis (Li and Zhao, 2014), but the specific effects of FMRP knockdown have varied with model system and developmental stage.
FMR1 is highly conserved between fruit flies, fish, frogs, rodents, and humans (Verkerk et al., 1991; Ashley et al., 1993; Wan et al., 2000; Lim et al., 2005; van 't Padje et al., 2005), suggesting that FMRP may play similar roles in brain development and circuit function in diverse experimental systems. Indeed, many studies have demonstrated that the basic cellular processes underlying deficits in neural function in FXS are highly conserved from fruit flies to humans (Bolduc et al., 2008; Doll and Broadie, 2014). Xenopus laevis provides several advantages for studying vertebrate brain development. Notably, tadpoles’ external development facilitates observation of neurogenesis in early developmental stages, in contrast to mammalian species in which comparable stages of development occur in utero. Additionally, Xenopus tadpoles are transparent, which allows direct visualization of the developing brain. The tadpole visual system has been extensively studied to elucidate mechanisms underlying neurogenesis and circuit development (Sin et al., 2002; Ruthazer et al., 2006; Manitt et al., 2009; Sharma and Cline, 2010; Bestman et al., 2012; Ghiretti et al., 2014). Fmr1 mRNA is expressed throughout development of Xenopus laevis embryos and tadpoles and increases in expression with brain development (Lim et al., 2005; Gessert et al., 2010), suggesting that FMRP may play a role in aspects of visual system development, including neurogenesis and neuronal maturation.
Here we investigate the role of FMRP in neural progenitor cell (NPC) proliferation, survival, and differentiation in the optic tectum of intact Xenopus laevis tadpoles. We use translation-blocking antisense morpholino oligonucleotides to decrease FMRP expression and electroporation of an FMRP expression construct to rescue or overexpress FMRP in stage 46 − 47 tadpoles. We observe neurogenesis over time by collecting in vivo time-lapse confocal and two-photon images of eGFP-expressing NPCs and their neuronal progeny. This highly sensitive time-lapse approach reveals the cumulative effects of cell proliferation and survival over the course of several days. We find that NPC proliferation, survival, differentiation, and neuronal dendritic arbor development are regulated by FMRP and are highly sensitive to the level of FMRP expression.
Materials and Methods
Animals
Albino Xenopus laevis tadpoles of either sex were obtained by in-house breeding or purchased from Xenopus Express. Tadpoles were reared in 0.1X Steinberg’s solution in a 12 h light/12 h dark cycle at 22 − 23 °C and used for experiments beginning at stage 46 (Nieuwkoop and Faber, 1956). During time-lapse imaging experiments, animals were housed individually in the wells of a six-well tissue culture plate containing 0.1X Steinberg’s. Animals were anesthetized in 0.02% MS222 prior to electroporation and imaging. All animal procedures were performed in accordance with The Scripps Research Institute’s animal care committee’s regulations.
Plasmids and morpholinos
A Xenopus laevis homolog of FMR1, fmr1a, was knocked down using a 3′ lissamine-tagged translation-blocking antisense morpholino oligonucleotide (GeneTools) with the sequence 5′-AGCTCCTCCATGTTGCGTCCGCACA-3′ (start codon underlined), referred to as fmr1a MO. Control lissamine-tagged oligonucleotides had the sequences 5′-TAACTCGCATCGTAGATTGACTAAA-3′ or 5′-CCTCTTACCTCAGTTACAATTTATA-3′, referred to as CMO. Morpholinos were dissolved in water.
To visualize neural progenitors and their progeny, we used a Sox2-driven expression construct to express fluorescent proteins and proteins of interest. This construct contains the Sox2 and Oct3/4-binding domain of the FGF minipromoter (Sox2bd) and requires the binding of endogenous Sox2 to drive expression (Bestman et al., 2012). This restricts expression to neural progenitor cells and their neuronal progeny, which retain the expressed protein but do not have any new protein expression from the plasmid. Expression was amplified using the gal4/UAS system. Using this construct, we expressed eGFP alone (Sox2bd::gal4-UAS::eGFP, referred to as Sox2bd::eGFP) or with Xenopus fmr1b (Open Biosystems, Clone ID no. 4755584). In order to assay the effectiveness of the fmr1a MO (Fig. 2), we generated a chimeric reporter construct in which 14 nucleotides from the 5′ UTR of fmr1a (5′-TGTGCGGACGCAAC-3′) were added upstream of the fmr1b sequence to render it sensitive to knockdown by fmr1a MO. In addition, we added an eGFP to the 3′ end of fmr1b separated by a t2A sequence, producing two discrete proteins from a single transcript (Sox2bd::gal4-UAS::fmr1-t2A-eGFP, referred to as fmr1-t2A-eGFP). For rescue experiments, we made silent mutations in the morpholino-binding region of fmr1-t2A-eGFP, making it MO insensitive (TGTGCGGACGCAACATGGAGGAGCT to TGTGtGGcCGgAAtATGGAaGAGCT), generating the construct Sox2bd::gal4-UAS::Δfmr1-t2A-eGFP, referred to as Δfmr1-t2A-eGFP. This construct was also used for overexpression experiments. In some experiments, we used plasmids with UAS-driven turbo RFP tagged with a nuclear localization sequence (UAS::tRFPnls) or UAS-driven eGFP (UAS::eGFP). Plasmids and morpholinos were injected into the brain ventricle, then platinum electrodes were placed on each side of the midbrain and voltage pulses were applied across the midbrain to electroporate optic tectal cells in stage 46 tadpoles.
Figure 2.

Validation of fmr1a morpholino-mediated knockdown. Antibody-dependent and -independent strategies to validate knockdown of FMRP by translation-blocking antisense morpholinos. A, Confocal Z-projections of FMRP immunoreactivity in 40 μm sections through optic tectum. B, HIGH (0.1 mM) fmr1a MO results in a 60% decrease in FMRP immunoreactivity (***p < 0.001). C, Antibody-independent strategy to validate in vivo knockdown by morpholinos. Animals are co-electroporated with Sox2bd::gal4-UAS::fmr1-t2A-eGFP and UAS::tRFPnls plasmids and either control morpholino (CMO) or fmr1a MO. In the presence of CMO, the electroporated plasmids will all be translated resulting in expression of FMRP, eGFP, and tRFPnls. In the presence of fmr1a MO, translation is inhibited resulting in a lack of FMRP and eGFP, while tRFPnls is expressed. The fluorescence intensity of eGFP is correlated with the expression of FMRP. D, Confocal Z-projections of optic tecta electroporated with the expression constructs and morpholinos in C and imaged in vivo show that LOW (0.05 mM) fmr1a MO and HIGH (0.1 mM) fmr1a MO decrease the expression of eGFP. Dashed lines outline the optic tectum and inset shows a schematic of the optic tectum. E, fmr1a MO significantly increases the percentage of cells in which only tRFPnls is detected (**p < 0.01, ***p < 0.001). F, eGFP/tRFP ratios in cells that had detectable eGFP. fmr1a MO significantly reduced the eGFP/tRFP ratio compared to CMO and the decrease with HIGH fmr1a MO is larger than that of LOW fmr1a MO (*p < 0.05, ***p < 0.001). Scale bars, 50 μm.
FMRP Western blot and immunohistochemistry
For Western blots of endogenous FMRP, stage 47 − 48 tadpole midbrains and adult rat brain were dissected and homogenized in RIPA buffer or 0.2% SDS in PBS and boiled for 5 − 10 min before brief sonication. Small aliquots were taken to measure protein concentration using the BCA Protein Assay Kit (Thermo Scientific, 23227). Then, 1× sample buffer was added to the remaining sample and boiled for 10 − 15 min. Fifteen micrograms of each lysate was separated on an SDS−polyacrylamide gel and proteins were transferred to a nitrocellulose membrane. The membrane was incubated in 1:500 mouse anti-FMRP (Millipore, MAB2160) or 1:500 rabbit anti-FMRP (AbCam, ab69815) primary antibody overnight at 4 °C, followed by goat anti-mouse or goat anti-rabbit HRP-conjugated secondary (BioRad) at room temperature. For quantification of FMRP overexpression, optic tecta of stage 46 tadpoles were electroporated with 1 μg/μl Sox2bd::eGFP or 1 μg/μl Δfmr1-t2A-eGFP (HIGH FMRP OE). Two days later, midbrains were dissected and Western blots were performed as described above on samples from two independent experiments. Different exposure periods were used for the same blots to avoid saturation. The blots were scanned and band intensities were measured from nonsaturating exposures with ImageJ. For comparisons, the intensity of each FMRP band was first normalized to its β-tubulin loading control band (which was obtained after stripping the same membrane) and then that value was normalized to the control value in each experiment.
For immunohistochemistry, stage 47 tadpoles were anesthetized with 0.02% MS222, immersed in 4% paraformaldehyde, and fixed using two bouts of microwave fixation at 150 W for 1 min followed by overnight fixation at 4 °C. Brains were dissected and sectioned at 40 μm on a vibratome. Sections were blocked and permeabilized in 5% normal donkey serum and 1% triton X-100 for 1 h at room temperature. Then sections were incubated in 1:200 mouse anti-FMRP (Millipore, MAB2160) overnight at 4 °C, followed by 2 h in 1:200 anti-mouse Alexa Fluor 488 (Life Technologies) at room temperature. Sections were mounted in Gel mount (Accurate) and imaged with an Olympus FluoView500 confocal microscope with a 20× (0.8 NA), 40× (1.0 NA), or 60× (1.4 NA) oil immersion lens. To quantify MO-mediated knockdown of endogenous FMRP, stage 46 animals were electroporated with CMO, 0.05 mM (LOW) fmr1a MO, or 0.1 mM (HIGH) fmr1a MO. Two days later, animals were fixed and brains were processed for FMRP immunohistochemistry as described above. Brain sections of comparable depths from animals in each of the three groups were imaged at 40× using identical imaging parameters. Image stacks were Z-projected and the average FMRP fluorescence intensity of the entire optic tectum was measured and then normalized to the average FMRP fluorescence intensity of CMO animals for each batch of animals that were electroporated and imaged together.
In vivo knockdown assay imaging and quantification
The optic tecta of stage 46 − 47 tadpoles were electroporated with 2 μg/μl fmr1-t2A-eGFP, 1 μg/μl UAS::tRFPnls, and either CMO, 0.05 mM (LOW) fmr1a MO or 0.1 mM (HIGH) fmr1a MO. Two days later, we performed in vivo imaging of labeled cells using a Perkin-Elmer Ultraview Vox spinning-disk confocal microscope with a 25× Nikon water immersion objective lens (1.1 NA). Volocity 3D image analysis software (Perkin Elmer) was used to automatically detect and outline tRFP-labeled cells, followed by manual confirmation and removal of incorrectly detected objects. Then, tRFP and eGFP fluorescence intensities throughout each outlined volume were determined and summed for each cell. To identify cells as tRFP-only, a cut off fluorescence intensity for eGFP was determined: we measured the minimum eGFP fluorescence intensity within the outlined volume for each cell and found the average minimum eGFP fluorescence for control cells in each experiment. We set a cut off at the average minimum eGFP fluorescence −0.5 SD. We required that the eGFP fluorescence intensity within the outlined volume of each cell be above that value to call the cell eGFP+. Then, the percentage of cells that were tRFP-only (eGFP−) for each animal was calculated. Next, for cells that were eGFP+, we calculated the eGFP/tRFP ratio for each cell and then normalized it to the average eGFP/tRFP ratio for the control cells for each batch of animals that were electroporated and imaged together.
In vivo quantification of FMRP overexpression
The optic tecta of stage 46 tadpoles were electroporated with 0.5 μg/μl Δfmr1-t2A-eGFP (LOW FMRP OE) or 1 μg/μl Δfmr1-t2A-eGFP (HIGH FMRP OE). Two days later, animals were imaged on a custom-built two-photon microscope with a 25× water immersion lens (1.05 NA). Images were Z-projected and the eGFP fluorescence intensity of each eGFP-labeled cell soma was measured. The average fluorescence intensity of all labeled cells was calculated and normalized to LOW FMRP OE.
In vivo time-lapse imaging of proliferation and differentiation
Stage 46 tadpole optic tecta were electroporated with plasmids and MOs as follows: control/CMO: 1 μg/μl UAS::tRFPnls with either 1 μg/μl Sox2bd::eGFP or 0.5 μg/μl Sox2bd::eGFP supplemented with 0.7 μg/μl UAS::eGFP, and CMO. FMRP knockdown: 1 μg/μl Sox2bd::eGFP with 1 μg/μl UAS::tRFPnls and either 0.05 mM (LOW) fmr1a MO, or 0.1 mM (HIGH) fmr1a MO. FMRP overexpression: 1 μg/μl UAS::tRFPnls with either 1 μg/μl Δfmr1-t2A-eGFP (HIGH FMRP OE) or 0.5 μg/μl Δfmr1-t2A-eGFP supplemented with 0.7 μg/μl UAS::eGFP (LOW FMRP OE). Rescue: 1 μg/μl Δfmr1-t2A-eGFP with 1 μg/μl UAS::tRFPnls and either 0.05 mM fmr1a MO (LOW MO HIGH Δfmr1 Rescue) or 0.1 mM fmr1a MO (HIGH MO HIGH Δfmr1 Rescue). Animals were imaged on a Perkin-Elmer Ultraview Vox spinning disk confocal microscope with a 25× water-immersion lens (1.1 NA) or a custom-built two-photon microscope with a 25× water-immersion lens (1.05 NA) at 1, 2, and 3 d following electroporation. Image analysis was performed using either Volocity 3D image analysis software using the measurement function or the ImageJ Cell Counter plugin. Analysis consisted of counting the total number of labeled cells per brain hemisphere in every tadpole and characterizing counted cells as either mature neurons or neural progenitor cells based on established morphological features. Neurons possess a pear-shaped or round soma with elaborated dendritic arbors and an axon, whereas neural progenitor cells are characterized by a triangular cell body and a long radial process extending from the ventricular zone to the pial surface, ending in an elaborated endfoot. Cells without processes that were not obviously undergoing cell death were counted as unidentifiable. Only animals with more than 10 labeled cells on the first day of imaging were included in the analysis. To examine cell proliferation and survival, we calculated the total number of cells present on each day of imaging and the percent change in cell number from days 1 to 3 [(day 3 − day 1)/day 1]. To examine cell differentiation, we calculated the total number of each cell type that was present on each day of imaging and what percentage of each cell type comprised the total cell population on each day of imaging.
