

Abstract
Although SHIP is a well-established suppressor of IgE plus Ag-induced degranulation and cytokine production in bone marrow-derived mast cells (BMMCs), little is known about its role in connective tissue (CTMCs) or mucosal (MMCs) mast cells. In this study, we compared SHIP’s role in the development as well as the IgE plus Ag and TLR-induced activation of CTMCs, MMCs, and BMMCs and found that SHIP delays the maturation of all three mast cell subsets and, surprisingly, that it is a positive regulator of IgE-induced BMMC survival. We also found that SHIP represses IgE plus Ag-induced degranulation of all three mast cell subsets and that TLR agonists do not trigger their degranulation, whether SHIP is present or not, nor do they enhance IgE plus Ag-induced degranulation. In terms of cytokine production, we found that in MMCs and BMMCs, which are poor producers of TLR-induced cytokines, SHIP is a potent negative regulator of IgE plus Ag-induced IL-6 and TNF-α production. Surprisingly, however, in splenic or peritoneal derived CTMCs, which are poor producers of IgE plus Ag-induced cytokines, SHIP is a potent positive regulator of TLR-induced cytokine production. Lastly, cell signaling and cytokine production studies with and without LY294002, wortmannin, and PI3Kα inhibitor-2, as well as with PI3K p85α−/− BMMCs and CTMCs, are consistent with SHIP positively regulating TLR-induced cytokine production via an adaptor-mediated pathway while negatively regulating IgE plus Ag-induced cytokine production by repressing the PI3K pathway.
Mast cells (MCs), which play critical roles in both allergic inflammation and host defense against microbial infections, can be subclassified based on distribution and granule contents in mice into long-lived connective tissue MCs (CTMCs) and short-lived mucosal MCs (MMCs) (1, 2). In terms of distribution, CTMCs are located in sterile tissues such as the connective tissues of the skin and peritoneum as well as the submucosa of the gastrointestinal tract and the lung, whereas MMCs are present in nonsterile tissues, such as the mucosal linings of the gastrointestinal tract and the lungs. In terms of granule contents, CTMC granules contain serotonin and the proteoglycan, heparin (which is stained by safranin), whereas MMCs do not contain serotonin and possess the proteoglycan, chondrotin sulfate E, instead of heparin, and so do not stain with safranin (1, 3). As well, CTMC granules contain higher levels of histamine and different proteases than that present in MMC granules. Because it is difficult to get sufficient numbers of these CTMCs and MMCs for biochemical studies, most of what we know about MCs comes from experiments with in vitro derived bone marrow MCs (BMMCs), which are typically generated by culturing mouse bone marrow (BM) with IL-3. This culturing regimen results, after 4–6 wk, in a large number of uniform mature MCs, as assessed by the cell surface expression of FcεRI and c-kit. These cells stain with alcian blue, but not with safranin, and are thought to resemble immature MMCs (4, 5). Interestingly, though, not only does there appear to be considerable plasticity between CTMCs and MMCs (6, 7), but introduction of BMMCs into MC-deficient mice results in the generation of both CTMCs and MMCs, depending on the tissues where these BMMCs lodge (7, 8).
MCs play a central role in type I hypersensitivity reactions, which occur following multivalent allergen-induced cross-linking of MC-bound IgE, and this mechanism of MC activation has been the focus of MC research for many years. A great deal has been learned about the intracellular signaling pathways involved (6, 9–11). However, because MCs are located at the portals between self and nonself and because of the more recent discovery that they express pathogen recognition receptors, like TLRs, they are now thought to be one of the first cells to recognize invading micro-organisms and trigger an appropriate immune response. In keeping with this, MC-deficient mice have been shown to be far more susceptible to septic peritonitis and other bacterial infections than their MC-reconstituted counterparts or wild-type (WT) mice (12, 13).
SHIP (also known as SHIP1) is well known for its role as a master negative regulator of FcεR1-mediated BMMC activation (14, 15). This is attributed to SHIP’s ability to hydrolyze the 5′-phosphate of the PI3K-generated second messenger phosphatidylinositol-3,4,5-trisphosphate, making SHIP a negative regulator of the PI3K pathway. However, very little is known about its role in CTMCs or MMCs. We therefore compared SHIP’s role in the maturation, proliferation, and survival of CTMCs, MMCs, and BMMCs, as well as in IgE plus Ag-induced and TLR-induced activation of these MC subsets, and that is the focus of this study. Our findings are consistent with SHIP’s established role as a negative regulator of the PI3K pathway. In addition to that, however, our results using PI3K inhibitors and p85α−/− MCs also suggest that SHIP’s reported adaptor function(s) may be responsible for SHIP being a positive regulator of TLR-induced cytokine production.
Materials and Methods
Reagents
All cytokines and tissue culture media were purchased from StemCell Technologies (Vancouver, BC, Canada). LY294002 (LY) and wortmannin (W) were from Calbiochem (San Diego, CA), whereas the PI3Kα inhibitor-2 was from Cayman Chemical (Ann Arbor, MI). LPS serotype O127:B8 was from Sigma-Aldrich (St. Louis, MO). CpG, with the sequence 5′-TCCATGACGTTCCT-GACGTT-3′, was synthesized and HPLC purified by Invitrogen (Burlington, ON, Canada). The dsRNA used was a synthetic analog of polyinosine:cytosine from Sigma-Aldrich. Peptidoglycan (PGN) from Staphylococcus aureus was from Fluka (Buchs, Switzerland). IgE (anti-DNP, clone SPE-7) and DNP30–40-human serum albumin (HSA) were from Sigma-Aldrich.
Mice
SHIP+/+ and SHIP−/− F2 mice, on a mixed C57BL/6 × 129Sv background, and p85α+/+ and p85α−/− C57BL/6 mice were used between 6 and 12 wk of age and housed in the Animal Resource Centre of the British Columbia Cancer Research Centre under specific pathogen-free conditions and according to approved and ethical treatment of animal standards of the University of British Columbia. Animals were euthanized by CO2 as-phyxiation.
Generation of different MC subsets
BMMCs were generated, as described previously (16). To generate MMCs, mouse spleens were cut into small pieces and pressed through a 100-μm nylon cell strainer, and the resulting single-cell suspension was set up at 1 × 106 cells/ml in MMC medium (IMDM, 10% FCS, 10 ng/ml IL-3). After 7 d, nonadherent cells were kept between 3 × 105 and 1 × 106 cells/ml in MMC medium. To generate CTMCs, spleen cells were cultured at 1 × 106 cells/ml in CTMC medium (IMDM, 10% FCS, 50 ng/ml stem cell factor [SCF]) and, after 7 d, nonadherent cells were kept between 3 × 105 and 1 × 106 cells/ml in CTMC medium. Alternatively, IMDM was injected into the peritoneal cavity, and 1 × 106 nucleated cells/ml were plated in Opti-MEM I (Invitrogen), 10% FCS, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 4% conditioned medium from murine SCF-secreting Chinese hamster ovary cells (CHO-KL). After 24 h, nonadherent cells were discarded, and fresh culture medium was added to the adherent cells. After 3 d, nonadherent and adherent cells, detached with trypsin-EDTA, were resuspended at 3 × 105 cells/ml in fresh culture medium. This was repeated twice per week. The resulting peritoneal CTMCs (pCTMCs) were used for experiments at 3–9 wk of culture (17). BM-derived connective tissue-like MCs (CT-like BMMCs) were also derived, in a fashion similar to BMMCs (16), but with the addition of 25 ng/ml SCF plus 1 ng/ml IL-4 (18).
MC survival assay
For MC survival studies, MCs were washed, resuspended at 1.5 × 106 cells/ml in IMDM plus 0.1% BSA ± 10 μg/ml SPE-7 IgE, and 100 μl cells were seeded in a 96-well plate. Viable cell counts were determined by trypan blue exclusion.
MC degranulation assay
Degranulation assays were carried out, as previously described (18), in Tyrode’s buffer at 6.25 × 106 cells/ml. Cells were seeded at 10 μl/well in 96-well V-bottom plates, and degranulation was triggered with 10 μl 2× concentrated stimulus, prepared in Tyrode’s buffer. After 1 h at 37°C, cells were centrifuged for 5 min at 335 × g and 10 μl cell-free supernatant, and the 0.5% Triton X-100 lysed cell pellet was analyzed for β-hexosaminidase levels (19). Percentage of degranulation was calculated by dividing the absorbance of the supernatant by the sum of the absorbencies of the supernatant and cell lysate.