CldU cell proliferation analysis
Stage 46 tadpole optic tecta were electroporated with CMO or fmr1a MO. One, two, or three days later, animals were incubated in 3.8 mM CldU (MP Biomedicals, 0210547880) in Steinberg’s solution for 2 h. Immediately thereafter, animals were anesthetized in 0.02% MS222 and fixed using either two bouts of microwave fixation at 150 W for 1 min followed by 2 h fixation at room temperature or overnight fixation at 4 °C. Brains were dissected and incubated in 2N HCl at 37 °C for 1 h, then blocked and permeabilized in 2.5% normal goat serum and 0.1% triton-X 100. Brains were incubated in 1:500 rat anti-CldU (Accurate, OBT0030G) overnight at 4 °C, followed by 2 h in 1:400 anti-rat Alexa Fluor 488 (Life Technologies) at room temperature. Brains were mounted in Gel mount (Accurate) and the dorsal 30 μm of the whole-mount brain was imaged with an Olympus FluoView500 confocal microscope with a 20× oil-immersion lens (0.8 NA). CldU+ cells located along the ventricular wall between the anterior commissure and the rostral portion of the third ventricle were counted manually using the ImageJ Cell Counter plugin. The volume of the ventricular region did not differ between groups and we reported the average total number of CldU+ cells within the ventricular region for each group.
Cell death analysis
For SYTOX staining of electroporated brains, the optic tecta of stage 46 animals were electroporated with CMO or fmr1a MO. One day following electroporation, animals were anesthetized in 0.02% MS222 and fixed by immersion in 4% paraformaldehyde overnight at 4 °C. Brains were dissected and immersed in 1:1000 SYTOX green nucleic acid stain (Life Technologies, S7020) in PBS for 20 min. For SYTOX staining combined with caspase-3 immunohistochemistry, stage 47 tadpoles were anesthetized in 0.02% MS222 and then injected with PBS or 50 mM staurosporine (Tocris Biosciences) to induce apoptosis. Twenty-four hours later, tadpoles were anesthetized in 0.02% MS222 and fixed by immersion in 4% paraformaldehyde overnight at 4 °C. Brains were dissected, permeabilized in 2% Triton-X 100, and then blocked in 2.5% normal goat serum and 0.1% Triton-X 100. Then, brains were incubated in 1:200 rabbit anti-caspase3 (AbCam, ab13847) overnight at 4 °C, followed by 3 h in 1:400 anti-rabbit Alexa Fluor 488 (Life Technologies, A11008) at room temperature. Next, brains were incubated in 1:1000 SYTOX orange nucleic acid stain (Life Technologies, S11368) in PBS for 15 min. Brains were imaged whole-mount on an Olympus FluoView500 confocal microscope with a 20× (0.8 NA) or 60× (1.4 NA) oil-immersion lens. Analysis was performed on the first 30 optical sections using the ImageJ Cell Counter plugin. SYTOX+ cells undergoing apoptosis have small, brightly stained nuclei. The total number of brightly SYTOX stained, apoptotic nuclei was counted. For SYTOX/caspase-3 analysis, the number of cells that were caspase-3 immunolabeled was also counted. Then, colocalization between the two channels was quantified. Given that fluorescently labeled objects decrease in brightness in the deeper optical sections in a confocal stack, we analyzed the intensity of the bright apoptotic SYTOX+ cells relative to their presumably healthy, dimmer nearest neighbors in the same optical section. We found that even though the absolute fluorescence intensity of a dying SYTOX+ cell was lower in deeper optical sections, the intensity of dying SYTOX+ cells was approximately double the intensity of their healthy neighbors. Regression analysis of depth within the tissue compared to the ratio of the intensity of SYTOX+ cells relative to their neighbors showed no correlation (R2 = 0.022).
In vivo time-lapse imaging of dendritic morphology
The optic tecta of stage 46 animals were electroporated with plasmids and MOs as follows: CMO/control: 1 μg/μl Sox2bd::eGFP or 0.5 μg/μl Sox2bd::eGFP supplemented with 0.7 μg/μl UAS::eGFP, and CMO. FMRP knockdown: 1 μg/μl Sox2bd::eGFP with 0.05 mM (LOW) fmr1a MO or 0.1 mM (HIGH) fmr1a MO. FMRP overexpression: 1 μg/μl Δfmr1-t2A-eGFP (HIGH FMRP OE) or 0.5 μg/μl Δfmr1-t2A-eGFP supplemented with 0.7 μg/μl UAS::eGFP (LOW FMRP OE). Rescue: 0.5 μg/μl Δfmr1-t2A-eGFP supplemented with 0.7 μg/μl UAS::eGFP and 0.1 mM fmr1a MO (HIGH MO LOW Δfmr1 Rescue). Animals were imaged on a custom-built two-photon microscope with a 20× (0.95 NA) or 25× (1.05 NA) water-immersion lens at 2 and/or 3 d following electroporation. The dendrites of well isolated single neurons were traced and reconstructed using Imaris software (Bitplane). Total dendritic length and total dendritic branch tip number were quantified.
Statistical analysis
All experiments were conducted with a randomized experimental design. Statistical tests are listed in Table 1.
Table 1.
Statistical table
| Data structure | Type of test | Power | |
| a | Normally distributed | ANOVA with post hoc Tukey | 1.00 |
| b | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 1.00 |
| c | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 1.00 |
| d | Not normally distributed | Mann−Whitney | 0.15 |
| e | Not normally distributed | Mann−Whitney | 0.56 |
| f | Not normally distributed | Mann−Whitney | 0.46 |
| g | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 1.00 |
| h | Normally distributed | ANOVA with post hoc Tukey | 1.00 |
| i | Normally distributed | ANOVA with post hoc Tukey | 0.66 |
| j | Normally distributed | t test | 0.23 |
| k | Not normally distributed | Mann−Whitney | 0.88 |
| l | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.99 |
| m | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.85 |
| n | Normally distributed | ANOVA with post hoc Tukey | 0.97 |
| o | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.92 |
| p | Normally distributed | ANOVA with post hoc Tukey | 0.63 |
| q | Normally distributed | ANOVA with post hoc Tukey | 0.90 |
| r | Normally distributed | ANOVA with post hoc Tukey | 0.65 |
| s | Normally distributed | ANOVA with post hoc Tukey | 0.61 |
| t | Not normally distributed | Kruskal-Wallis with post hoc Mann−Whitney | 0.60 |
| u | Normally distributed | ANOVA with post hoc Tukey | 0.58 |
| v | Not normally distributed | Mann−Whitney | 0.06 |
| w | Normally distributed | t test | 0.09 |
| x | Not normally distributed | Mann−Whitney | 0.17 |
| y | Normally distributed | t test | 0.12 |
| z | Not normally distributed | Mann−Whitney | 0.33 |
| aa | Not normally distributed | Mann−Whitney | 0.32 |
| bb | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.99 |
| cc | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 1.00 |
| dd | Normally distributed | ANOVA with post hoc Tukey | 0.71 |
| ee | Not normally distributed | Mann−Whitney | 0.07 |
| ff | Normally distributed | t test | 0.11 |
| gg | Normally distributed | t test | 0.09 |
| hh | Normally distributed | t test | 0.48 |
| ii | Normally distributed | t test | 0.12 |
| jj | Normally distributed | t test | 0.07 |
| kk | Normally distributed | ANOVA with post hoc Tukey | 0.99 |
| ll | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.99 |
| mm | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.74 |
| nn | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.68 |
| oo | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.86 |
| pp | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.70 |
| Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.84 | |
| rr | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.95 |
| ss | Not normally distributed | Mann−Whitney | 0.98 |
| tt | Not normally distributed | Mann−Whitney | 1.00 |
| uu | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.66 |
| vv | Not normally distributed | Kruskal−Wallis with post hoc Mann−Whitney | 0.60 |
For each statistical test run in the study, the data structure, statistical test, and power are listed.
Results
FMRP is highly expressed in progenitor cells and neurons
While it is known that fmr1 mRNA is expressed throughout Xenopus laevis embryonic development (Lim et al., 2005; Gessert et al., 2010), the expression pattern of FMRP in the optic tectum during visual system development is unknown. To examine expression of FMRP, we first performed Western blot of stage 47 − 48 tadpole midbrain labeled with FMRP antibody, which revealed a band at approximately 72 kD (data not shown and Fig. 3E ). We found that rat brain lysate labeled with FMRP antibody had a similar band (data not shown). To elucidate a more detailed expression pattern in the optic tectum, we performed immunohistochemistry for FMRP in stage 47 tadpoles. FMRP immunolabeling was detected in neural progenitor cells (NPCs) that line the brain ventricle and neurons that are located lateral to progenitors (Fig. 1A−C ). Furthermore, FMRP was expressed as punctate labeling throughout the tectal neuropil. This expression profile suggests that FMRP may regulate cell proliferation and/or differentiation of NPCs into neurons as well as aspects of neuronal development.
Figure 3.

Validation of FMRP rescue and overexpression. A, Strategy for validating that the Δfmr1 Rescue construct is morpholino-insensitive. Point mutations in the fmr1 expression construct prevent translational inhibition by fmr1a MO resulting in control levels of FMRP, eGFP, and tRFPnls. B, Confocal Z-projections of optic tecta electroporated with the 2 μg/μl Δfmr1-t2A-eGFP, 1 μg/μl UAS::tRFPnls and LOW (0.05 mM) fmr1a MO as depicted in A and imaged in vivo. Dashed lines outline the optic tectum and inset shows a schematic of the optic tectum. C, D, Quantification of the percentage of cells expressing tRFP-only (C) and the eGFP/tRFP ratio (D) were no different between CMO and LOW fmr1a MO. E, Western blots of Xenopus tadpole midbrain lysate labeled with anti-FMRP yields a band of approximately 72 kD, which is higher in intensity when FMRP is overexpressed with 1 μg/μl Δfmr1-t2A-eGFP (HIGH FMRP OE) compared to 1 μg/μl Sox2bd::eGFP (Ctrl) in two independent experiments. β-tubulin was used as a loading control. F, HIGH FMRP OE increases the intensity of the FMRP band by 1.6-fold compared to control. G, Z-projections from in vivo two-photon imaging of cells expressing 0.5 μg/μl Δfmr1-t2A-eGFP (LOW FMRP OE) or HIGH FMRP OE. H, eGFP fluorescence intensity is more than three times greater for HIGH FMRP OE compared to LOW FMRP OE (*p < 0.05). This reflects the difference in FMRP expression from these two construct concentrations since FMRP and eGFP are made from a single transcript. Scale bars, 50 μm.
Figure 1.
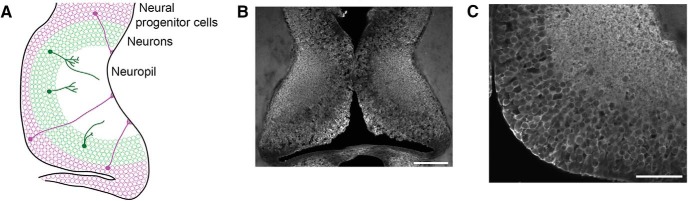
FMRP is expressed in Xenopus optic tectal progenitors and neurons. A, Schematic of the Xenopus tadpole optic tectum showing the location of neural progenitor cells (purple) and neurons (green) extending processes into the neuropil. B, A single optical confocal section of stage 47 Xenopus optic tectum shows widespread FMRP immunoreactivity. Scale bar, 100 μm. C, A higher magnification view from a single optical section in a different animal shows FMRP immunoreactivity across all cell layers and throughout the neuropil. Scale bar, 50 μm.
Validation of morpholino-mediated FMRP knockdown
To test the requirement of FMRP in neurogenesis and neuronal development, we knocked down FMRP in the optic tectum of stage 46 tadpoles by electroporating a morpholino (MO) against Xenopus fmr1a. MOs bind their complementary sequence on mRNA and prevent translation of proteins of interest. We used two independent assays to validate MO-mediated knockdown of FMRP. First, we electroporated animals with control MO (CMO), 0.05 mM (LOW) fmr1a MO, or 0.1 mM (HIGH) fmr1a MO and performed FMRP immunohistochemistry to assay knockdown of endogenous FMRP (Fig. 2A ). Two days following electroporation (dfe), HIGH fmr1a MO resulted in a 60% decrease in the fluorescence intensity of endogenous FMRP (Fig. 2B ; CMO: N = 6 animals; LOW fmr1a MO: N = 6 animals, p = 0.96a compared to CMO; HIGH fmr1a MO: N = 6 animals, p < 0.0001a compared to CMO). With FMRP immunohistochemistry, detection of knockdown by LOW fmr1a MO was variable across experiments, suggesting that this degree of knockdown is near the detection threshold using this assay.
Next, we developed a sensitive in vivo assay to assess the ability of MOs to block translation in Xenopus that does not require antibody detection. For this assay, we electroporated a reporter construct into the Xenopus optic tectum, which generates two discrete proteins from a single transcript: the protein of interest and a fluorescent protein reporter (FP) linked by a t2A sequence. When MO and the reporter construct are co-electroporated, the MO prevents translation of the transcript, decreasing expression of both the protein of interest and the FP. Measurements of FP intensity can be used as a proxy for knockdown of the protein of interest, in this case, FMRP. Here, we used a plasmid that contains a promoter with the Sox2 and Oct3/4-binding domain of the FGF minipromoter that requires binding of endogenous Sox2 to express eGFP and FMRP in Sox2-expressing NPCs and their neuronal progeny (Bestman et al., 2012). FMRP and eGFP are separated by a t2A sequence, producing two discrete proteins from a single transcript. Expression from this plasmid is amplified using the gal4/UAS system. This plasmid is called Sox2bd::gal4-UAS::fmr1-t2A-eGFP and will be referred to as fmr1-t2A-eGFP (Fig. 2C ). In addition, we co-expressed a UAS-driven turboRFP tagged with a nuclear localization sequence (UAS::tRFPnls) to visualize labeled cells. We anticipated that when CMO is co-electroporated with fmr1-t2A-eGFP and UAS::tRFPnls, CMO would not affect translation and FMRP, eGFP, and tRFPnls would all be expressed. In contrast, when fmr1a MO is co-electroporated, translation of FMRP and eGFP would be inhibited, but expression of tRFPnls would be unaffected. We electroporated stage 46 − 47 animals with fmr1-t2A-eGFP, UAS::tRFPnls, and either CMO, LOW fmr1a MO, or HIGH fmr1a MO and then imaged labeled cells in vivo using a spinning-disk confocal microscope (Fig. 2D ). When we imaged control cells 1 dfe, we found that cells expressed tRFPnls but very little eGFP (data not shown). This is most likely explained by differences in the timing of expression of tRFP and eGFP, because tRFP matures more rapidly than eGFP. When we imaged control cells at 2 dfe, we found that eGFP and tRFPnls were both highly expressed in electroporated cells (Fig. 2D ). Therefore, we imaged animals at 2 dfe to test the effectiveness of the two concentrations of fmr1a MO (Fig. 2D−F ). We quantified the percentage of cells that lacked detectable eGFP expression, an indicator of strong knockdown. Both concentrations of fmr1a MO yielded a higher percentage of cells that lacked detectable eGFP expression compared to CMO (Fig. 2E; CMO: N = 27 animals; LOW fmr1a MO: N = 30 animals, p < 0.0001b compared to CMO; HIGH fmr1a MO: N = 20 animals, p = 0.0016b compared to CMO). We did not detect any significant differences between LOW and HIGH fmr1a MO on the percentage of tRFP-only cells (p = 0.31b). However, animals electroporated with HIGH fmr1a MO tended to have fewer labeled cells and more debris from what we suspect are dying cells. Therefore, it is likely that cells with the most severe knockdown in the presence of HIGH fmr1a MO did not survive. We address the potential effect of FMRP knockdown on cell survival in Fig. 5. In cells where eGFP was visible, the ratio of eGFP/tRFP was significantly reduced with fmr1a MO compared to CMO, and HIGH fmr1a MO had a greater reduction than LOW fmr1a MO (Fig. 2F; CMO: N = 253 cells; LOW fmr1a MO: N = 275 cells, p < 0.0001c compared to CMO; HIGH fmr1a MO: N = 172 cells, p < 0.0001c compared to CMO, p = 0.020c compared to LOW fmr1a MO). Electroporation of a lower concentration of fmr1a MO (0.01 mM) resulted in no significant knockdown (data not shown). Together, these two assays demonstrate that fmr1a MO is effective at knocking down FMRP expression and that LOW and HIGH fmr1a MO reflect different levels of knockdown. We used both concentrations of fmr1a MO in our experiments to test how sensitive tectal cells are to the reduction in FMRP.
Figure 5.

Knockdown of FMRP increases cell death. A, Confocal Z-projections through five optical sections of tectum with caspase-3 (Casp3) immunoreactivity and SYTOX Orange staining. Twenty-four hour incubation in staurosporine (STS) increases the number of apoptotic cells that are immunoreactive for Casp3 and brightly stained for SYTOX Orange. Scale bar, 100 μm. B, High-magnification single-optical sections from different animals demonstrate the staining variations of apoptotic cells. The majority of positively labeled cells are stained for both Casp3 and SYTOX Orange (white arrows). The remaining cells are positive for only SYTOX (yellow arrow) or only Casp3 (blue arrow). Scale bar, 20 μm. C, Quantification of total apoptotic cells in the presence or absence of STS demonstrates that SYTOX Orange and Casp3 detect the STS-induced increase in cell death. SYTOX stains a larger dying cell population than Casp3. D, SYTOX Green staining in whole-mount optic tecta was used to identify cells undergoing apoptosis in the presence of fmr1a MO. Bright, apoptotic SYTOX Green+ cells are marked by blue and yellow arrows in confocal Z-projections through the dorsal 30 optical sections of tectum. Cells marked by blue arrows are shown at higher magnification (right) in single-optical sections of the areas highlighted to the left (yellow arrows in the Z-projection to the left are out of the plane of focus in the single-optical section to the right). Scale bars, 50 μm. E, Quantification of the total number of apoptotic SYTOX Green+ cells at 1 dfe shows that both concentrations of fmr1a MO increase cell death compared to CMO (**p < 0.01, ***p < 0.001).
Validation of FMRP rescue and overexpression
In order to test the specificity of knockdown by fmr1a MO, we generated a rescue construct with silent mutations in the MO-binding region to render it MO-insensitive (Sox2bd::gal4-UAS::Δfmr1-t2A-eGFP, referred to as Δfmr1-t2A-eGFP) and used the in vivo knockdown assay to confirm that it is MO-insensitive (Fig. 3A,B ). As expected, we found no change in the percentage of Δfmr1-t2A-eGFP-electroporated cells that expressed only tRFPnls in the presence of LOW fmr1a MO (Fig. 3C; CMO: N = 24 animals; LOW fmr1a MO: N = 20 animals, p = 0.65d). In addition, electroporation of LOW fmr1a MO did not reduce the eGFP/tRFP ratio (Fig. 3D; CMO: N = 310 cells; LOW fmr1a MO: N = 264 cells, p = 0.10e). Together, these results demonstrate that Δfmr1-t2A-eGFP is MO-insensitive and can be used for testing the specificity of fmr1a MO in rescue experiments.
We also used Δfmr1-t2A-eGFP to test the effect of FMRP overexpression on neurogenesis in vivo. We assayed FMRP overexpression using two independent methods. First, we electroporated the optic tectum with 1 μg/μl Sox2bd::eGFP (control) or 1 μg/μl Δfmr1-t2A-eGFP (HIGH FMRP OE) and performed Western blot analysis of FMRP expression in the midbrain (Fig. 3E ). Two days following electroporation, HIGH FMRP OE resulted in a 1.6-fold increase in FMRP expression in the midbrain compared to control (Fig. 3F ). Since electroporation of HIGH FMRP OE will label a subset of cells within the optic tectum and this assay reports the increase in FMRP expression throughout the entire midbrain, it will underestimate the extent of FMRP overexpression in electroporated cells. Next, we assayed FMRP overexpression specifically in cells that overexpress Δfmr1. We electroporated animals with 0.5 μg/μl Δfmr1-t2A-eGFP (LOW FMRP OE) or HIGH FMRP OE and performed in vivo two-photon imaging of eGFP-labeled cells (Fig. 3G ). HIGH FMRP OE resulted in brightly labeled eGFP+ cells, while cells expressing LOW FMRP OE were much dimmer. On average, eGFP fluorescence intensities were more than three times higher for cells expressing HIGH FMRP OE compared to cells expressing LOW FMRP OE (Fig. 3H ; LOW FMRP OE: N = 4 cells; HIGH FMRP OE: N = 28 cells, p = 0.017f). These differences in eGFP expression correlate with differences in FMRP expression since the two proteins are produced from a single transcript; therefore, these two concentrations of Δfmr1-t2A-eGFP produce a threefold difference in FMRP expression. Together, these results demonstrate that Δfmr1-t2A-eGFP can be used to overexpress FMRP and that LOW and HIGH FMRP OE result in different degrees of overexpression. Therefore, we used LOW and HIGH FMRP OE to test the sensitivity of neurogenesis to overexpression of FMRP. In subsequent experiments, eGFP expression with LOW FMRP OE was enhanced by coelectroporation of 0.7 μg/μl UAS::eGFP to facilitate imaging of labeled cells.
FMRP knockdown and overexpression reduce cell proliferation
Evidence from humans suggests that the gene dosage of FMR1 is tightly regulated and that both decreases and increases in FMRP can cause disease. Individuals with decreases in FMRP with Fragile X Syndrome and individuals with gene duplications of FMR1 both present with intellectual disability (Rio et al., 2010; Nagamani et al., 2012; Vengoechea et al., 2012; Hickey et al., 2013). To test whether FMRP regulates cell proliferation and/or survival in the optic tectum, we manipulated FMRP expression levels using knockdown and overexpression. We electroporated animals with Sox2bd::eGFP and UAS::tRFPnls to label tectal progenitors and their progeny, and either CMO, LOW fmr1a MO, or HIGH fmr1a MO. Then, we performed in vivo time-lapse imaging of eGFP+ cells at 1, 2, and 3 dfe (Fig. 4A ). We quantified the percent change in cell number between 1 and 3 dfe. CMO animals increased eGFP+ cell number from 1 − 3 dfe as NPCs proliferated. LOW fmr1a MO significantly reduced the normal increase in cell numbers seen in controls (Fig. 4B ; CMO: N = 20 animals; LOW fmr1a MO: N = 17 animals, p = 0.0012g compared to CMO). This LOW fmr1a MO-mediated decrease in cell number was rescued by coexpression of 1 μg/μl Δfmr1-t2A-eGFP (Fig. 4B ; LOW MO HIGH Δfmr1 Rescue: N = 10 animals, p = 0.035g compared to LOW fmr1a MO, p = 0.79g compared to CMO). When we knocked down FMRP using HIGH fmr1a MO, we found an even greater reduction in cell numbers, with a net loss of cells between 1-3 dfe (Fig. 4C,D ; CMO: N = 24 animals; HIGH fmr1a MO: N = 24 animals, p < 0.0001h). This result suggests that FMRP knockdown with HIGH fmr1a MO increases cell death, consistent with our observation from the in vivo knockdown assay. The HIGH fmr1a MO-mediated decrease in cell number was rescued by coexpression of 1 μg/μl Δfmr1-t2A-eGFP (Fig. 4C,D ; HIGH MO HIGH Δfmr1 Rescue: N = 17 animals, p = 0.0041h compared to HIGH fmr1 MO, p = 0.50h compared to CMO). These results demonstrate that fmr1a MO specifically knocks down FMRP since coexpression of MO-insensitive fmr1 was able to rescue the decrease in cell number.
Figure 4.

Knockdown and overexpression of FMRP decrease proliferation. A, Z-projections from in vivo confocal time-lapse images of cells expressing Sox2bd::eGFP + CMO (CMO) or 0.05 mM fmr1a MO (LOW fmr1a MO), and 1 μg/μl Δfmr1-t2A-eGFP + 0.05 mM fmr1a MO (LOW MO HIGH Δfmr1 Rescue) taken at 1 and 3 dfe. Dashed lines outline the optic tectum and inset shows a schematic of the optic tectum. B, The percent change in the number of eGFP+ cells increases over 3 d in CMO animals. FMRP knockdown with LOW fmr1a MO blocks the increase in cell number between 1 − 3 dfe. Coexpression of LOW fmr1a MO and HIGH Δfmr1 (LOW MO HIGH Δfmr1 Rescue) rescues the normal increase in cell number from 1 − 3 dfe (*p < 0.05, **p < 0.01). C, Z-projections from in vivo confocal time-lapse images of cells expressing Sox2bd::eGFP + CMO (CMO) or 0.1 mM fmr1a MO (HIGH fmr1a MO), and 1 μg/μl Δfmr1-t2A-eGFP + 0.1 mM fmr1a MO (HIGH MO HIGH Δfmr1 Rescue). Dashed lines outline the optic tectum. D, FMRP knockdown with HIGH fmr1a MO results in a negative percent change in cell number between 1 − 3 dfe, suggesting that proliferation and cell survival are affected with a higher concentration of morpholino. This decrease was rescued by co-electroporation of HIGH Δfmr1 (**p < 0.01, ***p < 0.001). E, A 2 h pulse of the thymidine analog CldU delivered at 3 dfe confirms that cell proliferation is decreased by HIGH fmr1a MO (*p < 0.05). F, Z-projections from in vivo confocal time-lapse images of Sox2bd::eGFP+ (Control) and 1 μg/μl Δfmr1-t2A-eGFP+ (HIGH FMRP OE) cells collected at 1 and 3 dfe. Dashed lines outline the optic tectum. G, H, The percent change in the number of eGFP+ cells increases over 3 d in control animals. HIGH FMRP OE significantly reduced the percent change in cell number between 1 − 3 dfe. Data from individual animals (G) and the mean ± SEM (H; ***p < 0.001). Scale bar, 50 μm.
The experiments described above indicate that FMRP knockdown decreases cell proliferation, however, our in vivo time-lapse imaging assay reports changes in both cell proliferation and survival. We therefore used acute incorporation of the thymidine analog CldU to test directly whether cell proliferation is affected with knockdown of FMRP. Animals were electroporated with MOs and incubated in CldU by bath application for 2 h at 1, 2, or 3 dfe. We did not detect changes in CldU incorporation at 1 or 2 dfe (data not shown). At 3 dfe, HIGH fmr1a MO significantly decreased the number of CldU+ proliferating cells in the optic tectum compared to CMO, but LOW fmr1a MO did not affect proliferation using this measure (Fig. 4E; CMO: N = 10 animals; LOW fmr1a MO: N = 11 animals, p = 0.38i compared to CMO; HIGH fmr1a MO: N = 13 animals, p = 0.025i compared to CMO). These results suggest that proliferation is differentially affected by different levels of fmr1a MO, with only a high concentration of MO being sufficient to decrease cell proliferation. In addition, the relatively modest decrease in proliferation detected with CldU incorporation demonstrates the utility of time-lapse imaging as a method to study cell proliferation. We found much more dramatic defects when we tracked a population of labeled cells over the course of 3 d with time-lapse imaging since effects are cumulative over time. While a decrease in CldU incorporation was not apparent until 3 dfe, we found a decrease in the total number of eGFP-labeled cells between 1 − 3 dfe with HIGH fmr1aMO using in vivo time-lapse imaging. This suggests that decreased proliferation with HIGH fmr1a MO is due to gradual depletion of the progenitor pool rather than an immediate quiescence of NPCs. This gradual decrease in the number of proliferating cells may be a result of increased NPC death or increased neuronal differentiation. These possibilities are explored in Figures 5 and 6.