Stimulation of MCs for cytokine production
To stimulate with TLR agonists ± IgE, MCs were incubated in MC starvation medium (IMDM, 10% FCS) at 1 × 106 cells/ml for 18 h (for BMMCs and MMCs) or 3 h (CTMCs and pCTMCs) at 37°C and then washed and resuspended in MC starvation medium at 1 × 106 cells/ml. The cells (750 μl) were then seeded in 12-well flat-bottom plates. A total of 750 μl MC starvation medium ± IgE (for nonstimulated [N/S] samples) or TLR ligands ± SPE-7 IgE, in MC starvation medium at twice the indicated final concentration, was added to the MCs for 24 h at 37°C. To stimulate with TLR ligands plus IgE plus Ag, MCs were washed and incubated at 1 × 106 cells/ml in MC starvation medium plus 0.1 μg/ml SPE-7 for 18 h (for BMMCs and MMCs) or 3 h (CTMCs and pCTMCs) at 37°C. MCs were then washed and resuspended at 1 × 106 cells/ml with MC starvation medium plus 2 ng/ml DNP-HSA, and 750 μl cells were seeded in 12-well flat-bottom plates. TLR ligands were made up in MC starvation medium at twice the indicated final concentration, and 750 μl was added to the MCs. For N/S samples, 750 μl MC starvation medium was added. For samples containing PI3K inhibitors, it was important to keep the final DMSO concentration ≤0.05% because of differential effects of higher DMSO concentrations on SHIP+/+ and SHIP−/− MCs. MCs were stimulated for 24 h at 37°C. Tissue culture supernatants were assayed for the concentration of IL-12p40, IL-6, IL-10, TNF-α, IFN-γ, and IL-4 by ELISA, according to the manufacturer’s instructions (BD Biosciences, Mississauga, ON, Canada).
Stimulation of MCs for SDS-PAGE and Western analysis
CTMCs, starved for 3 h, and MMCs, or BMMCs starved overnight, at 37°C in IMDM plus 10% FCS were washed, resuspended in IMDM, and pre-treated or not for 20 min at 37°C with LY, W, or PI3K p110α inhibitor 2. The cells were then treated with LPS, IL-3, or IgE plus Ag for the indicated times, lysed with SDS sample buffer, boiled for 2 min, and loaded onto 10% polyacrylamide gels, as described previously (20). The following Abs were used for Western blotting: anti–phospho-IKKα/β (Ser176/180, catalog 2697), anti–phospho-p38 MAPK (Thr180/Tyr182, catalog 9211), anti–phospho-JNK (Thr183/Tyr185, catalog 9251), and anti–phospho-ERK1/2 (Thr202/Tyr204, catalog 9106) all from Cell Signaling Technology (Pickering, ON, Canada); anti–phospho-Akt (S473, catalog 44-621G) from Invitrogen (Burlington, ON, Canada); and anti–Grb-2 (catalog sc-255) from Santa Cruz Biotechnology (Santa Cruz, CA).
FACS
To assess maturity, MCs at 1 × 106 cells/ml in HBSS-modified medium containing 2% FCS (HF) were incubated for 30 min at 4°C with anti-CD16/32 (2.4G2) (StemCell Technologies) to block FcRs, and then incubated for 45 min in the dark with FcεR1α-FITC and c-kit allophycocyanin Abs, added at 1:1000 and 1:200, respectively. Then they were washed with HF and subjected to FACS at 5 × 105 cells/ml. To assess TLR4 and CD14 levels, unconjugated Abs to TLR4 (Santa Cruz Bio-technology, Santa Cruz, CA) and CD14 (Pharmingen, Mississauga, ON, Canada) were used in conjunction with anti-rat PE secondary Ab. Data were collected using a FACSCalibur flow cytometer using CellQuest Pro software, and data were analyzed using FlowJo software.
Cytospin and granule staining of MCs
MCs (5 × 104 cells) in 0.5 ml PBS were centrifuged onto a microscope slide at 500 rpm for 5 min in a Cytospin 3 centrifuge and stained for 15 min in 0.5% alcian blue/0.3% acetic acid solution, followed by 20 min in 0.1% safranin/0.1% acetic acid. Cells were air dried, and pictures were taken using an Axioplan 2 imaging microscope.
Analysis of TLR expression by RT-PCR
Total RNAwas prepared from MCs with TRIzol reagent, and genomic DNA contaminants were removed using the TURBO DNA-free kit (Ambion), according to each supplier’s recommendations. To RNA samples obtained from CTMCs, heparinase 1 (Sigma-Aldrich) was added according to the supplier’s recommendations. First-strand synthesis was performed using Moloney murine leukemia virus reverse transcription. The reactions were performed as per the manufacturer’s instructions, except the reactions were linearly upscaled to 25 μl. An oligo(dT)18 primer was used. PCRs were performed using the Phusion High-Fidelity DNA Polymerase kit. Reactions were performed in a 25 μl total volume consisting of 1 μl template, 16.5 μl PCR-H2O, 5 μl 5× Phusion HF buffer (containing 7.5 mM MgCl2), 0.75 μl DMSO, 0.25 μl Phusion High-Fidelity DNA Polymerase (2 U/μl), 0.5 μl 10 μM forward primer solution, 0.5 μl 10 μM reverse primer solution, and 0.5 μl dNTP (10 mM each) solution. PCRs with specific primers for GAPDH were carried out with an annealing temperature of 63°C and with the oligonucleotide primers 5′-TTAGCCCCCC-TGGCCAAGG-3′ and 5′-CTTACTCCTTGGAGGCCATG-3′, amplifying a 541-bp fragment. PCRs for mouse TLRs were performed with primers from Invivogen (version 07H09-SV) and carried out with an annealing temperature of 58°C. PCR amplifications were performed at 98°C for 75 s (initial denaturing step), followed by 35 cycles at 98°C for 15 s, 58°C or 63°C for 20 s, and 72°C for 30 s, followed by a final step at 72°C for 10 min. All reactions were run in a GeneAmp PCR System 9700 thermo cycler.
Results
SHIP is a negative regulator of MC maturation
To compare SHIP’s role in the maturation and proliferation of CTMCs and MMCs, SHIP+/+ and SHIP−/− mouse spleens were cultured in CTMC medium (containing SCF) or MMC medium (containing IL-3), respectively, as described previously (21). SHIP+/+ and SHIP−/− BM cultures were also initiated for comparison (16). As shown in Fig. 1A, SHIP−/− MCs matured faster than WT MCs, as assessed by cell surface expression of c-kit and FcεR1α, regardless of the subtype examined, and this was most pronounced with CTMCs and BMMCs. Whereas the SHIP−/− MMC maturation rate was only slightly faster than WT MMCs, it was consistently observed in five independent experiments. Of note, both SHIP+/+ and SHIP−/− CTMCs matured faster than MMCs or BMMCs, with >80% of SHIP+/+ and >95% of SHIP−/− spleen-derived cells becoming mature CTMCs by 3 wk of culture. By week 5, >90% of the cells in all cultures were mature MCs. Also of note, mature CTMCs were substantially smaller than MMCs or BMMCs, as assessed by flow cytometry, and the absence of SHIP resulted in slightly larger MMCs and BMMCs (Supplemental Fig. 1A).
FIGURE 1.

SHIP negatively regulates MC maturation, but positively regulates IgE-induced BMMC survival. SHIP+/+ and SHIP−/− CTMC, MMC, and BMMC cultures were initiated, as described in Materials and Methods, and (A) their maturity was assessed by surface expression of c-kit and FcεR1α. Cells that were doubly positive were considered mature. (B) Proliferation was assessed by counting trypan blue excluding cells. Shown is the absolute cell number divided by the starting number of cells at week 0. (A and B) SHIP +/+ MCs = ▲ with solid lines; SHIP−/− MCs = △ with dashed lines. (C) SHIP+/+ and SHIP−/− MC subsets were incubated in 0.1% BSA medium (SHIP+/+ = ▲; SHIP−/− = △, solid lines) or +5 μg/ml SPE-7 IgE (SHIP+/+ = #x025BC;; SHIP−/− = ▽, dashed lines), and viable cells were counted. Values are the mean ± SD from two independent experiments in triplicate. Significant differences (p < 0.05) are indicated (*).