To test the effect of FMRP overexpression on cell proliferation and survival in the tadpole brain, we electroporated animals with either Sox2bd::eGFP (control) or Δfmr1-t2A-eGFP (FMRP OE) to label tectal progenitors and their progeny and performed in vivo time-lapse imaging of eGFP+ cells between 1 − 3 dfe (Fig. 4F ). Control animals tended to increase the number of eGFP+ cells from 1 − 3 dfe as labeled NPCs proliferated in the tectum (Fig. 4G ). Animals with LOW FMRP OE had a similar increase in cell number from 1 − 3 dfe compared to control (Control: 58.5% ± 8.5%, N = 22 animals; LOW FMRP OE: 42.3% ± 9.9%, N = 20 animals, p = 0.22j). In contrast, HIGH FMRP OE tended to decrease the number of eGFP+ cells from 1 − 3 dfe (Fig. 4G ). On average, HIGH FMRP OE significantly reduced the number of eGFP+ cells generated from 1 − 3 dfe compared to controls (Fig. 4H ; CMO: N = 37 animals; HIGH FMRP OE: N = 25 animals, p < 0.0001k). This experiment indicates that overexpression of FMRP in the optic tectum can affect cell proliferation and/or cell survival. Combined with the results from our knockdown experiments, these results demonstrate that tectal cell proliferation and/or survival are sensitive to both increases and decreases in the level of FMRP.
FMRP knockdown increases cell death
We found that LOW fmr1a MO reduced the change in cell number from 1 − 3 dfe with in vivo time-lapse imaging without affecting cell proliferation as measured by CldU incorporation. While time-lapse imaging is a more sensitive assay and may be picking up proliferation defects not detected by CldU incorporation, this result suggests that decreased cell survival may be the primary defect with LOW fmr1a MO. Furthermore, the net loss of cells from 1 − 3 dfe with HIGH fmr1a MO suggests that loss of FMRP leads to cell death. Therefore, we tested the role of FMRP in cell survival. To test measures of cell death, we incubated tadpoles in staurosporine (STS) for 24 h to induce apoptosis. Then, we performed immunohistochemistry for caspase-3 (Casp3) and stained with SYTOX. Casp3 is an executioner caspase that is activated during the late phase of apoptosis (Kumar, 2007). SYTOX is a nucleic acid stain that brightly labels cells undergoing chromatin condensation at the end of apoptosis. STS dramatically increased the number of Casp3+ and SYTOX+ cells undergoing apoptosis (Fig. 5A−C ). About 50% of the labeled apoptotic cells were positive for both Casp3 and SYTOX, demonstrating that they label cells during a similar phase of cell death (Fig. 5C ). Of the remaining apoptotic cells we detected, a larger fraction were positive for SYTOX alone than for Casp3 alone. In addition, cells that were Casp3−SYTOX+ appeared to have even smaller pyknotic nuclei than those that were Casp3+SYTOX+. This suggests that SYTOX stains a larger proportion of the apoptotic cells than Casp3 and that it stains cells within and further along the cell death cascade compared to Casp3. Therefore, we used SYTOX staining to assess the role of FMRP in cell death.
We electroporated animals with MOs, then fixed and stained for SYTOX at 1, 2, or 3 dfe. At 1 dfe, the number of apoptotic SYTOX+ cells was significantly increased with fmr1a MO (Fig. 5D,E; CMO: N = 39 animals; LOW fmr1a MO: N = 43 animals, p = 0.0046l compared to CMO; HIGH fmr1a MO: N = 41 animals, p < 0.0001l compared to CMO). There was a trend toward HIGH fmr1a MO increasing the number of apoptotic SYTOX+ cells to a greater extent than LOW fmr1a MO (p = 0.078l). The large loss in cell number with HIGH fmr1a MO over 3 d of live imaging lends more support to HIGH fmr1a MO increasing cell death to a larger extent than LOW fmr1a MO. This increase in cell death was transient, as the number of SYTOX+ cells was similar between CMO and fmr1a MO at 2 and 3 dfe (data not shown). Taken together with the time-lapse imaging and CldU results, our experiments demonstrate that both cell proliferation and cell survival are regulated by FMRP. Furthermore, cell survival appears to be more sensitive to the level of FMRP since lower concentrations of MO were able to increase cell death without affecting proliferation. When FMRP was knocked down with a higher MO concentration, our data suggest that cell death increased further and a decrease in proliferation became apparent.
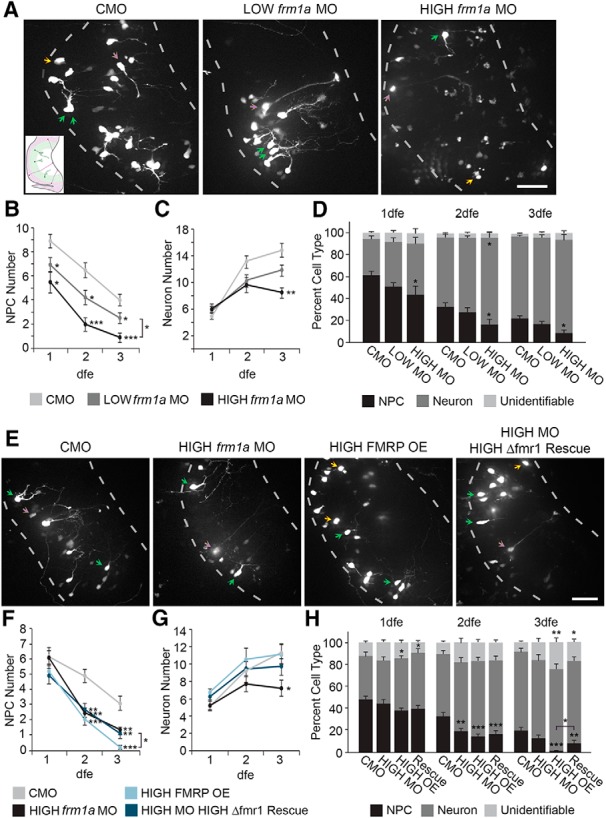
FMRP regulates neuronal differentiation
The experiments described above show that fmr1a MO decreases cell proliferation and survival, however, it is not clear whether one cell type, NPCs or neurons, is more sensitive to FMRP knockdown than another. We therefore investigated whether FMRP knockdown has different effects on the NPCs and neurons within our labeled population. We categorized the labeled cells from the in vivo time-lapse imaging as either NPCs or neurons based on morphology. NPCs are characterized by a triangular cell body and a long radial process extending from the ventricular zone to the pial surface, ending in an elaborated endfoot. Neurons possess a pear-shaped or round soma with elaborated dendritic arbors and an axon. Any cell that lacked a process was categorized as unidentifiable. We quantified the number of NPCs, neurons, and unidentifiable cells to analyze the effect of knockdown on each cell type (Fig. 6A −C). LOW and HIGH fmr1a MO significantly reduced the number of NPCs on all 3 d of imaging compared to CMO (Fig. 6B ; 1 dfe NPCs: CMO N = 27 animals; LOW fmr1a MO N = 17 animals, p = 0.027m; HIGH fmr1a MO N = 8 animals, p = 0.011m; 2 dfe NPCs: CMO N = 27 animals; LOW fmr1a MO N = 17 animals, p = 0.030n; HIGH fmr1a MO N = 8 animals, p = 0.0006n; 3 dfe NPCs: CMO N = 27 animals; LOW fmr1a MO N = 17 animals, p = 0.041o; HIGH fmr1a MO N = 8 animals, p = 0.0007o). The reduction of NPC number at 3 dfe was significantly larger for HIGH fmr1a MO compared to LOW fmr1a MO (p = 0.032o). There was a trend toward reduced neuron number with LOW fmr1a MO at 2 and 3 dfe (Fig. 6C; 2 dfe neurons: CMO N = 27 animals; LOW fmr1a MO N = 17 animals, p = 0.080p; 3 dfe neurons: CMO N = 27 animals; LOW fmr1a MO N = 17 animals, p = 0.088q). Combined with the significant decrease in NPCs with LOW fmr1a MO, these results suggest that the increase in cell death detected at 1 dfe in the presence of LOW fmr1a MO may preferentially affect NPCs and nonsignificant reductions in neuron number that appear later are due to depletion of the progenitor pool. HIGH fmr1a MO produced a trend toward reducing neuron number at 2 dfe and significantly reduced the number of neurons at 3 dfe (Fig. 6C ; 2 dfe neurons: CMO N = 27 animals; HIGH fmr1a MO N = 8 animals, p = 0.11p; 3 dfe neurons: CMO N = 27 animals; HIGH fmr1a MO N = 8 animals, p = 0.0031q). The decrease in neuron number may be due in part to death of neurons with a higher degree of knockdown, and a decrease in NPC proliferation (Fig. 4C−E ) and increased death of NPCs (Fig. 5D,E ) likely also contribute to the decreased number of neurons though depletion of the progenitor pool with HIGH fmr1a MO.
Figure 6.
FMRP regulates differentiation. In vivo confocal time-lapse images of cells expressing Sox2bd::eGFP and CMO or fmr1a MO collected at 3 dfe and quantification of the changes in neural progenitor cells (NPCs) and neurons over time. A, Confocal Z-projections show the numbers of NPCs (purple arrows), neurons (green arrows), and unidentifiable cells (yellow arrows) in optic tecta expressing CMO, LOW (0.05 mM) fmr1a MO, and HIGH (0.1 mM) fmr1a MO. Dashed lines outline the optic tectum and inset shows a schematic of the optic tectum. B, C, Over 3 d of imaging, there is a decrease in the number of NPCs (B) and an increase in the number of neurons (C) in control animals. LOW and HIGH fmr1a MO decrease the number of NPCs, and HIGH fmr1a MO also decreases the number of neurons (*p < 0.05, **p < 0.01, ***p < 0.001). D, Knockdown of FMRP with HIGH fmr1a MO decreases the proportion of NPCs and increases the proportion of neurons (*p < 0.05). E, Z-projections from in vivo confocal time-lapse images of cells expressing Sox2bd::eGFP + CMO (CMO) or 0.1 mM fmr1a MO (HIGH fmr1a MO), or 1 μg/μl Δfmr1-t2A-eGFP alone (HIGH FMRP OE) or with 0.1 mM fmr1a MO (HIGH MO HIGH Δfmr1 Rescue) at 3 dfe. Dashed lines outline the optic tectum. F, HIGH fmr1a MO and HIGH FMRP OE decrease the number of NPCs and co-electroporation of 1 μg/μl Δfmr1-t2A-eGFP and HIGH fmr1a MO (HIGH MO HIGH Δfmr1 Rescue) partially rescues the defect at 3 dfe with HIGH FMRP OE alone, but does not rescue to control levels (*p < 0.05, **p < 0.01, ***p < 0.001). G, Neuron numbers decrease with HIGH fmr1a MO and this decrease is rescued by co-electroporation of 1 μg/μl Δfmr1-t2A-eGFP (HIGH MO HIGH Δfmr1 Rescue; *p < 0.05). H, HIGH fmr1a MO and HIGH FMRP OE both decrease the proportion of NPCs, and HIGH FMRP OE also increases the proportion of neurons and unidentifiable cells. At 3 dfe, coexpression of HIGH fmr1a MO and 1 μg/μl Δfmr1 partially rescues the HIGH FMRP OE-mediated decrease in NPC proportion, but other defects are not rescued (*p < 0.05, **p < 0.01, ***p < 0.001). Scale bars, 50 μm.
We quantified the proportion of NPCs and neurons present within the labeled cell population to determine whether loss of FMRP affects differentiation of progenitors into neurons. The proportions of labeled NPCs and neurons were unchanged with LOW fmr1a MO compared to CMO for all 3 d of imaging (Fig. 6D ). By contrast, HIGH fmr1a MO decreased the percent of NPCs on all 3 d of imaging (Fig. 6D; 1 dfe %NPCs: CMO N = 27 animals; HIGH fmr1a MO N = 8 animals, p = 0.050r; 2 dfe %NPCs: CMO N = 27 animals; HIGH fmr1a MO N = 8 animals, p = 0.033s; 3 dfe %NPCs: CMO N = 27 animals; HIGH fmr1a MO N = 8 animals, p = 0.024t). In addition, HIGH fmr1a MO increased the proportion of neurons at 2 dfe (2 dfe %neurons: CMO N = 27 animals; HIGH fmr1a MO N = 8 animals, p = 0.041u). At 1 and 3 dfe, the decrease in percent NPCs was accompanied by nonsignificant increases in percentages of both unidentifiable cells and neurons. We suspect that unidentifiable cells are immature neurons that lack processes, although we cannot rule out the possibility that they are dying cells that still have normal cell body morphology.
Next, we assessed the effect of FMRP overexpression on the numbers of NPCs and neurons and whether expression of MO-insensitive FMRP might rescue the decreases in NPC and neuron number with HIGH fmr1a MO. LOW FMRP OE had no effect on NPC or neuron number (1 dfe NPC: Control 12.9 ± 1.0, N = 22 animals; LOW FMRP OE 12.5 ± 1.2, N = 20 animals, p = 0.69v; 2 dfe NPC: Control 13.1 ± 1.1, N = 22 animals; LOW FMRP OE 12.1 ± 1.3, N = 20 animals, p = 0.56w; 3 dfe NPC: Control 11.2 ± 1.3, N = 22 animals; LOW FMRP OE 9.6 ± 1.0, N = 20 animals, p = 0.43x; 1 dfe neurons: Control 16.4 ± 1.5, N = 22 animals; LOW FMRP OE 14.5 ± 2.0, N = 20 animals, p = 0.44y; 2 dfe neurons: Control 28.9 ± 2.2, N = 22 animals; LOW FMRP OE 24.2 ± 2.1, N = 20 animals, p = 0.073z; 3 dfe neurons: Control 36.1 ± 2.5, N = 22 animals; LOW FMRP OE 30.8 ± 2.4, N = 20 animals, p = 0.21aa). HIGH FMRP OE decreased the number of NPCs at 2 and 3 dfe, without affecting neuron number (Fig. 6E−G ; 2 dfe NPCs: CMO N = 17 animals; HIGH FMRP OE N = 15 animals, p = 0.0002bb; 3 dfe NPCs: CMO N = 17 animals; FMRP OE N = 15 animals, p < 0.0001cc). This suggests that asymmetric, self-renewing divisions are decreased and the loss of NPCs is due in large part to direct differentiation of NPCs into neurons when FMRP is overexpressed. We found that coexpression of HIGH fmr1a MO and 1 μg/μl Δfmr1-t2A-eGFP increased the number of NPCs at 3 dfe compared to HIGH FMRP OE alone, but was unable to rescue decreases in NPC number seen with HIGH FMRP OE or HIGH fmr1a MO alone back to control levels (Fig. 6F ; 2 dfe NPCs: HIGH fmr1a MO N = 16 animals, p = 0.0004bb compared to CMO; HIGH MO HIGH Δfmr1 Rescue N = 17 animals, p = 0.0058bb compared to CMO; 3 dfe NPCs: HIGH fmr1a MO N = 16 animals, p = 0.0080cc compared to CMO; HIGH MO HIGH Δfmr1 Rescue N = 17 animals, p = 0.020cc compared to HIGH FMRP OE, p = 0.0049cc compared to CMO). Coexpression of HIGH fmr1a MO and 1 μg/μl Δfmr1-t2A-eGFP rescued the decrease in neuron number seen with HIGH fmr1a MO alone (Fig. 6G ; 3 dfe neurons: CMO N = 17 animals; HIGH fmr1a MO N = 16 animals, p = 0.037dd compared to CMO; HIGH MO HIGH Δfmr1 Rescue N = 17 animals, p = 0.72dd compared to CMO).
When we quantified the proportion of cell types present within the labeled population to assess the effect of FMRP overexpression on differentiation, we found no change with LOW FMRP OE (1 dfe %NPCs: Control 38.8% ± 1.9%, N = 18 animals; LOW FMRP OE 40.8% ± 4.5%, N = 20 animals, p = 0.53ee; 2 dfe %NPCs: Control 28.9% ± 1.9%, N = 22 animals; LOW FMRP OE 31.9% ± 3.9%, N = 20 animals, p = 0.48ff; 3 dfe %NPCs: Control 21.5% ± 1.6%, N = 22 animals; LOW FMRP OE 23.2% ± 2.4%, N = 20 animals, p = 0.56gg; 1 dfe %neurons: Control 47.1% ± 2.0%, N = 22 animals; LOW FMRP OE 40.1% ± 3.0%, N = 20 animals, p = 0.06hh; 2 dfe %neurons: Control 60.1% ± 1.7%, N = 22 animals; LOW FMRP OE 57.4% ± 3.2%, N = 20 animals, p = 0.45ii; 3 dfe %neurons: Control 69.8% ± 1.7%, N = 22 animals; LOW FMRP OE 68.7% ± 2.1%, N = 20 animals, p = 0.68jj). HIGH FMRP OE significantly decreased the proportion of NPCs at 2 and 3 dfe accompanied by a significant increase in unidentifiable cells at 3 dfe (Fig. 6H ; 2 dfe %NPCs: CMO N = 17 animals; HIGH FMRP OE N = 15 animals, p = 0.0001kk; 3 dfe %NPCs: CMO N = 17 animals; HIGH FMRP OE N = 15 animals, p < 0.0001ll; 3 dfe %unidentifiable: CMO N = 17 animals; HIGH FMRP OE N = 15 animals, p = 0.0048mm). In addition, HIGH FMRP OE increased the proportion of neurons at 1 dfe (Fig. 6H ; CMO N = 17 animals; HIGH FMRP OE N = 15 animals, p = 0.043nn). Coexpression of HIGH fmr1a MO and 1 μg/μl Δfmr1-t2A-eGFP partially rescued the decrease in NPC proportion at 3 dfe by HIGH FMRP OE alone, but failed to rescue the remaining defects from HIGH fmr1a MO or HIGH FMRP OE alone (Fig. 6H ; 2 dfe %NPCs: HIGH fmr1a MO N = 16 animals, p = 0.0048kk compared to CMO; HIGH MO HIGH Δfmr1 Rescue p = 0.0006kk compared to CMO; 3 dfe %NPCs: HIGH MO HIGH Δfmr1 Rescue N = 16 animals, p = 0.018ll compared to HIGH FMRP OE, p = 0.0084ll compared to CMO; 1 dfe %neurons: HIGH MO HIGH Δfmr1 Rescue N = 17 animals, p = 0.026nn compared to CMO; 3 dfe %unidentifiable: HIGH MO HIGH Δfmr1 Rescue p = 0.026mm compared to CMO).
FMRP regulates dendritic morphology
The in vivo imaging experiments above suggested that neuronal dendrite arbor development might be abnormal with knockdown or overexpression of FMRP. Defects in spine morphology have been widely reported in Fragile X patients and animal models (Hinton et al., 1991; Comery et al., 1997; Irwin et al., 2001; Nimchinsky et al., 2001; Cruz-Martín et al., 2010), but reports of defects in dendritic morphology have been mixed (Irwin et al., 2002; Galvez et al., 2003; Lee et al., 2003; Castrén et al., 2005; Koekkoek et al., 2005; Thomas et al., 2008; Guo et al., 2011; Scotto-Lomassese et al., 2011; Sheridan et al., 2011; Guo et al., 2012; Till et al., 2012; Telias et al., 2013; Doers et al., 2014). While Xenopus tectal neurons lack dendritic spines, we analyzed dendritic arbor morphology to assess whether FMRP plays a role in dendritic development. We imaged tectal neurons in vivo in animals sparsely electroporated with Sox2bd::eGFP and either CMO, LOW fmr1a MO, or HIGH fmr1a MO at 2 and 3 dfe using a two-photon microscope (Fig. 7A ). We reconstructed the dendritic arbors of imaged neurons and quantified total dendritic branch length and total dendritic branch tip number (Fig. 7A−C ). At 2 dfe, HIGH fmr1a MO decreased total dendritic branch tip number (Fig. 7C ; 2 dfe Branch tip number: CMO N = 66 cells; HIGH fmr1a MO N = 46 cells, p = 0.018oo). At 2 dfe there were also noticeable decreases in total dendritic branch length with both MO concentrations and in total dendritic branch tip number with LOW fmr1a MO, but these did not reach significance (Fig. 7B,C ; 2 dfe Length: CMO N = 66 cells; LOW fmr1a MO N = 60 cells, p = 0.34pp compared to CMO; HIGH fmr1a MO N = 46 cells, p = 0.20pp compared to CMO; 2 dfe Branch tip number: LOW fmr1a MO N = 60 cells, p = 0.12oo compared to CMO). At 3 dfe, HIGH fmr1a MO decreased total dendritic branch length and total dendritic branch tip number (Fig. 7B,C ; 3 dfe Length: CMO N = 68 cells; HIGH fmr1a MO N = 49 cells, p = 0.0097qq; 3 dfe Branch tip number: CMO N = 68 cells; HIGH fmr1a MO N = 49 cells, p = 0.0014rr). We calculated branch density as the ratio of total dendritic branch tip number/total dendritic branch length and found no change in branch density with FMRP knockdown (Fig. 7D ). This suggests that neurons lacking FMRP follow the same branching rule as control cells, they are just smaller overall.
Figure 7.

FMRP regulates dendritic development. In vivo two-photon time-lapse images of cells expressing Sox2bd::eGFP and CMO or fmr1a MO collected at 2 and 3 dfe. A, Two-photon Z-projections of imaged cells and their reconstructed dendritic arbors at 2 and 3 dfe for cells with FMRP knockdown compared to control. B, HIGH (0.1 mM) fmr1a MO decreased total dendritic length at 3 dfe (**p < 0.01). C, HIGH fmr1a MO decreased total dendritic branch tip number at 2 and 3 dfe (*p < 0.05, **p < 0.01). D, Branch density was unchanged between the groups. E, Two-photon Z-projection and reconstructed dendritic arbor of a cell expressing 1 μg/μl Δfmr1-t2A-eGFP (HIGH FMRP OE) at 3 dfe. F, G, HIGH FMRP OE decreased total dendritic length (F) and total dendritic branch tip number (G) compared to control (Sox2bd::eGFP; ***p < 0.001). H, Two-photon Z-projections of imaged cells and their reconstructed dendritic arbors at 3 dfe for cells when FMRP is knocked down (HIGH fmr1a MO), overexpressed with 0.5 μg/μl Δfmr1-t2A-eGFP (LOW FMRP OE), and rescued (HIGH MO LOW Δfmr1 Rescue) compared to control (CMO). I, J, Co-electroporation of LOW Δfmr1-t2A-eGFP rescued HIGH fmr1a MO-mediated decreases in total dendritic length (I) and dendritic branch tip number (J) (*p < 0.05, **p <0.01). Scale bars, 20 μm.
Next, we electroporated animals with Δfmr1-t2A-eGFP alone or with HIGH fmr1a MO to overexpress or rescue FMRP expression and performed in vivo two-photon imaging at 3 dfe (Fig. 7E−J ). HIGH FMRP OE decreased total dendritic length and total dendritic branch tip number compared to control (Fig. 7E−G ; Length: Control N = 45 cells; HIGH FMRP OE N = 44 cells, p < 0.0001ss; Branch number: Control N = 45 cells; HIGH FMRP OE N = 44 cells, p < 0.0001tt). LOW FMRP OE resulted in a trend toward decreased total dendritic branch length and no change in total dendritic branch tip number (Fig. 7H−J ; Length: CMO N = 38 cells; LOW FMRP OE N = 32 cells, p = 0.10uu; Branch tip number: CMO N = 38 cells; LOW FMRP OE N = 32 cells, p = 0.28vv). Coexpression of HIGH fmr1a MO and 0.5 μg/μl Δfmr1-t2A-eGFP rescued defects in total dendritic branch length and total dendritic branch tip number caused by HIGH fmr1a MO alone (Fig. 7H−J ; Length: HIGH fmr1a MO N = 20 cells, p = 0.011uu compared to CMO; HIGH MO LOW Δfmr1 Rescue N = 31 cells, p = 0.027uu compared to HIGH fmr1a MO, p = 0.75uu compared to CMO; Branch tip number: HIGH fmr1a MO N = 20 cells, p = 0.0087vv compared to CMO; HIGH MO LOW Δfmr1 Rescue N = 31 cells, p = 0.024vv compared to HIGH fmr1a MO, p = 0.68vv compared to CMO). These results demonstrate that both increases and decreases in FMRP interfere with normal dendritic arbor development.
Discussion
We used in vivo time-lapse imaging to investigate the functions of FMRP in NPC proliferation, survival, and differentiation in Xenopus tadpole optic tectum. This highly sensitive experimental strategy tracks a labeled cell population over time, thereby revealing cumulative effects of manipulating FMRP on neurogenesis. Increasing or decreasing FMRP decreased proliferation and/or increased apoptosis of NPCs and their progeny. FMRP knockdown also decreases cell proliferation and survival detected with CldU incorporation and SYTOX staining. These experimental strategies assess outcomes at single time points, which helped elucidate the timing and roles of FMRP knockdown on proliferation and apoptosis. In addition, increasing or decreasing FMRP expression increases NPC differentiation into neurons and the resulting neurons have simpler dendritic arbors. These findings suggest that dysregulation of neurogenesis during embryonic development contributes to the pathogenesis of FXS.
We knocked down FMRP using translation-blocking MOs in tadpole brain to recapitulate loss of FMRP during human fetal development. FMRP is expressed in full mutation carrier FXS human fetuses until about 12.5 weeks of gestation (Willemsen et al., 2002) and in embryonic stem cells derived from full mutation human embryos prior to differentiation (Eiges et al., 2007; Urbach et al., 2010). These findings suggest that models of FXS in which FMRP is expressed early in embryonic development and then eliminated through conditional knockout or knockdown methods will most closely mirror loss of FMRP expression in the disease state.
Neurogenesis is sensitive to FMRP levels
Our in vivo time-lapse imaging approach followed a GFP-labeled population of Sox2-expressing NPCs and their progeny over 3 d to evaluate several distinct cellular events contributing to neurogenesis, including NPC proliferation and survival, the rate of differentiation of progenitors into neurons, and dendritic arbor elaboration in neurons. Over 3 d, the number of eGFP-labeled cells in control animals increases as labeled NPCs proliferate. In addition, the proportion of NPCs decrease and the proportion of neurons increase within the labeled population as neurons differentiate. Finally, as neurons mature, their dendritic arbors become more elaborate. The level of FMRP is critical to each of these processes during neurogenesis and the degree to which FMRP is knocked down or overexpressed changes the phenotypic outcome (Fig. 8). For example, with respect to FMRP knockdown, LOW fmr1a MO does not affect NPC proliferation, but increases NPC cell death compared to CMO. The proportions of NPCs and neurons within the labeled population do not change, suggesting that differentiation is normal; however, the neurons tend to have deficient dendritic arbor development. A greater degree of FMRP knockdown with HIGH fmr1a MO produces a trend toward more cell death than seen with LOW fmr1a MO. In addition, NPC proliferation decreases and neuronal differentiation increases. Together, this results in a smaller cell population but a higher proportion of neurons. Furthermore, the resulting neurons have a more severe and lasting deficit in dendritic arbor growth and branching than seen with lower FMRP knockdown. These data indicate that higher levels of FMRP knockdown affect some of the cellular events contributing to neurogenesis, such as NPC proliferation and neuronal differentiation, whereas lower levels of knockdown affect other cellular events, such as NPC apoptosis, suggesting that these processes are differentially sensitive to different levels of FMRP.
Figure 8.