Interestingly, SHIP’s influence on cell proliferation depended on the MC subtype (Fig. 1B). Whereas SHIP−/− CTMCs proliferated substantially faster than WT CTMCs, there was no significant difference in the growth of SHIP+/+ and SHIP−/− MMCs, and SHIP−/− BMMCs actually proliferated far slower than WT BMMCs. Of note, staining of the different SHIP+/+ and SHIP−/− mature MC subtypes with safranin, which stains heparin red (1, 3), and with alcian blue, which stains chondroitin sulfate blue (1, 3), confirmed that the derived CTMCs were indeed CTMCs, that the MMCs and BMMCs were mucosal-like MCs. These staining studies also showed that the absence of SHIP did not prevent the appearance of these granular components (Supplemental Fig. 1B). We also compared the cell surface expression of c-kit and FcεR1α on week 5 cultures of SHIP+/+ and SHIP−/− CTMCs, MMCs, and BMMCs by flow cytometry and found they all expressed similar levels of these two maturation markers (Supplemental Fig. 1C).
SHIP enhances IgE-induced BMMC survival
Because IgE, in the absence of Ag, has been shown to enhance BMMC survival [with some IgEs, e.g., SPE-7, being much more potent (cytokinergic) than others (22–24)], we asked whether IgE was also capable of enhancing the survival of MMCs and CTMCs when deprived of their growth factors. As shown in the left panel of Fig. 1C, IgE substantially enhanced the survival of CTMCs, with no difference being observed between SHIP+/+ and SHIP−/− MCs. The survival of SHIP+/+ and SHIP−/− MMCs, however, was only slightly increased with IgE (middle panel, Fig. 1C), although this was mainly because MMCs were substantially more capable of surviving in the absence of IgE than CTMCs. Once again, the presence of SHIP made no difference to survival, with or without IgE. Lastly, as shown in the right panel of Fig. 1C, SHIP+/+ and SHIP−/− BMMCs survived equally well, and the best of the three MC subtypes, in the absence of IgE. Interestingly, however, whereas IgE increased the survival of both SHIP+/+ and SHIP−/− BMMCs over the 4 d of the study, it was far more effective at promoting the proliferation/survival of SHIP+/+ BMMCs. Thus, surprisingly, SHIP appears to be a positive regulator of BMMC proliferation/survival in the presence of a highly cytokinergic IgE.
SHIP is a negative regulator of CTMC, MMC, and BMMC degranulation
To explore SHIP’s role following the activation of mature CTMCs and MMCs, we first asked whether SHIP, which is an established negative regulator of IgE plus Ag-induced BMMC degranulation (18), also negatively regulated CTMC and MMC degranulation. Specifically, we compared the ability of SHIP+/+ and SHIP−/− CTMCs, MMCs, and BMMCs to degranulate in response to IgE plus Ag by preloading the MCs with 0.1 μg/ml SPE-7 IgE and then stimulating with increasing concentrations of DNP-HSA. As shown in Fig. 2A, all subsets of SHIP−/− MCs degranulated significantly more than SHIP+/+ MCs at all Ag concentrations tested, establishing that SHIP negatively regulates IgE plus Ag-induced MMC and CTMC degranulation. However, the SHIP−/− BMMCs and MMCs were more sensitive to IgE plus Ag-induced degranulation than SHIP−/− CTMCs because they degranulated to a significant extent with as little as 1 ng/ml Ag.
FIGURE 2.

IgE plus Ag-induced degranulation of CTMCs, MMCs, and BMMCs is repressed by SHIP. (A) SHIP+/+ (black bars) and SHIP−/− (white bars) CTMCs, MMCs, and BMMCs were preloaded with 0.1 μg/ml IgE for 18 h at 37°C and exposed to the indicated concentration of Ag for 1 h at 37°C, and degranulation was assessed. (B) SHIP+/+ (black bars) and SHIP−/−(white bars) CTMCs, MMCs, and BMMCs were treated N/S, or exposed to 100 ng/ml LPS, 50 μg/ml dsRNA, 0.3 μM CpG, or 10 μg/ml PGN for 1 h, and degranulation was assessed. (C and D) MCs were preloaded with IgE, as in (A), and then either N/S or treated with TLR ligands [same concentrations as in (B)] plus 2 ng/ml Ag (B) or 0.2 ng/ml Ag (C) for 1 h. (E) SHIP+/+ (black bars) and SHIP−/−(white bars) BMMCs, MMCs, and CTMCs were treated with 10 μg/ml SPE-7 IgE or 400 ng/ml SCF for 1 h at 37°C, and degranulation was assessed. As well, as a positive control, SHIP+/+ (black bars) and SHIP−/−(white bars) CTMCs were preloaded with 0.1 μg/ml SPE-7 IgE for 18 h at 37°C and exposed to 20 ng/ml DNP-HSA for 1 h at 37°C, and degranulation was assessed. Data shown are the means ±SD from at least two independent experiments carried out in duplicate (A–C) or one experiment in triplicate (D) or duplicate (E). Similar results were obtained in at least two independent experiments.
TLR agonists neither trigger nor synergize with IgE plus Ag in triggering SHIP+/+ or SHIP−/− CTMC, MMC, or BMMC degranulation
Because MCs are among the first cells to recognize invading microorganisms, and they do so, in large part, via their TLRs, we asked whether TLR agonists triggered degranulation of any of the three MC subsets and, if so, whether SHIP influenced the extent of degranulation. Specifically, SHIP+/+ and SHIP−/− MMCs, CTMCs, and BMMCs were stimulated with concentrations of TLR agonists that gave maximal cytokine production from BMMCs in preliminary studies, that is, 100 ng/ml Escherichia coli LPS, 50 μg/ml dsRNA, 0.3 μM phosphorothioate-modified CpG-containing oligodeoxynucleotide (CpG), and 10 μg/ml PGN. However, there was no degranulation in any MC subset with these TLR agonists whether SHIP was present or not (Fig. 2B). This is consistent with other reports showing TLR agonists do not induce BMMC degranulation (25, 26), with the exception of PGN, which has been reported to induce BMMC degranulation under certain circumstances (27).
Because synergistic interactions between TLR agonists and IgE plus Ag have been reported for cytokine production from BMMCs (26, 28, 29), we next asked whether the same TLR agonists could augment IgE plus Ag-induced degranulation, that is, SHIP+/+ or SHIP−/− MMCs, CTMCs, and BMMCs were preloaded with 0.1 μg/ml SPE-7 IgE and then stimulated with 2 ng/ml Ag in the presence or absence of TLR ligands for 1 h. As shown in Fig. 2C, none of the TLR agonists increased degranulation. These studies were repeated with the SHIP−/− MMCs and BMMCs at a lower Ag concentration (0.2 ng/ml) because these cells maximally degranulated with 2 ng/ml Ag. However, the addition of TLR ligands once again did not increase degranulation (Fig. 2D), confirming that TLR stimulation did not affect IgE plus Ag-induced degranulation.
Because we showed in previous studies that SHIP−/− BMMCs, unlike WT BMMCs, degranulated in the presence of SCF (30) or following addition of highly cytokinergic IgEs [without subsequent Ag-induced cross-linking (18)], we asked whether this held true for SHIP−/− MMCs and CTMCs as well. As shown in Fig. 2E, SHIP−/− MMCs did indeed degranulate to some extent in the presence of SPE-7 IgE or with SCF, but SHIP−/− CTMCs did not, perhaps reflecting their lower responsiveness to IgE plus Ag-induced degranulation.