Neurogenesis is sensitive to FMRP levels. Summary diagram showing the consequences of perturbing FMRP levels on the labeled cell population. The numbers and proportions of neural progenitor cells (purple) and neurons (green), as well as the dendritic arbor morphology of neurons are altered in the presence of fmr1a MO or overexpression of FMRP. LOW fmr1a MO increases NPC apoptosis, leading to a reduction in the progenitor pool and a lower total number of cells present at 3 dfe compared to control. The neurons generated with LOW fmr1a MO have a trend toward deficient dendritic arbor development. HIGH fmr1a MO increases apoptosis compared to LOW fmr1a MO. In addition, HIGH fmr1a MO decreases proliferation and increases NPC differentiation into neurons. This leads to a greater reduction of the progenitor pool, a lower total number of cells present at 3 dfe, and a larger proportion of neurons among the cell population at 3 dfe. In addition, those neurons have a persistent defect in dendrite arbor elaboration. LOW FMRP overexpression does not result in defects in cell proliferation, cell death, differentiation, or dendritic morphology. HIGH FMRP overexpression increases cell death, decreases proliferation, and increases differentiation leading to a complete loss of the progenitor pool at 3 dfe. Neuron numbers are at control levels at 3 dfe because of the dramatic increase in differentiation, but those neurons have a defect in dendritic arbor development.
A low level of FMRP overexpression does not produce any defects in cell proliferation, differentiation, or dendritic morphology. In contrast, a high level of FMRP overexpression reduces the normal increase in cell number over 3 d, suggesting that NPC proliferation and cell survival are decreased. This effect on NPCs is accompanied by a greater increase in differentiation into neurons than with HIGH fmr1a MO and leads to a near total loss of the labeled NPC pool. The resulting neurons also have simpler dendritic arbors. Consistent with the results above about fmr1a knockdown, these results demonstrate that different levels of FMRP regulate different processes contributing to neurogenesis.
Co-electroporating LOW or HIGH fmr1a MO and HIGH Δfmr1 rescued defects in cell proliferation and survival. In addition, co-electroporating HIGH fmr1a MO and LOW Δfmr1 rescued dendritic arbor development. MO electroporation results in a widespread MO distribution and likely decreases FMRP throughout the tectum, whereas plasmid electroporation results in more sparsely distributed Δfmr1 expression. The rescue seen under these conditions suggests that FMRP functions cell-autonomously to regulate cell proliferation, survival, and dendritic arbor morphology. In contrast, co-electroporating HIGH fmr1a MO and HIGH Δfmr1 only partially rescued defects in neuronal differentiation, possibly because of non-cell-autonomous circuit-wide effects of FMRP knockdown on differentiation. Alternately, the level of FMRP expressed under the rescue condition may not be within the physiological range. In support of this, the differentiation phenotype in the rescue condition closely mirrored that of HIGH FMRP OE. Therefore, the data suggest that a combination of HIGH fmr1a MO and HIGH Δfmr1 results in a higher than normal FMRP expression. Interestingly, this experimental condition did rescue the change in cell number between 1 − 3 dfe, suggesting that proliferation and survival may be less sensitive to FMRP levels than neuronal differentiation.
Our studies show that increasing or decreasing FMRP levels can have similar outcomes with respect to cell proliferation and neuronal differentiation (Fig. 8). FMRP functions as a translational repressor (Waung and Huber, 2009) and, as such, increasing or decreasing FMRP levels would be expected to decrease or increase protein levels of its target mRNAs, respectively. Hundreds of FMRP target mRNAs have been identified (Darnell et al., 2011) and these targets may include both enhancers and repressors of neuronal development. The combined effect of protein dysregulation of these various targets may ultimately lead to strikingly similar phenotypes when FMRP is increased and decreased.
FMRP regulates cell proliferation
Along with dendritic spine abnormalities, postmortem FXS brains commonly display macrocephaly, dilation of the ventricles, and cortical atrophy (Sabaratnam, 2000). Imaging studies show both increases and decreases in the size of brain regions (Lightbody and Reiss, 2009). These findings suggest that cell numbers may be affected in FXS, which could arise from defects in neurogenesis, including cell survival, proliferation, and differentiation, which can expand or deplete the progenitor pool. Recent studies have implicated FMRP in the control of neurogenesis both in vivo and in vitro, but the effect of loss of FMRP varies with experimental conditions. Cell proliferation in adult Fmr1 knockout (KO) mouse hippocampus in vivo and in vitro has been reported to increase (Luo et al., 2010; Guo et al., 2011) or remain unchanged (Eadie et al., 2009; Guo et al., 2012) in 2 − 3 month old animals, and to decrease in 9 − 12 month old animals (Lazarov et al., 2012). Similarly, Drosophila dFmr1 mutant neuroblast cultures and embryonic Fmr1 KO mouse cortex have increased cell proliferation (Castrén et al., 2005; Callan et al., 2010). In FXS human embryonic stem cells (ESCs) and embryonic and early postnatal mouse cortex, loss of functional FMRP does not appear to alter cell proliferation (Castrén et al., 2005; Bhattacharyya et al., 2008; Tervonen et al., 2009).
Our experiments show that loss or overexpression of FMRP in the Xenopus tadpole optic tectum decreases cell proliferation. Knockdown and overexpression of FMRP both prevent the normal increase in cell number detected over 3 d of imaging. We used CldU incorporation to test proliferation at discrete time points between 1 − 3 dfe, and detected a decrease in CldU incorporation at 3 dfe. This suggests that during our 3 d imaging window, increases in apoptosis and differentiation early on led to a gradual depletion of the progenitor pool resulting in a decrease in proliferating cells by 3 dfe. The decrease in accumulation of cells over 3 d of live imaging was quite large with perturbation of FMRP levels, but the decrease in CldU incorporation with fmr1a MO was much more modest. This suggests that small decreases in cell proliferation as detected by CldU incorporation can have profound impacts on the numbers of NPCs and neurons that are generated over time. Many of the previous experiments investigating the role of FMRP in cell proliferation used incorporation of the thymidine analog BrdU, which may not have the sensitivity to reveal small changes that are present. In addition, experiments in embryonic or early postnatal mouse that failed to detect changes in cell proliferation were conducted in KO animals. KO animals may have compensatory changes that mask alterations in cell proliferation but are apparent with acute knockdown of FMRP, as in our experiments. In fact, Saffary and Xie (2011) found that depletion of the neural progenitor pool induced by the loss of FMRP is much more substantial with shRNA-mediated knockdown of FMRP than in KO animals.
FMRP regulates cell survival
Loss of FMRP has variable effects on cell survival during development of different organisms. Cell survival was unaffected in Drosophila dFmr1 mutant neuroblast cultures and following acute FMRP knockdown in vivo in embryonic mouse cortex (Callan et al., 2010; Saffary and Xie, 2011). However, the normal cell death of peptidergic neurons during Drosophila development decreased in dFmr1 mutants (Gatto and Broadie, 2011). A similar decrease in cell death was observed in early postnatal cortex and hippocampus of Fmr1 KO mice (Cheng et al., 2013). FMRP overexpression in Drosophila increases cell death (Wan et al., 2000). However, in cultured ESCs and embryonic hippocampal neurons from Fmr1 KO mouse, as well as in rat embryonic cortical neuron cultures and in vivo in juvenile rat striatum with acute FMRP knockdown, loss of FMRP during development increased apoptosis (Castrén et al., 2005; Jacobs and Doering, 2010; Jeon et al., 2012). While FMRP reportedly has both pro- and anti-apoptotic roles in the developing brain, studies have consistently shown increased apoptosis in the hippocampus of adult Fmr1 KO mice (Eadie et al., 2009; Luo et al., 2010; Guo et al., 2011; Lazarov et al., 2012). Furthermore, healthy cells upregulate FMRP in response to apoptosis-inducing stimuli and the loss of FMRP renders cells more vulnerable to death (Jeon et al., 2011; Jeon et al., 2012; Liu et al., 2012; Zhang et al., 2014).
We evaluated apoptosis in NPCs and neurons with knockdown and overexpression of FMRP. Acute FMRP knockdown increases apoptosis and NPC survival is preferentially sensitive to FMRP knockdown. LOW fmr1a MO increased apoptosis, measured by SYTOX staining, and imaging showed that NPCs were the only cell type that is significantly decreased in number. Furthermore, SYTOX+ labeling shows that most dying cells are within or close to the proliferative zone. Together, the data indicate that NPCs are the primary apoptotic cell type with LOW fmr1a MO, consistent with previous studies suggesting the role of FMRP in apoptosis may be cell-type specific (Castrén et al., 2005; Lazarov et al., 2012). With HIGH fmr1a MO, apoptosis was increased compared to LOW fmr1a MO and SYTOX staining showed that apoptosis included both NPCs and neurons. Thus, HIGH fmr1a MO decreases the number of neurons generated during the 3 d imaging window indirectly by depleting the progenitor pool through NPC apoptosis and directly through neuronal apoptosis. Many animals with HIGH FMRP OE had a small loss of labeled tectal cells between 1 − 3 dfe, suggesting that FMRP overexpression leads to apoptosis. The total number of neurons is normal while NPC numbers are decreased with FMRP overexpression, suggesting that NPCs are preferentially lost to apoptosis. However, NPC differentiation into neurons is also increased under these conditions. Therefore, neuronal apoptosis that is offset by increased neuronal differentiation is also consistent with our data. We could not directly test the cell-type specificity of apoptosis with FMRP OE because it would require assessing apoptosis in response to FMRP OE in a cell-autonomous manner. In contrast, MO electroporation is more widespread and apoptosis can be assessed using a global measure like SYTOX staining. Together, these results demonstrate that both increasing and decreasing FMRP expression increase apoptosis and that, under some conditions, NPCs are preferentially sensitive to apoptosis.
FMRP regulates differentiation
Loss of FMRP decreased neuronal differentiation in adult hippocampus in some experiments (Luo et al., 2010; Guo et al., 2011; Guo et al., 2012), while it increased neuronal differentiation in other studies (Eadie et al., 2009; Lazarov et al., 2012). Likewise, mixed results were reported in human FXS ESCs (Castrén et al., 2005; Bhattacharyya et al., 2008; Telias et al., 2013). Loss of FMRP consistently increases NPC differentiation into intermediate progenitor cells and/or neurons in embryonic and early postnatal mouse cortex both in vivo and in vitro (Castrén et al., 2005; Tervonen et al., 2009; Saffary and Xie, 2011) and in postmortem fetal human brain (Tervonen et al., 2009).
In our experiments, loss or overexpression of FMRP increases NPC differentiation into neurons, resulting in an increased proportion of neurons and decreased proportion of NPCs within the labeled cell population. NPCs are also more susceptible to apoptosis with perturbed FMRP levels, which contributes to the decrease in NPCs. However, if neuronal differentiation is normal, a loss of NPCs due solely to apoptosis should reduce the progenitor pool and decrease the number of resulting neurons proportionally, as we found with LOW fmr1a MO. In contrast, HIGH fmr1a MO decreased the proportion of NPCs and increased the proportion of neurons within the labeled cell population, indicating an increase in neuronal differentiation. Depletion of the progenitor pool due to the combination of apoptosis and increased differentiation with HIGH fmr1a MO decreased the number of neurons generated, but those neurons make up a higher percentage of the labeled cell population. Interestingly, overexpression with HIGH Δfmr1 completely depleted the progenitor pool, suggesting that all NPCs differentiated into neurons. Perturbations in FMRP levels that decreased NPCs often produced nonsignificant increases in unidentifiable cells, which are likely immature neurons that lack processes used to categorize them as neurons. In control animals, unidentifiable cell numbers were highest at 1 dfe and decreased in subsequent days as distinctive neuronal morphology developed. Previous in vivo time-lapse imaging of tectal progenitors at intervals of 2 − 19 h over several days demonstrated that ∼50% of labeled NPCs differentiate directly into neurons without dividing, whereas other NPCs undergo classical symmetric or asymmetric divisions, after which one or both progeny rapidly adopts neuronal morphology (Bestman et al., 2012). Here, HIGH fmr1a MO and HIGH FMRP OE resulted in larger proportions of neurons and smaller proportions of NPCs compared to CMO without increasing cell number over the 3 d imaging period. This suggests that the increased proportion of neurons did not result from NPC proliferation and differentiation, but may have occurred through direct differentiation of NPCs into neurons. Given our 24 h time-lapse imaging interval, division followed by apoptosis is also supported by our data.
FMRP regulates dendritic arbor development
The role of FMRP in dendritic arbor development has been studied in human stem cell preparations and in various brain regions of Fmr1 KO mouse in vitro and in vivo. While defects in dendritic arbor morphology have not been observed in adult Fmr1 KO visual cortex, cerebellum, and olfactory bulb (Irwin et al., 2002; Koekkoek et al., 2005; Scotto-Lomassese et al., 2011), reduced dendritic length and/or complexity has been observed in developing brain regions. Newborn neurons in adult Fmr1 KO hippocampus have decreased dendritic length, complexity, and branch tip number in vivo and in vitro (Guo et al., 2011; Guo et al., 2012). Decreased dendrite number, length, and branching have been noted in vitro in differentiated human FXS ESCs and iPSCs (Castrén et al., 2005; Sheridan et al., 2011; Doers et al., 2014), with one exception (Telias et al., 2013). Cultured neurons from embryonic Fmr1 KO mouse cortex have decreased dendrite number and length, while those from hippocampus have decreased dendrite length and area (Castrén et al., 2005; Jacobs and Doering, 2010). In vivo, modest and/or transient defects in dendritic orientation in somatosensory cortex and decreased dendritic branch length in spinal cord of Fmr1 KO mice have also been observed (Galvez et al., 2003; Thomas et al., 2008; Till et al., 2012).
Here, FMRP loss or overexpression decreased total dendritic branch length and arbor complexity, again demonstrating the sensitivity of neuronal phenotypes to FMRP expression levels. The defects in dendritic arbor development may result from loss of FMRP specifically in the imaged neurons, or their defective history during proliferation and differentiation could contribute. HIGH Δfmr1 expression rescued the cell proliferation/survival phenotype, but failed to rescue the differentiation and dendritic arbor phenotypes. We systematically decreased the concentration of overexpressed Δfmr1 plasmid until it did not produce a dendritic arbor defect when expressed on its own. Only then were we able to rescue the defect in dendritic morphology resulting from HIGH fmr1a MO, again suggesting that neuronal phenotypes are exquisitely sensitive to FMRP levels.
Summary
In summary, we have shown that FMRP regulates neuronal proliferation, survival, differentiation, and dendritic arbor development in vivo in the Xenopus tadpole. These processes are highly sensitive to FMRP levels and both increases and decreases in FMRP affect neurogenesis. Our ability to uncover these phenotypes is based on our in vivo time-lapse imaging strategy, which is optimized to detect cumulative effects of perturbing FMRP levels compared to outcome measures based on single time points. Ambiguities in the literature with respect to FMRP’s role in neuronal proliferation and survival may be present, in part, because traditional assays lack the sensitivity to resolve these changes. The use of different aged animals and different means of knockdown may also account for this variability in neurogenesis phenotypes. We modeled the loss of FMRP in the human FXS fetus using acute FMRP knockdown at a developmental stage in Xenopus similar to mammalian fetal development. These studies demonstrate promise in using Xenopus to identify fundamental features of FXS. At present, clinical and preclinical research for FXS focus on the development of drugs that modulate glutamatergic synaptic signaling, and therefore target events that occur much later in brain development than neurogenesis. Yet, as suggested in this paper, disorders like FXS may result from developmental events that have gone awry prenatally when most cell proliferation occurs in mammals. Some of the neuronal phenotypes that current interventions are trying to ameliorate could result from a defective history during the genesis of those neurons. Therefore, the development of interventions that target early events in brain development such as cell proliferation, differentiation, and survival may prove to be of great therapeutic benefit.
Synthesis
The decision was a result of the Reviewing Editor Alfonso Represa and the peer reviewers coming together and discussing their recommendations until a consensus was reached. A fact-based synthesis statement explaining their decision and outlining what is needed to prepare a revision is listed below. The following reviewers agreed to reveal their identity: Ingrid Bureau
The authors took the original approach to knock-down FMRP expression with morpholinos to study the effects on neurogenesis in the tadpole. This model has the merit to match the onset of the disease better than the constitutional ko because Fmr1 is silenced only after the 12.5 weeks of gestation in humans and because FMRP remain in embryonic stem cells. However, the knock-down of FMRP by morpholinos is not complete, but 60 % maximum. They monitored with time lapse two-photon imaging the rate of differentiation from neuroprogenitors (NP) to neurons, cell death and the neuron dendritic complexity over the course of 3 days. They also studied the effects induced by an overexpression of FMRP which was delivered through the electroporation of plasmids. The knock-down and the over expression were both tested at two doses. They found that knocking down or over-expressing FMRP had a dose-dependent effect on NP survival, on neuron differentiation and on dendritic growth. Interestingly, the two procedures had the same overall outcome: an increased NP apoptosis, an increased differentiation of NP into neurons and a simpler dendritic arbor for neurons. Over-expressing FMRP produced a more severe phenotype. These effects could be rescued by re-introducing FMRP into ko cells provided that the right dose was delivered: the rescue of NP number needed high doses of ΔFmr1 while the rescue of the dendritic arbor could only be seen with low doses (higher concentration of ΔFmr1 most likely resulted in an over-expression of FMRP). These results demonstrate that several developmental processes are exquisitely regulated by FMRP expression levels.
Authors stated in the discussion that previous time-lapse imaging analysis of tectal progenitors (Bestman et al 2012) allowed demonstrating that around 50% of NPCs in the optic tectum directly differentiate into neurons. This approach would be used here to evaluate the differentiation actions of FMRP and so provide a better demonstration that FMRP levels influence neuronal differentiation. In fact the conclusion raised in the present version of the manuscript is based on the analysis of ratios of cell progenitor vs. neuron numbers, what is convincing but indirect. Would these experiments be too much time consuming, I would accept instead a comment in the manuscript pondering the observation.
Figure 2D High fmr1 a MO: I wonder if this is a really representative figure of this condition. Other than a reduction on the number of positive cells, "red" cells are grouped on a border of the preparation rising concerns about the electroporation.
Experiments illustrated in Figure 7D-High MO condition- demonstrate a decrease of dendritic branch tip numbers but no change on branch density (unclear to me how this density is measured). Authors propose that this reflects a reduction of the length of dendritic branches. This later parameter must be measured and included in the figure.
Line 712. I don't agree with the statement: "These results demonstrate that FMRP regulates several processes.... And that these processes are differentially sensitive to changing levels of FMRP."
The overall effects of the knock-down and the OE are the same. "differentially sensitive" in the sentence suggests otherwise.
The discussion would benefit from an additional section dealing with the question "why two opposite procedures (decreasing and increasing FMRP level) have the same overall outcomes on NP survival, neuron differentiation and maturation?"
Author Response
The authors took the original approach to knock-down FMRP expression with morpholinos to study the effects on neurogenesis in the tadpole. This model has the merit to match the onset of the disease better than the constitutional ko because Fmr1 is silenced only after the 12.5 weeks of gestation in humans and because FMRP remain in embryonic stem cells. However, the knock-down of FMRP by morpholinos is not complete, but 60 % maximum. They monitored with time lapse two-photon imaging the rate of differentiation from neuroprogenitors (NP) to neurons, cell death and the neuron dendritic complexity over the course of 3 days. They also studied the effects induced by an overexpression of FMRP which was delivered through the electroporation of plasmids. The knock-down and the over expression were both tested at two doses. They found that knocking down or over-expressing FMRP had a dose-dependent effect on NP survival, on neuron differentiation and on dendritic growth. Interestingly, the two procedures had the same overall outcome: an increased NP apoptosis, an increased differentiation of NP into neurons and a simpler dendritic arbor for neurons. Over-expressing FMRP produced a more severe phenotype. These effects could be rescued by re-introducing FMRP into ko cells provided that the right dose was delivered: the rescue of NP number needed high doses of ΔFmr1 while the rescue of the dendritic arbor could only be seen with low doses (higher concentration of ΔFmr1 most likely resulted in an over-expression of FMRP). These results demonstrate that several developmental processes are exquisitely regulated by FMRP expression levels. Authors stated in the discussion that previous time-lapse imaging analysis of tectal progenitors (Bestman et al 2012) allowed demonstrating that around 50% of NPCs in the optic tectum directly differentiate into neurons. This approach would be used here to evaluate the differentiation actions of FMRP and so provide a better demonstration that FMRP levels influence neuronal differentiation. In fact the conclusion raised in the present version of the manuscript is based on the analysis of ratios of cell progenitor vs. neuron numbers, what is convincing but indirect. Would these experiments be too much time consuming, I would accept instead a comment in the manuscript pondering the observation.
Comment to editor: The frequent imaging protocol used in Bestman et al 2012 allowed us to observe that some radial glial progenitors differentiate directly into neurons, whereas other radial glial progenitors undergo classical symmetric or asymmetric mitoses, after which one or both progeny rapidly adopts neuronal morphology. Although this prior work provides the foundation for the work presented here, observations collected at daily intervals, as used in this study, capture the same neurogenic events as seen with the more frequent imaging intervals. We have added the following text in lines ##846-855, in response to the comment above:
“Previous in vivo time-lapse imaging of tectal progenitors at intervals of 2-19 hours over several days demonstrated that ∼ 50% of labeled NPCs differentiate directly into neurons without dividing, whereas other NPCs undergo classical symmetric or asymmetric divisions, after which one or both progeny rapidly adopts neuronal morphology (Bestman et al., 2012). Here, HIGH fmr1 MO and HIGH FMRP OE resulted in larger proportions of neurons and smaller proportions of NPCs compared to CMO, without increasing cell number over the 3 day imaging period. This suggests that the increased proportion of neurons did not result from NPC proliferation and differentiation, but may have occurred through direct differentiation of NPCs into neurons. Given our 24 hour time-lapse imaging interval, division followed by apoptosis is also supported by our data. “
It would require more than a year to repeat the studies with FMRP MO and OE using the experimental protocol and analysis presented in Bestman et al 2012. We hope that the revised text addressed the reviewers concerns.
Figure 2D High fmr1 a MO: I wonder if this is a really representative figure of this condition. Other than a reduction on the number of positive cells, "red" cells are grouped on a border of the preparation rising concerns about the electroporation.
Response: While conditions for electroporation are held constant between groups, we often find variability in the locations and numbers of electroporated cells. In the experiments presented in Figure 2, the numbers and locations of cells that are electroporated does not need to be equivalent from animal to animal. Instead, for the cells that are electroporated, the key parameters are (1) how many of them have detectable eGFP expression (quantified in Figure 2E) and (2) of those with detectable eGFP expression, how intense is that eGFP expression relative to tRFP (quantified in Figure 2F). With respect to these parameters, the images that are presented in Figure 2D are representative of the data quantified in Figures 2E-F. We mention in the text concerning Figure 2 that the fewer labeled cells in the HIGH fmr1a MO condition (Fig 2D, right panel) is likely due to cell death, which we document in Figure 5.
Experiments illustrated in Figure 7D-High MO condition- demonstrate a decrease of dendritic branch tip numbers but no change on branch density (unclear to me how this density is measured). Authors propose that this reflects a reduction of the length of dendritic branches. This later parameter must be measured and included in the figure.
Response: we now define branch density in the text Line 636), as the ratio of total dendritic branch tip number / total dendrite branch length. Both total dendritic branch tip number and total dendrite branch length were measured and presented in Figure 7B and C.
Line 712. I don't agree with the statement: "These results demonstrate that FMRP regulates several processes.... And that these processes are differentially sensitive to changing levels of FMRP." The overall effects of the knock-down and the OE are the same. "differentially sensitive" in the sentence suggests otherwise.
Response: Thanks for pointing out this confusion. We have changed the text (lines ##683-685) to state that the design of the experiment allowed us “to evaluate several distinct cellular events contributing to neurogenesis, including NPC proliferation and survival, the rate of differentiation of progenitors into neurons, as well as dendritic arbor elaboration in neurons”. Then, in lines 699-703, we state more clearly that “higher levels of FMRP knockdown affect some of the cellular events contributing to neurogenesis, such as NPC proliferation and neuronal differentiation, whereas lower levels of knockdown affect other cellular events, such as NPC cell death, suggesting that these processes are differentially sensitive to different levels of FMRP.” The comment refers to a separate observation, that knockdown and overexpression of FMRP have similar overall outcomes with respect to neurogenesis, as schematized in Figure 8. We address this comment, along with the comment below, in a new paragraph starting on line 730.
The discussion would benefit from an additional section dealing with the question "why two opposite procedures (decreasing and increasing FMRP level) have the same overall outcomes on NP survival, neuron differentiation and maturation?"
Response: We have added a new paragraph discussing this point, starting on line 730.
Other changes in the text shown in track changes were made to maintain the discussion at 3000 words despite the increased text required to respond to reviewers’ comments.