SHIP positively regulates TLR-induced, but negatively regulates IgE plus Ag-induced cytokine production from MCs
Activated MCs not only degranulate, releasing preformed mediators of inflammation, but also secrete de novo synthesized mediators, and this release can occur in the absence of degranulation (31, 32). Because TLR stimulation has been shown to be a major trigger of this de novo mediator release from BMMCs (13, 33), we compared the ability of SHIP+/+ and SHIP−/− CTMCs, MMCs, and BMMCs to secrete the cytokines IL-6, TNF-α, IL-12, and IL-10 in response to dsRNA, which binds TLR3 and acts through a MyD88-independent pathway; CpG, which binds TLR9 and signals via the MyD88-dependent pathway; and LPS, which binds TLR4 and stimulates both MyD88-dependent and independent pathways (34). As shown in Fig. 3, when stimulated with TLR agonists alone, WT CTMCs were by far the most robust producers of IL-6, TNF-α, IL-12, and IL-10. In marked contrast, SHIP−/− CTMCs produced little or no cytokines in response to these TLR agonists. These results suggest, quite surprisingly, that SHIP is a potent positive regulator of TLR-induced cytokine production from CTMCs. Both SHIP+/+ and SHIP−/− MMCs, in contrast, produced little to no cytokines in response to TLR stimulation. Lastly, BMMCs, like MMCs, were poor responders, but WT BMMCs were slightly better than their SHIP−/− counterparts at producing IL-6, again suggesting SHIP is a positive regulator of TLR-induced cytokine production, but that MMCs and BMMCs are far less responsive to TLR ligands than CTMCs. Consistent with this, TNF-α, IL-12, and IL-10 were not detectable from TLR-stimulated SHIP+/+ or SHIP−/− MMCs or BMMCs.
FIGURE 3.

TLR-activated WT CTMCs secrete the highest levels of cytokines. SHIP+/+ (black bars) and SHIP−/− (white bars) CTMCs, MMCs, and BMMCs were incubated for 24 h with buffer alone (N/S), 100 ng/ml LPS, 50 μg/ml dsRNA, or 0.3 μM CpG, and cell-free supernatants were assessed for IL-6, TNF-α, IL-12, and IL-10 levels by ELISA. Values shown are the mean ± SD from one representative experiment, measured in duplicate. Similar results were obtained in three independent experiments.
When MCs were stimulated for 24 h with TLR ligands in the presence of 0.1 μg/ml SPE-7 IgE, there was no change in the level of cytokine production from either SHIP+/+ or SHIP−/− CTMCs (compare the top panels of Fig. 4 with those of Fig. 3). Also, IgE alone (the N/S columns) was incapable of stimulating cytokine secretion from WT or SHIP−/− CTMCs. Interestingly, however, SHIP−/− MMCs and SHIP−/− BMMCs now produced higher levels of IL-6 and TNF-α than their WT counterparts (compare middle and bottom panels of Fig. 4 with those of Fig. 3), and IgE alone triggered a substantial amount of IL-6 and TNF-α from SHIP−/− MMCs and BMMCs. However, IgE plus TLR ligands, like TLR ligands alone, were incapable of triggering the secretion of detectable levels of IL-12 or IL-10 from these cells. These results suggest that SHIP is a negative regulator of IgE-induced cytokine secretion from MMCs and BMMCs, consistent with our previous studies showing that IgE stimulates more IL-6 from SHIP−/− than from WT BMMCs (35).
FIGURE 4.

IgE synergizes with TLR ligands to trigger TNF-α and IL-6 production from SHIP−/− MMCs and BMMCs, but not from CTMCs. SHIP+/+ (black bars) and SHIP−/− (white bars) CTMCs, MMCs, and BMMCs were treated for 24 h with 0.1 μg/ml SPE-7 IgE alone (N/S) or plus 100 ng/ml LPS, plus 50 μg/ml dsRNA, or plus 0.3 μM CpG, and cell-free supernatants were assessed for IL-6, TNF-α, IL-12, and IL-10 levels by ELISA. Values shown are the mean ± SD from one representative experiment, measured in duplicate. Similar results were obtained in two independent experiments.
When the different MC subsets were stimulated with TLR agonists in the presence of IgE plus Ag, there was a slight increase in cytokine production from WT CTMCs, but still no detectable production of cytokines from SHIP−/− CTMCs (top panels of Fig. 5). Interestingly, of the TLR ligands tested, only CpG was capable of triggering detectable levels of the anti-inflammatory cytokine, IL-10, from WT CTMCs, and IgE ±Ag had no effect on this level (compare Figs. 3–5). Importantly, there was a dramatic increase in the levels of IL-6 and TNF-α produced by SHIP−/− MMCs and BMMCs, even without TLR ligand stimulation (N/S indicates IgE plus Ag without TLR stimulation in Fig. 5), and the levels increased with the addition of TLR agonists. However, IgE plus Ag ± TLR ligands stimulated far less IL-6 and TNF-α from WT MMCs and WT BMMCs, consistent with SHIP being a potent negative regulator of IgE plus Ag-induced cytokine production. Interestingly, however, the absence of SHIP did not increase IL-10 or IL-12 secretion to detectable levels.
FIGURE 5.

IgE plus Ag dramatically synergizes with TLR ligands to trigger TNF-α and IL-6 production from SHIP−/− MMCs and BMMCs, but not from WT or SHIP−/− CTMCs. SHIP+/+ (black bars) and SHIP−/− (white bars) CTMCs, MMCs, and BMMCs were treated for 24 h with 0.1 μg/ml SPE-7 IgE plus 1 ng/ml Ag alone (N/S) or plus 100 ng/ml LPS, plus 50 μg/ml dsRNA, or plus 0.3 μM CpG, and cell-free supernatants were assessed for IL-6, TNF-α, IL-12, and IL-10 levels by ELISA. Values shown are the means ± SD from one representative experiment, measured in duplicate. Similar results were obtained in three independent experiments.
To confirm the unexpected finding that SHIP was a potent positive regulator of TLR-induced cytokines from CTMCs, we also derived CTMCs from SHIP+/+ and SHIP−/− peritoneal cavity cells (pCTMCs), according to Malbec et al. (17), and found, as shown in Fig. 6A, that LPS once again triggered far more IL-6 and TNF-α from these SHIP+/+ than SHIP−/− CTMCs.
FIGURE 6.

(A) WT peritoneal cell-derived CTMCs also secrete substantially more IL-6 and TNF-α than SHIP−/− cells in response to LPS. SHIP+/+ (black bars) and SHIP−/− (white bars) BMMCs and MMCs were stimulated with 0.1 μg/ml SPE-7 for 18 h at 37°C, washed, and resuspended in MC starvation medium (N/S) ± 2 ng/ml DNP-HSA. SHIP+/+ (black bars) and SHIP−/− (white bars) pCTMCs were stimulated ± 100 ng/ml LPS for 24 h at 37°C. Cell-free supernatants were assessed for IL-6 (left panel) and TNF-α (right panel) levels by ELISA. Values shown are the mean ± SD from one representative experiment, measured in duplicate. Similar results were obtained in three independent experiments. (B) TLR expression of SHIP+/+ and SHIP−/− CTMCs, MMCs, and BMMCs. For CTMCs, 200 ng cDNA was used for RT-PCR, whereas for MMCs and BMMCs 160 ng cDNA was used. As a loading control, 100 ng cDNA was used as template in a PCR with primers specific for GAPDH. Genomic DNA (10 ng) was used as a positive control. (C) SHIP+/+ (black bars) and SHIP−/− (white bars) CTMCs were incubated for 24 h with buffer alone (N/S) or with 10 μg/ml PGN, and cell-free supernatants were assessed for IL-6, TNF-α, IL-12, and IL-10 levels by ELISA. Values shown are the mean ± SD from one representative experiment, measured in duplicate. Similar results were obtained in three independent experiments.
SHIP influences TLR expression in MCs
To gain some insight into why TLR-stimulated WT CTMCs secreted far higher cytokine levels than either SHIP−/− CTMCs or SHIP+/+ and SHIP−/− MMCs or BMMCs, we first examined the expression levels of their TLRs. RT-PCR analysis of the SHIP+/+ and SHIP−/− CTMCs, MMCs, and BMMCs revealed that the levels of TLR3 and TLR9 in SHIP−/− CTMCs were actually higher than in their WT counterparts, whereas TLR4 levels were about the same in the presence or absence of SHIP (Fig. 6B). Because we found TLR2 expression was also far higher in SHIP−/− than SHIP+/+ CTMCs (Fig. 6B, top left panel), we compared cytokine production following PGN (a TLR2 agonist) stimulation of these cells and found that, even though WT CTMCs expressed far lower TLR2 levels, they secreted substantially higher levels of IL-6, TNF-α, IL-12, and IL-10 (Fig. 6C). Thus, because the SHIP+/+ CTMCs produced substantially higher levels of IL-6, TNF-α, IL-12, and IL-10, rather than lower levels, when TLR2, 3, 4, and 9 were stimulated, it was unlikely that this could be explained by differences in TLR expression. In contrast, the undetectable levels of TLR3 and TLR9 observed with SHIP+/+ and SHIP−/− MMCs and BMMCs were consistent with the relatively low response levels of these cells to dsRNA and CpG, respectively. The similar TLR expression pattern seen with MMCs and BMMCs reinforces the notion that these two MC subsets are closely related.
Because mRNA levels do not necessarily reflect protein levels and certainly not cell surface protein levels, we then assessed the cell surface levels of TLR4 and its coreceptor, CD14, on the different MC subsets. As shown in Supplemental Fig. 2, the levels were similar on all MC subsets. Thus, the different responses of the MC subsets to LPS were not due to different cell surface receptor levels.
IgE plus Ag or LPS stimulation of SHIP−/− MCs triggers higher PI3K pathway activity than in WT MCs
To gain some insight into why SHIP was a positive regulator of TLR-induced cytokine secretion but a negative regulator of IgE plus Ag-induced cytokine secretion, we carried out signaling studies with SHIP+/+ and SHIP−/− BMMCs. This was done not only because it was extremely difficult to obtain reproducible cell signaling results with CTMCs, but also to ensure that the same signaling pathways were available (i.e., same cellular context) to respond to LPS (which, as shown in the bottom panel of Fig. 3, triggers more IL-6 from WT BMMCs) and to IgE plus Ag (which, as shown in the N/S lanes of the bottom panel of Fig. 5, triggers more IL-6 from SHIP−/− BMMCs). As shown in Fig. 7A, activation of the PI3K pathway (i.e., phosphorylation of Akt) by either IgE plus Ag (left panel) or by LPS (right panel) was consistently higher in SHIP−/− than WT BMMCs. This suggested that the PI3K pathway was a positive regulator of IgE plus Ag-induced cytokines, consistent with earlier studies with BMMCs (11, 30, 36), and perhaps a negative regulator of TLR-induced cytokine secretion. Because the JNK and p38 pathways have been implicated in previous MC studies involving TLR-induced cytokines (26, 37, 38), we examined the activation of these pathways as well. Interestingly, whereas the phosphorylation of p38 may have been slightly more prolonged in SHIP−/− BMMCs (Fig. 7A), we were unable to detect any phospho-JNK in response to IgE plus Ag or LPS stimulation (data not shown). An examination of the phosphorylation of the third MAPK, ERK, suggested no difference with or without SHIP in response to IgE plus Ag, but perhaps more prolonged phosphorylation in the absence of SHIP following LPS stimulation (Fig. 7A). Because the NF-κB pathway has also been shown to play an important positive role in cytokine production from MCs (35), we also assessed the activation of its upstream activator, IKK, and found a similar IgE plus Ag-induced phosphorylation/activation of this Iκ kinase in the presence and absence of SHIP (Fig. 7A), but could not detect its phosphorylation in response to LPS (data not shown).
FIGURE 7.

SHIP positively regulates cytokine production from LPS-stimulated MCs via an adaptor pathway. (A) SHIP+/+ and SHIP−/− BMMCs were stimulated with IgE plus Ag or LPS for the indicated times, and SDS-lysed total cell lysates were subjected to Western analysis, as indicated. Short and long refer to exposure times of the x-ray film. (B) SHIP+/+ and SHIP−/− BMMCs were pretreated for 30 min with or without the indicated concentration of LY, W, or PI3Kα inhibitor-2, and then stimulated with IgE plus Ag (left panel) or LPS (right panel), and SDS-lysed total cell lysates were subjected to Western analysis, as indicated. The numbers under the IgE plus Ag- and LPS-induced pAkt blots in (B) refer to the levels of pAkt, relative to Grb2 (the loading control), determined via densitometry. Similar results were obtained in two other experiments. (C) SHIP+/+ (black bars) and SHIP−/− (white bars) BMMCs were stimulated with IgE plus Ag (left panel), or SHIP+/+ and SHIP−/− pCTMCs were stimulated with LPS (right panel) for 24 h with the indicated concentrations of LY, W, or PI3Kα inhibitor-2, and cell-free supernatants were assessed for IL-6 levels by ELISA. Values shown are the means ± SD from one representative experiment, measured in duplicate. *p < 0.05 compared with no inhibitor. Similar results were obtained in three independent experiments for (A and B) and five independent experiments for (C).
Reducing PI3K activity markedly reduces IgE plus Ag-induced, but does not increase LPS-induced cytokine production from MCs
Given that SHIP is a negative regulator of the PI3K pathway (39), our results to this point suggested that the PI3K pathway was a positive regulator of IgE plus Ag-induced, but a negative regulator of TLR-induced cytokine production from MCs. However, because SHIP can also act as an adaptor (40–43), we wanted to rule in or out SHIP adaptor effects on cytokine production. To test this, we first used the PI3K inhibitors LY and W and PI3Kα inhibitor-2 (because preliminary studies with different p110 isoform-specific inhibitors established that p110α inhibitors were the most potent at inhibiting IgE plus Ag-induced IL-6 production [data not shown]). We employed three different PI3K inhibitors rather than just one because all PI3K inhibitors possess some off-target effects. However, because these off-target effects tend to differ from one inhibitor to another (44–46), we felt we could be more confident that they were exerting their effects via inhibition of PI3K if they all elicited the same biological response. As shown in Fig. 7B, we found that low concentrations of these three PI3K inhibitors consistently reduced IgE plus Ag-induced pAkt in SHIP+/+ (left panel) and LPS-induced pAkt in SHIP−/− (right panel) BMMCs, but did not consistently reduce pAkt levels in IgE plus Ag-stimulated SHIP−/− or LPS-induced SHIP+/+ BMMCs, unless we went to higher concentrations of inhibitors (data not shown). We then tested these inhibitors on IgE plus Ag-induced and LPS-induced IL-6 production from SHIP+/+ and SHIP−/− BMMCs and CTMCs, respectively. Of note, LY, W, and PI3Kα inhibitor-2, at the concentrations used, had no deleterious effects on cell viability in these cells (data not shown). As shown in the left panel of Fig. 7C, these three inhibitors inhibited IgE plus Ag-induced IL-6 production from both SHIP+/+ and SHIP−/− BMMCs (and MMCs; data not shown), and these findings were consistent with the PI3K pathway being a positive regulator of IgE plus Ag-induced cytokine production from MCs. However, looking at their effects on LPS-induced cytokine production, none of the inhibitors increased IL-6 production above control, LPS-induced levels, and LY significantly reduced them (Fig. 7C, right panel). Importantly, we tested LY, W, and PI3Kα inhibitor-2 at various concentrations and for various times (from 3 to 24 h) for their effects on cytokine production and found that only W, at very high concentrations (above 100 nM, where nontarget effects are observed), stimulated IL-6 production significantly above control levels (data not shown). Importantly, significant stimulation above control IL-6 levels with high concentrations of W was not seen with LPS-stimulated SHIP−/− BMMCs. Because an increase in IL-6 production with low levels of PI3K inhibition would be expected, especially in SHIP−/− BMMCs, if the PI3K pathway was negatively regulating LPS-induced IL-6 production, we therefore tentatively concluded that SHIP positively regulates LPS-induced cytokine production from MCs primarily via an adaptor-mediated pathway.
To confirm this, we made numerous attempts to express WT and phosphatase-dead forms of SHIP in SHIP−/− CTMCs. However, although we could express these SHIP constructs in Phoenix Ampho 293T producer cell lines, we were unsuccessful in achieving detectable expression, using either lipofectamine or retroviral infection, in mature SHIP−/− CTMCs, BMMCs, or BM progenitors. While engaged in these studies, Ma et al. (47) reported that IL-3–stimulated BMMCs that were deficient in PI3K p85α were far less capable of activating Akt than WT BMMCs. We therefore investigated whether Akt was also less activated in response to LPS in these p85α−/− BMMCs. As shown in Fig. 8A, this was indeed the case. This then allowed us to test whether SHIP was promoting LPS-induced cytokine production by lowering PI3K/Akt activation or via its role as an adaptor using a genetic approach rather than via small molecule inhibitors, with their associated off-target effects. If SHIP was promoting LPS-induced IL-6 production by lowering PI3K/Akt activation, we would predict that the p85α−/− CTMCs and BMMCs would secrete more IL-6 than WT cells. As shown in Fig. 8B, however, this was not the case. In fact, the WT CTMCs and BMMCs secreted substantially more IL-6. This is consistent with the PI3K pathway being a positive regulator of LPS-induced IL-6 production, but that SHIP’s adaptor function(s) overrides its negative enzymatic effects on the PI3K pathway.