References
- Abitbol M, Menini C, Delezoide AL, Rhyner T, Vekemans M, Mallet J (1993) Nucleus basalis magnocellularis and hippocampus are the major sites of FMR-1 expression in the human fetal brain. Nat Genet 4:147–153. 10.1038/ng0693-147 [DOI] [PubMed] [Google Scholar]
- Ashley CT, Sutcliffe JS, Kunst CB, Leiner HA, Eichler EE, Nelson DL, Warren ST (1993) Human and murine FMR-1: alternative splicing and translational initiation downstream of the CGG-repeat. Nat Genet 4:244–251. 10.1038/ng0793-244 [DOI] [PubMed] [Google Scholar]
- Bestman JE, Lee-Osbourne J, Cline HT (2012) In vivo time-lapse imaging of cell proliferation and differentiation in the optic tectum of xenopus laevis tadpoles. J Comp Neurol 520:401–433. 10.1002/cne.22795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Dölen G, Bear MF (2012) The pathophysiology of fragile X (and what it teaches us about synapses). Annu Rev Neurosci 35:417–443. 10.1146/annurev-neuro-060909-153138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, McMillan E, Wallace K, Tubon TC Jr, Capowski EE, Svendsen CN (2008) Normal neurogenesis but abnormal gene expression in human fragile X cortical progenitor cells. Stem Cells Dev 17:107–117. 10.1089/scd.2007.0073 [DOI] [PubMed] [Google Scholar]
- Bolduc FV, Bell K, Cox H, Broadie KS, Tully T (2008) Excess protein synthesis in drosophila fragile X mutants impairs long-term memory. Nat Neurosci 11:1143–1145. 10.1038/nn.2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callan MA, Cabernard C, Heck J, Luois S, Doe CQ, Zarnescu DC (2010) Fragile X protein controls neural stem cell proliferation in the drosophila brain. Hum Mol Genet 19:3068–3079. 10.1093/hmg/ddq213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrén M, Tervonen T, Kärkkäinen V, Heinonen S, Castrén E, Larsson K, Bakker CE, Oostra BA, Akerman K (2005) Altered differentiation of neural stem cells in fragile X syndrome. Proc Natl Acad Sci U S A 102:17834–17839. 10.1073/pnas.0508995102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Corbin JG, Levy RJ (2013) Programmed cell death is impaired in the developing brain of FMR1 mutants. Dev Neurosci 35:347–358. 10.1159/000353248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT (1997) Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A 94:5401–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Martín A, Crespo M, Portera-Cailliau C (2010) Delayed stabilization of dendritic spines in fragile X mice. J Neurosci 30:7793–7803. 10.1523/JNEUROSCI.0577-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146:247–261. 10.1016/j.cell.2011.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL (1993) The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet 4:335–340. 10.1038/ng0893-335 [DOI] [PubMed] [Google Scholar]
- Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, Zhang SC, Abbeduto L, Bhattacharyya A (2014) iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev 23:1777–1787. 10.1089/scd.2014.0030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll CA, Broadie K (2014) Impaired activity-dependent neural circuit assembly and refinement in autism spectrum disorder genetic models. Front Cell Neurosci 8:30.[PMC] [ 10.3389/fncel.2014.00030 ] [] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eadie BD, Zhang WN, Boehme F, Gil-Mohapel J, Kainer L, Simpson JM, Christie BR (2009) Fmr1 knockout mice show reduced anxiety and alterations in neurogenesis that are specific to the ventral dentate gyrus. Neurobiol Dis 36:361–373. 10.1016/j.nbd.2009.08.001 [DOI] [PubMed] [Google Scholar]
- Eiges R, Urbach A, Malcov M, Frumkin T, Schwartz T, Amit A, Yaron Y, Eden A, Yanuka O, Benvenisty N, Ben-Yosef D (2007) Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 1:568–577. 10.1016/j.stem.2007.09.001 [DOI] [PubMed] [Google Scholar]
- Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG Jr, Warren ST, Oostra BA,Nelson DL, Caskey CT (1991) Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the sherman paradox. Cell 67:1047–1058. [DOI] [PubMed] [Google Scholar]
- Galvez R, Gopal AR, Greenough WT (2003) Somatosensory cortical barrel dendritic abnormalities in a mouse model of the fragile X mental retardation syndrome. Brain Res 971:83–89. 10.1016/S0006-8993(03)02363-1 [DOI] [PubMed] [Google Scholar]
- Gatto CL, Broadie K (2011) Fragile X mental retardation protein is required for programmed cell death and clearance of developmentally-transient peptidergic neurons. Dev Biol 356:291–307. 10.1016/j.ydbio.2011.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessert S, Bugner V, Tecza A, Pinker M, Kühl M (2010) FMR1/FXR1 and the miRNA pathway are required for eye and neural crest development. Dev Biol 341:222–235. 10.1016/j.ydbio.2010.02.031 [DOI] [PubMed] [Google Scholar]
- Ghiretti AE, Moore AR, Brenner RG, Chen LF, West AE, Lau NC, Van Hooser SD, Paradis S (2014) Rem2 is an activity-dependent negative regulator of dendritic complexity in vivo. J Neurosci 34:392–407. 10.1523/JNEUROSCI.1328-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Allan AM, Zong R, Zhang L, Johnson EB, Schaller EG, Murthy AC, Goggin SL, Eisch AJ, Oostra BA, Nelson DL, Jin P, Zhao X (2011) Ablation of fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat Med 17:559–565. 10.1038/nm.2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Murthy AC, Zhang L, Johnson EB, Schaller EG, Allan AM, Zhao X (2012) Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum Mol Genet 21:681–691. 10.1093/hmg/ddr501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He CX, Portera-Cailliau C (2013) The trouble with spines in fragile X syndrome: density, maturity and plasticity. Neuroscience 251:120–128. 10.1016/j.neuroscience.2012.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey SE, Walters-Sen L, Mosher TM, Pfau RB, Pyatt R, Snyder PJ, Sotos JF, Prior TW (2013) Duplication of the Xq27.3-q28 region, including the FMR1 gene, in an X-linked hypogonadism, gynecomastia, intellectual disability, short stature, and obesity syndrome. Am J Med Genet A 161A:2294–2299. 10.1002/ajmg.a.36034 [DOI] [PubMed] [Google Scholar]
- Hinds HL, Ashley CT, Sutcliffe JS, Nelson DL, Warren ST, Housman DE, Schalling M (1993) Tissue specific expression of FMR-1 provides evidence for a functional role in fragile X syndrome. Nat Genet 3:36–43. 10.1038/ng0193-36 [DOI] [PubMed] [Google Scholar]
- Hinton VJ, Brown WT, Wisniewski K, Rudelli RD (1991) Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet 41:289–294. 10.1002/ajmg.1320410306 [DOI] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT (2000) Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10:1038–1044. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT (2001) Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet 98:161–167. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Idupulapati M, Gilbert ME, Harris JB, Chakravarti AB, Rogers EJ, Crisostomo RA, Larsen BP, Mehta A, Alcantara CJ, Patel B, Swain RA, Weiler IJ, Oostra BA, Greenough WT (2002) Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet 111:140–146. 10.1002/ajmg.10500 [DOI] [PubMed] [Google Scholar]
- Jacobs S, Doering LC (2010) Astrocytes prevent abnormal neuronal development in the fragile x mouse. J Neurosci 30:4508–4514. 10.1523/JNEUROSCI.5027-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SJ, Seo JE, Yang SI, Choi JW, Wells D, Shin CY, Ko KH (2011) Cellular stress-induced up-regulation of FMRP promotes cell survival by modulating PI3K-akt phosphorylation cascades. J Biomed Sci 18:17[PMC] [ 10.1186/1423-0127-18-17 ] [] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SJ, Han SH, Yang SI, Choi JW, Kwon KJ, Park SH, Kim HY, Cheong JH, Ryu JH, Ko KH, Wells DG, Shin CY (2012) Positive feedback regulation of Akt-FMRP pathway protects neurons from cell death. J Neurochem 123:226–238. 10.1111/j.1471-4159.2012.07886.x [DOI] [PubMed] [Google Scholar]
- Koekkoek SK et al. (2005) Deletion of FMR1 in purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in fragile X syndrome. Neuron 47:339–352. 10.1016/j.neuron.2005.07.005 [DOI] [PubMed] [Google Scholar]
- Kumar S (2007) Caspase function in programmed cell death. Cell Death Differ 14:32–43. 10.1038/sj.cdd.4402060 [DOI] [PubMed] [Google Scholar]
- Lazarov O, Demars MP, Zhao Kda T, Ali HM, Grauzas V, Kney A, Larson J (2012) Impaired survival of neural progenitor cells in dentate gyrus of adult mice lacking fMRP. Hippocampus 22:1220–1224. 10.1002/hipo.20989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Li W, Xu K, Bogert BA, Su K, Gao FB (2003) Control of dendritic development by the drosophila fragile X-related gene involves the small GTPase Rac1. Development 130:5543–5552. 10.1242/dev.00792 [DOI] [PubMed] [Google Scholar]
- Li Y, Zhao X (2014) Concise review: fragile X proteins in stem cell maintenance and differentiation. Stem Cells 32:1724–1733. 10.1002/stem.1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightbody AA, Reiss AL (2009) Gene, brain, and behavior relationships in fragile X syndrome: evidence from neuroimaging studies. Dev Disabil Res Rev 15:343–352. 10.1002/ddrr.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JH, Luo T, Sargent TD, Fallon JR (2005) Developmental expression of xenopus fragile X mental retardation-1 gene. Int J Dev Biol 49:981–984. 10.1387/ijdb.052070jl [DOI] [PubMed] [Google Scholar]
- Liu W, Jiang F, Bi X, Zhang YQ (2012) Drosophila FMRP participates in the DNA damage response by regulating G2/M cell cycle checkpoint and apoptosis. Hum Mol Genet 21:4655–4668. 10.1093/hmg/dds307 [DOI] [PubMed] [Google Scholar]
- Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, Pfeiffer RL, Szulwach KE, Duan R, Barkho BZ, Li W, Liu C, Jin P, Zhao X (2010) Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet 6:e1000898[PMC] [ 10.1371/journal.pgen.1000898 ] [] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manitt C, Nikolakopoulou AM, Almario DR, Nguyen SA, Cohen-Cory S (2009) Netrin participates in the development of retinotectal synaptic connectivity by modulating axon arborization and synapse formation in the developing brain. J Neurosci 29:11065–11077. 10.1523/JNEUROSCI.0947-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamani SC, Erez A, Probst FJ, Bader P, Evans P, Baker LA, Fang P, Bertin T, Hixson P, Stankiewicz P, Nelson D, Patel A, Cheung SW (2012) Small genomic rearrangements involving FMR1 support the importance of its gene dosage for normal neurocognitive function. Neurogenetics 13:333–339. 10.1007/s10048-012-0340-y [DOI] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J (1956) Normal table of Xenopus laevis (Daudin); a systematical and chronological survey of the development from the fertilized egg till the end of metamorphosis. Amsterdam: North-Holland Pub. Co. [Google Scholar]
- Nimchinsky EA, Oberlander AM, Svoboda K (2001) Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci 21:5139–5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlé I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boué J, Bertheas MF, Mandel JL (1991) Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252:1097–1102. [DOI] [PubMed] [Google Scholar]
- Pacey LK, Doering LC (2007) Developmental expression of FMRP in the astrocyte lineage: implications for fragile X syndrome. Glia 55:1601–1609. 10.1002/glia.20573 [DOI] [PubMed] [Google Scholar]
- Rio M, Malan V, Boissel S, Toutain A, Royer G, Gobin S, Morichon-Delvallez N, Turleau C, Bonnefont JP, Munnich A, Vekemans M, Colleaux L (2010) Familial interstitial Xq27.3q28 duplication encompassing the FMR1 gene but not the MECP2 gene causes a new syndromic mental retardation condition. Eur J Hum Genet 18:285–290. 10.1038/ejhg.2009.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthazer ES, Li J, Cline HT (2006) Stabilization of axon branch dynamics by synaptic maturation. J Neurosci 26:3594–3603. 10.1523/JNEUROSCI.0069-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabaratnam M (2000) Pathological and neuropathological findings in two males with fragile-X syndrome. J Intellect Disabil Res 44 :81–85. 10.1046/j.1365-2788.2000.00261.x [DOI] [PubMed] [Google Scholar]
- Saffary R, Xie Z (2011) FMRP regulates the transition from radial glial cells to intermediate progenitor cells during neocortical development. J Neurosci 31:1427–1439. 10.1523/JNEUROSCI.4854-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro MR, Bray SM, Warren ST (2012) Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol 7:219–245. 10.1146/annurev-pathol-011811-132457 [DOI] [PubMed] [Google Scholar]
- Scotto-Lomassese S, Nissant A, Mota T, Néant-Féry M, Oostra BA, Greer CA, Lledo PM, Trembleau A, Caillé I (2011) Fragile X mental retardation protein regulates new neuron differentiation in the adult olfactory bulb. J Neurosci 31:2205–2215. 10.1523/JNEUROSCI.5514-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Cline HT (2010) Visual activity regulates neural progenitor cells in developing xenopus CNS through musashi1. Neuron 68:442–455. 10.1016/j.neuron.2010.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, Loring JF, Haggarty SJ (2011) Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One 6:e26203[PMC] [ 10.1371/journal.pone.0026203 ] [] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin WC, Haas K, Ruthazer ES, Cline HT (2002) Dendrite growth increased by visual activity requires NMDA receptor and rho GTPases. Nature 419:475–480. 10.1038/nature00987 [DOI] [PubMed] [Google Scholar]
- Telias M, Segal M, Ben-Yosef D (2013) Neural differentiation of fragile X human embryonic stem cells reveals abnormal patterns of development despite successful neurogenesis. Dev Biol 374:32–45. 10.1016/j.ydbio.2012.11.031 [DOI] [PubMed] [Google Scholar]
- Tervonen TA, Louhivuori V, Sun X, Hokkanen ME, Kratochwil CF, Zebryk P, Castrén E, Castrén ML (2009) Aberrant differentiation of glutamatergic cells in neocortex of mouse model for fragile X syndrome. Neurobiol Dis 33:250–259. 10.1016/j.nbd.2008.10.010 [DOI] [PubMed] [Google Scholar]
- Thomas CC, Combe CL, Dyar KA, Inglis FM (2008) Modest alterations in patterns of motor neuron dendrite morphology in the Fmr1 knockout mouse model for fragile X. Int J Dev Neurosci 26:805–811. 10.1016/j.ijdevneu.2008.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till SM, Wijetunge LS, Seidel VG, Harlow E, Wright AK, Bagni C, Contractor A, Gillingwater, TH Kind PC (2012) Altered maturation of the primary somatosensory cortex in a mouse model of fragile X syndrome. Hum Mol Genet 21:2143–2156. 10.1093/hmg/dds030 [DOI] [PubMed] [Google Scholar]
- Urbach A, Bar-Nur O, Daley GQ, Benvenisty N (2010) Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 6:407–411. 10.1016/j.stem.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van 't Padje S, Engels B, Blonden L, Severijnen LA, Verheijen F, Oostra BA, Willemsen R (2005) Characterisation of fmrp in zebrafish: evolutionary dynamics of the fmr1 gene. Dev Genes Evol 215:198–206. 10.1007/s00427-005-0466-0 [DOI] [PubMed] [Google Scholar]
- Vengoechea J, Parikh AS, Zhang S, Tassone F (2012) De novo microduplication of the FMR1 gene in a patient with developmental delay, epilepsy and hyperactivity. Eur J Hum Genet 20:1197–1200. 10.1038/ejhg.2012.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheij C, Bakker CE, de Graaff E, Keulemans J, Willemsen R, Verkerk AJ, Galjaard H, Reuser AJ, Hoogeveen AT, Oostra BA (1993) Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature 363:722–724. 10.1038/363722a0 [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, van Ommen GJ, Blonden LA, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Caskey CT, Nelson DL, Oostra BA, Warren ST (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65:905–914. [DOI] [PubMed] [Google Scholar]
- Wan L, Dockendorff TC, Jongens TA, Dreyfuss G (2000) Characterization of dFMR1, a drosophila melanogaster homolog of the fragile X mental retardation protein. Mol Cell Biol 20:8536–8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, Huber KM (2009) Protein translation in synaptic plasticity: mGluR-LTD, fragile X. Curr Opin Neurobiol 19:319–326. 10.1016/j.conb.2009.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijetunge LS, Chattarji S, Wyllie DJ, Kind PC (2013) Fragile X syndrome: from targets to treatments. Neuropharmacology 68:83–96. 10.1016/j.neuropharm.2012.11.028 [DOI] [PubMed] [Google Scholar]
- Willemsen R, Bontekoe CJ, Severijnen LA, Oostra BA (2002) Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum Genet 110:601–605. 10.1007/s00439-002-0723-5 [DOI] [PubMed] [Google Scholar]
- Zhang W, Cheng Y, Li Y, Chen Z, Jin P, Chen D (2014) A feed-forward mechanism involving drosophila fragile X mental retardation protein triggers a replication stress-induced DNA damage response. Hum Mol Genet 23:5188–5196. 10.1093/hmg/ddu241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Bear MF (2012) Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol 4:a009886. 10.1101/cshperspect.a009886 [DOI] [PMC free article] [PubMed] [Google Scholar]