FIGURE 8.
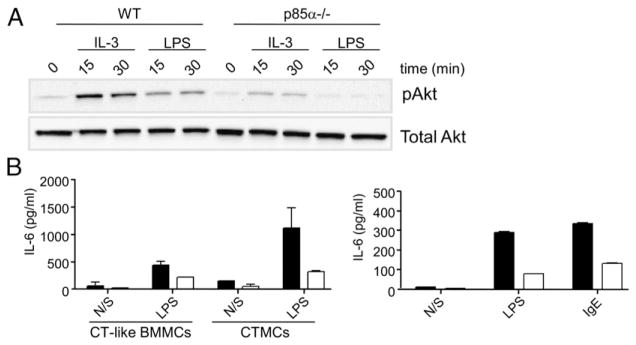
PI3K p85α−/− BMMCs and CTMCs secrete less IL-6 than their WT counterparts. (A) BMMCs from WT or p85α−/− mice were starved overnight in serum- and cytokine-free media and stimulated with IL-3 (10 ng/ml) or LPS (10 ng/ml) for 15 and 30 min. Equal amounts of protein lysates were subjected to Western blot analysis using an anti–phospho-Akt or total Akt Ab. Similar results were obtained in two independent experiments. (B) p85α+/+ (black bars) and p85α−/− (white bars) CT-like BMMCs and CTMCs were stimulated with or without 100 ng/ml LPS for 24 h (left panel), or p85α+/+ (black bars) and p85α−/− (white bars) BMMCs were stimulated with or without 100 ng/ml LPS or 5 μg/ml IgE (as a positive control) for 24 h (right panel), and cell-free supernatants were assessed for IL-6 levels by ELISA. Values shown are the means ± SD from one representative experiment, measured in duplicate. Similar results were obtained in two independent experiments with CT-like BMMCs, two with CTMCs and four with BMMCs.
Discussion
Although BMMCs have been used extensively as a surrogate for in vivo derived MCs, they are phenotypically distinct from either MMCs or CTMCs (48), and we have therefore compared, in this work, SHIP’s role in the development and activation of CTMCs, MMCs, and BMMCs. For the majority of our studies, CTMCs and MMCs were derived from SHIP+/+ and SHIP−/− spleen cells (21), because this resulted in robust, homogeneous populations of safranin-positive CTMCs and safranin-negative/alcian blue-positive MMCs. However, we also derived pCTMCs from SHIP+/+ and SHIP−/− peritoneal cells, as described by Malbec et al. (17), and obtained similar results to those obtained with spleen-derived CTMCs, both in degranulation (data not shown) and cytokine production studies. However, our degranulation results differed from those of Malbec et al. (17), who found that pCTMCs degranulated more robustly than BMMCs and that the absence of SHIP did not increase their degranulation. Of note, whereas we found that spleen cells yielded far more CTMCs upon in vitro culturing than BM cells, it was the cytokine mixture used during in vitro expansion that determined the type of mature MC obtained.
In terms of SHIP’s role in the maturation, proliferation, and survival of the three MC subsets, they all matured faster in the absence of SHIP, in keeping with our previous results with BMMCs (14), but major differences were observed in proliferation, with the absence of SHIP enhancing the proliferation of CTMCs, reducing that of BMMCs and having no effect on MMCs. We also found differences in IgE-induced survival, with IgE prolonging the survival of CTMCs and MMCs, but the presence of SHIP making no difference to this survival. The presence of SHIP in BMMCs, however, actually promoted their proliferation, whereas in its absence the BMMCs simply survived. This difference could be related to the far more rapid maturation of the SHIP−/− BMMCs, which could explain their subsequent reduced proliferation rate and, perhaps, reduced proliferation potential with IgE. However, an important caveat to keep in mind when comparing the proliferation and/or survival of SHIP+/+ and SHIP−/− BM cells is that SHIP−/− C57BL/6 mice rapidly become systemically inflamed, and this might affect hematopoietic precursor frequencies and cytokine exposure in vivo. Whereas we used young mice to try and keep these effects to a minimum, we cannot rule out that these in vivo conditions could affect the cell types that grow out in vitro.
Examining SHIP’s role in degranulation, we found that SHIP is a potent negative regulator of IgE plus Ag-induced degranulation of CTMCs and MMCs, consistent with our earlier studies with BMMCs (18). Importantly, TLR ligands alone did not induce degranulation, in keeping with previous reports (25, 26), nor did they enhance IgE plus Ag-induced degranulation from any MCs, even those lacking SHIP. This contrasts with one report claiming that PGN induces the degranulation of BMMCs (27), but this may be because they derived their BMMCs with 10% PWM-stimulated spleen cell-conditioned medium rather than IL-3 and/or because they derived their BMMCs from C3H/HeN rather than C57BL/6 mice.
In contrast to our degranulation results, we found substantial differences in cytokine production between CTMCs and MMCs/BMMCs. For example, we found much higher TLR-induced cytokine production from WT CTMCs than from WT BMMCs, consistent with a recent report showing higher cytokine production from TLR2-stimulated pCTMCs than from BMMCs (49). Also, although the responses were weak, we found that WT BMMCs produced higher levels of IL-6 in response to LPS and dsRNA than SHIP−/− BMMCs, suggesting that SHIP is a positive regulator of TLR stimulation in BMMCs as well as in CTMCs (Fig. 3). In contrast, IgE plus Ag stimulated far higher cytokine production from SHIP−/− MMCs/BMMCs than SHIP−/− CTMCs, and this low IgE plus Ag-induced cytokine production from SHIP−/− CTMCs, relative to SHIP−/− BMMCs, cannot be explained by a simple lack of response to IgE plus Ag because SHIP−/− CTMCs degranulate in response to this stimulus (Fig. 2A). Consistent with our results using IgE+/− TLR ligands (Fig. 4), highly cytokinergic IgE alone, in conjunction with TLR2 or TLR4 stimulation, has been reported to increase inflammatory cytokines from BMMCs (50). As well, TLR2 and TLR4 agonists have been reported to synergize with IgE plus Ag in triggering cytokine secretion from BMMCs (26, 28, 51). In contrast, however, TLR2 activation has also been reported to reduce both IgE plus Ag-induced BMMC degranulation and TNF-α production (52).
To explore why WT MMCs and BMMCs responded so poorly to TLR stimulation and so strongly, in the absence of SHIP, to IgE plus Ag, whereas WT CTMCs responded so strongly to TLR ligands and so weakly, in the absence of SHIP, to IgE plus Ag, we first compared TLR mRNA levels by RT-PCR. We found the expression pattern was more limited in WT MMCs and BMMCs (i.e., TLR1, 2, 4, 6) than in WT CTMCs (TLR1, 2, 4, 6, 8, 9) and that this is similar, in general, to that reported previously (25, 32, 53). Interestingly, we found that the absence of SHIP resulted in higher expression of TLR1, 2, 3, 7, 8, and 9 in CTMCs. One caveat, of course, with RT-PCR studies is that they may not reflect cell surface protein levels, and this certainly appeared to be the case for TLR9 because RT-PCR studies suggested no TLR9 expression in BMMCs or MMCs, whereas we found that CpG significantly increased IgE plus Ag-induced IL-6 and TNF-α production from these MCs (Fig. 5). Because of this, we honed in on LPS-stimulated events because RT-PCR studies suggested similar TLR4 expression levels and subsequent flow cytometric analyses confirmed equal cell surface expression of TLR4 and its co-receptor, CD14, on SHIP+/+ and SHIP−/− MCs (Supplemental Fig. 2).
Because our subsequent cell signaling studies suggested that the PI3K pathway was more activated in the absence of SHIP when stimulated with either LPS or IgE plus Ag, we examined the effects of PI3K inhibition and found that LY, W, and PI3Kα inhibitor-2 reduced IgE plus Ag-induced IL-6 production from SHIP+/+ and SHIP−/− BMMCs/MMCs, consistent with the PI3K pathway being a positive regulator of IgE plus Ag-induced cytokine production. However, we found that even though SHIP was a positive regulator of LPS-induced IL-6 production, PI3K inhibition by three different PI3K inhibitors did not increase the levels of this cytokine. The only exception to this finding was when very high levels of W were employed and, even then, elevated IL-6 was only seen with WT CTMCs. To avoid the off-target effects of PI3K inhibitors, we also compared WT and PI3K p85α−/− BMMCs and CTMCs and obtained results consistent with our small molecule PI3K inhibitor results, that is, that SHIP may be acting as a positive regulator via its adaptor function(s). Our results are consistent with very nice studies showing that W does not inhibit LPS-induced IL-6 production from WT BMMCs (54) and that SHIP promotes TLR2- and TLR4-induced cytokine production from murine macrophages (55). However, in the latter study, Keck et al. (55) concluded, based on results obtained with high doses of W, that PI3K inhibition increased LPS-induced cytokine production in macrophages. Our results also contrast with those of Luyendyk et al. (56), who carried out some very elegant genetic studies showing that the PI3K/Akt pathway negatively regulates LPS-induced TNF-α and IL-6 production in macrophages. However, other studies in macrophages suggest that PI3K plays a positive role during LPS-induced cytokine production. For example, Kuo et al. (57) found that transfecting RAW264.7 cells with a class I PI3K p110 dominant-negative mutant inhibited LPS-induced TNF-α secretion. Our data with p85α−/− CTMCs suggest that, at least in MCs, PI3K appears to be playing a positive role. We cannot say at this time whether this is a difference between MCs and macrophages or not. Of note, whereas our data are consistent with SHIP acting as a positive regulator of TLR-induced cytokines via its adaptor function(s), we cannot rule out that it may still be acting as a positive regulator via its enzymatic activity by hydrolyzing another substrate (e.g., inositol tetraphosphate) that inhibits TLR agonist-induced cytokine production.
The differences we have observed between CTMCs and MMCs/BMMCs are in keeping with several previous studies showing significant differences between these MC subsets, including differences in TLR expression (49), in responsiveness to IL-18 (which induces BMMCs/MMCs, but not CTMCs to secrete the chemokine, Ccll, to recruit Th2 cells) (58), in expression of STAT4 (in CTMCs) and STAT6 (in MMCs/BMMCs) (59) and in TLR-induced cytokines and chemokines (higher with CTMCs) (25). Differences have also been reported in IgE plus Ag-induced survival, with survival being more pronounced with CTMCs (60), and this may explain why IgE is a more potent survival enhancer of CTMCs than MMCs (Fig. 1C).
In terms of in vivo significance, our finding that TLR agonists trigger far higher levels of inflammatory cytokines from WT CTMCs than from WT MMCs makes sense because CTMCs are in sterile sites of the body and should be more vigilant toward microbes than MMCs, which are in “dirty” environments. As far as MMCs are concerned, our work, coupled with recent studies by Haddon et al. (61) showing that it is the absence of SHIP in MCs that is responsible for both the increased IL-6 and TNF-α serum levels and the chronic lung inflammation that is characteristic of SHIP−/− mice, suggests that it is the high levels of IL-6 and TNF-α specifically produced by IgE ± Ag-induced SHIP−/− MMCs that most likely drive this chronic lung inflammation (62–64), by attracting and activating other immune cells (27, 65, 66). As well, our finding that TLR stimulation augments IgE plus Ag-induced TNF-α and IL-6 production from MMCs (Fig. 5) might explain the exacerbation of IgE-mediated allergic episodes by infectious agents (26) and suggests that activating SHIP specifically in MMCs might be useful for treating chronic inflammatory diseases like asthma.
Supplementary Material
Acknowledgments
This work was supported by the Terry Fox Foundation and the Canadian Cancer Society, with core support from the British Columbia Cancer Foundation and the British Columbia Cancer Agency. F.A. was supported in this work by Michael Smith Foundation for Health Research and Natural Sciences & Engineering Research Council studentships.
We thank Drs. Odile Malbec and Marc Daeron for SCF-secreting Chinese hamster ovary (CHO-KL) cells and Christine Kelly for preparing the manuscript.
Abbreviations used in this article
- BM
bone marrow
- BMMC
BM-derived mast cell
- CT-like BMMC
BM-derived connective tissue-like MC
- CTMC
connective tissue mast cell
- HF
HBSS-modified medium containing 2% FCS
- HSA
human serum albumin
- LY
LY294002
- MC
mast cell
- MMC
mucosal MC
- N/S
nonstimulated
- pCTMC
peritoneal CTMC
- PGN
peptidoglycan
- SCF
stem cell factor
- W
wortmannin
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures
G.K. is a founding member of Aquinox Pharmaceuticals, which is dedicated to identifying small molecule activators and inhibitors of SHIP.
References
- 1.Welle M. Development, significance, and heterogeneity of mast cells with particular regard to the mast cell-specific proteases chymase and tryptase. J Leukoc Biol. 1997;61:233–245. doi: 10.1002/jlb.61.3.233. [DOI] [PubMed] [Google Scholar]
- 2.Gurish MF, Boyce JA. Mast cells: ontogeny, homing, and recruitment of a unique innate effector cell. J Allergy Clin Immunol. 2006;117:1285–1291. doi: 10.1016/j.jaci.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 3.Beil WJ, Schulz M, McEuen AR, Buckley MG, Walls AF. Number, fixation properties, dye-binding and protease expression of duodenal mast cells: comparisons between healthy subjects and patients with gastritis or Crohn’s disease. Histochem J. 1997;29:759–773. doi: 10.1023/a:1026421303260. [DOI] [PubMed] [Google Scholar]
- 4.Razin E, Stevens RL, Akiyama F, Schmid K, Austen KF. Culture from mouse bone marrow of a subclass of mast cells possessing a distinct chondroitin sulfate proteoglycan with glycosaminoglycans rich in N-ace-tylgalactosamine-4,6-disulfate. J Biol Chem. 1982;257:7229–7236. [PubMed] [Google Scholar]
- 5.Razin E, Ihle JN, Seldin D, Mencia-Huerta JM, Katz HR, LeBlanc PA, Hein A, Caulfield JP, Austen KF, Stevens RL. Interleukin 3: a differentiation and growth factor for the mouse mast cell that contains chondroitin sulfate E proteoglycan. J Immunol. 1984;132:1479–1486. [PubMed] [Google Scholar]
- 6.Maltby S, Khazaie K, McNagny KM. Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochim Biophys Acta. 2009;1796:19–26. doi: 10.1016/j.bbcan.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sonoda S, Sonoda T, Nakano T, Kanayama Y, Kanakura Y, Asai H, Yonezawa T, Kitamura Y. Development of mucosal mast cells after injection of a single connective tissue-type mast cell in the stomach mucosa of genetically mast cell-deficient W/Wv mice. J Immunol. 1986;137:1319–1322. [PubMed] [Google Scholar]
- 8.Otsu K, Nakano T, Kanakura Y, Asai H, Katz HR, Austen KF, Stevens RL, Galli SJ, Kitamura Y. Phenotypic changes of bone marrow-derived mast cells after intraperitoneal transfer into W/Wv mice that are genetically deficient in mast cells. J Exp Med. 1987;165:615–627. doi: 10.1084/jem.165.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metcalfe DD, Peavy RD, Gilfillan AM. Mechanisms of mast cell signaling in anaphylaxis. J Allergy Clin Immunol. 2009;124:639–646. doi: 10.1016/j.jaci.2009.08.035. quiz 647–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryan JJ, Fernando JF. Mast cell modulation of the immune response. Curr Allergy Asthma Rep. 2009;9:353–359. doi: 10.1007/s11882-009-0052-z. [DOI] [PubMed] [Google Scholar]
- 11.Gilfillan AM, Rivera J. The tyrosine kinase network regulating mast cell activation. Immunol Rev. 2009;228:149–169. doi: 10.1111/j.1600-065X.2008.00742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Echtenacher B, Männel DN, Hültner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. 1996;381:75–77. doi: 10.1038/381075a0. [DOI] [PubMed] [Google Scholar]
- 13.Metz M, Maurer M. Mast cells: key effector cells in immune responses. Trends Immunol. 2007;28:234–241. doi: 10.1016/j.it.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Rauh MJ, Kalesnikoff J, Hughes M, Sly L, Lam V, Krystal G. Role of Src homology 2-containing-inositol 5′-phosphatase (SHIP) in mast cells and macrophages. Biochem Soc Trans. 2003;31:286–291. doi: 10.1042/bst0310286. [DOI] [PubMed] [Google Scholar]
- 15.Antignano F, Ruschmann J, Hamilton M, Ho V, Lam V, Kuroda E, Sly LM, Krystal G. The Src homology 2 containing inositol 5′ phosphatases. In: Bradsaw RA, Dennis EA, editors. Handbook of Cell Signalling. Elsevier; San Diego, CA: 2009. pp. 1065–1084. [Google Scholar]
- 16.Sly LM, Kalesnikoff J, Lam V, Wong D, Song C, Omeis S, Chan K, Lee CWK, Siraganian RP, Rivera J, Krystal G. IgE-induced mast cell survival requires the prolonged generation of reactive oxygen species. J Immunol. 2008;181:3850–3860. doi: 10.4049/jimmunol.181.6.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malbec O, Roget K, Schiffer C, Iannascoli B, Dumas AR, Arock M, Daëron M. Peritoneal cell-derived mast cells: an in vitro model of mature serosal-type mouse mast cells. J Immunol. 2007;178:6465–6475. doi: 10.4049/jimmunol.178.10.6465. [DOI] [PubMed] [Google Scholar]
- 18.Huber M, Helgason CD, Damen JE, Liu L, Humphries RK, Krystal G. The src homology 2-containing inositol phosphatase (SHIP) is the gatekeeper of mast cell degranulation. Proc Natl Acad Sci USA. 1998;95:11330–11335. doi: 10.1073/pnas.95.19.11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishizumi H, Yamamoto T. Impaired tyrosine phosphorylation and Ca2+ mobilization, but not degranulation, in lyn-deficient bone marrow-derived mast cells. J Immunol. 1997;158:2350–2355. [PubMed] [Google Scholar]
- 20.Damen JE, Wakao H, Miyajima A, Krosl J, Humphries RK, Cutler RL, Krystal G. Tyrosine 343 in the erythropoietin receptor positively regulates erythropoietin-induced cell proliferation and Stat5 activation. EMBO J. 1995;14:5557–5568. doi: 10.1002/j.1460-2075.1995.tb00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kataoka TR, Komazawa N, Morii E, Oboki K, Nakano T. Involvement of connective tissue-type mast cells in Th1 immune responses via Stat4 expression. Blood. 2005;105:1016–1020. doi: 10.1182/blood-2004-07-2811. [DOI] [PubMed] [Google Scholar]
- 22.Kalesnikoff J, Huber M, Lam V, Damen JE, Zhang J, Siraganian RP, Krystal G. Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity. 2001;14:801–811. doi: 10.1016/s1074-7613(01)00159-5. [DOI] [PubMed] [Google Scholar]
- 23.Asai K, Kitaura J, Kawakami Y, Yamagata N, Tsai M, Carbone DP, Liu FT, Galli SJ, Kawakami T. Regulation of mast cell survival by IgE. Immunity. 2001;14:791–800. doi: 10.1016/s1074-7613(01)00157-1. [DOI] [PubMed] [Google Scholar]
- 24.Kitaura J, Song J, Tsai M, Asai K, Maeda-Yamamoto M, Mocsai A, Kawakami Y, Liu FT, Lowell CA, Barisas BG, et al. Evidence that IgE molecules mediate a spectrum of effects on mast cell survival and activation via aggregation of the FcepsilonRI. Proc Natl Acad Sci USA. 2003;100:12911–12916. doi: 10.1073/pnas.1735525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsushima H, Yamada N, Matsue H, Shimada S. TLR3-, TLR7-, and TLR9-mediated production of proinflammatory cytokines and chemokines from murine connective tissue type skin-derived mast cells but not from bone marrow-derived mast cells. J Immunol. 2004;173:531–541. doi: 10.4049/jimmunol.173.1.531. [DOI] [PubMed] [Google Scholar]
- 26.Qiao H, Andrade MV, Lisboa FA, Morgan K, Beaven MA. FcepsilonR1 and Toll-like receptors mediate synergistic signals to markedly augment production of inflammatory cytokines in murine mast cells. Blood. 2006;107:610–618. doi: 10.1182/blood-2005-06-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Supajatura V, Ushio H, Nakao A, Akira S, Okumura K, Ra C, Ogawa H. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J Clin Invest. 2002;109:1351–1359. doi: 10.1172/JCI14704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fehrenbach K, Port F, Grochowy G, Kalis C, Bessler W, Galanos C, Krystal G, Freudenberg M, Huber M. Stimulation of mast cells via FcεR1 and TLR2: the type of ligand determines the outcome. Mol Immunol. 2007;44:2087–2094. doi: 10.1016/j.molimm.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 29.Nigo YI, Yamashita M, Hirahara K, Shinnakasu R, Inami M, Kimura M, Hasegawa A, Kohno Y, Nakayama T. Regulation of allergic airway inflammation through Toll-like receptor 4-mediated modification of mast cell function. Proc Natl Acad Sci USA. 2006;103:2286–2291. doi: 10.1073/pnas.0510685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huber M, Helgason CD, Scheid MP, Duronio V, Humphries RK, Krystal G. Targeted disruption of SHIP leads to Steel factor-induced degranulation of mast cells. EMBO J. 1998;17:7311–7319. doi: 10.1093/emboj/17.24.7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metz M, Grimbaldeston MA, Nakae S, Piliponsky AM, Tsai M, Galli SJ. Mast cells in the promotion and limitation of chronic inflammation. Immunol Rev. 2007;217:304–328. doi: 10.1111/j.1600-065X.2007.00520.x. [DOI] [PubMed] [Google Scholar]
- 32.Stelekati E, Orinska Z, Bulfone-Paus S. Mast cells in allergy: innate instructors of adaptive responses. Immunobiology. 2007;212:505–519. doi: 10.1016/j.imbio.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 33.Nakano N, Nishiyama C, Kanada S, Niwa Y, Shimokawa N, Ushio H, Nishiyama M, Okumura K, Ogawa H. Involvement of mast cells in IL-12/23 p40 production is essential for survival from polymicrobial infections. Blood. 2007;109:4846–4855. doi: 10.1182/blood-2006-09-045641. [DOI] [PubMed] [Google Scholar]
- 34.Carty M, Bowie AG. Recent insights into the role of Toll-like receptors in viral infection. Clin Exp Immunol. 2010;161:397–406. doi: 10.1111/j.1365-2249.2010.04196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalesnikoff J, Baur N, Leitges M, Hughes MR, Damen JE, Huber M, Krystal G. SHIP negatively regulates IgE + antigen-induced IL-6 production in mast cells by inhibiting NF-κB activity. J Immunol. 2002;168:4737–4746. doi: 10.4049/jimmunol.168.9.4737. [DOI] [PubMed] [Google Scholar]
- 36.Poderycki M, Tomimori Y, Ando T, Xiao W, Maeda-Yamamoto M, Sauer K, Kawakami Y, Kawakami T. A minor catalytic activity of Src family kinases is sufficient for maximal activation of mast cells via the high-affinity IgE receptor. J Immunol. 2010;184:84–93. doi: 10.4049/jimmunol.0901590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zorn CN, Keck S, Hendriks RW, Leitges M, Freudenberg MA, Huber M. Bruton’s tyrosine kinase is dispensable for the Toll-like receptor-mediated activation of mast cells. Cell Signal. 2009;21:79–86. doi: 10.1016/j.cellsig.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 38.Yang YJ, Chen W, Carrigan SO, Chen WM, Roth K, Akiyama T, Inoue J, Marshall JS, Berman JN, Lin TJ. TRAF6 specifically contributes to FcepsilonRI-mediated cytokine production but not mast cell degranulation. J Biol Chem. 2008;283:32110–32118. doi: 10.1074/jbc.M802610200. [DOI] [PubMed] [Google Scholar]
- 39.Hamilton MJ, Ho VW, Kuroda E, Ruschmann J, Antignano F, Lam V, Krystal G. Role of SHIP in cancer. Exp Hematol. 2011;39:2–13. doi: 10.1016/j.exphem.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 40.Koncz G, Tóth GK, Bökönyi G, Kéri G, Pecht I, Medgyesi D, Gergely J, Sármay G. Co-clustering of Fcgamma and B cell receptors induces dephosphorylation of the Grb2-associated binder 1 docking protein. Eur J Biochem. 2001;268:3898–3906. doi: 10.1046/j.1432-1327.2001.02295.x. [DOI] [PubMed] [Google Scholar]
- 41.Song M, Kim MJ, Ha S, Park JB, Ryu SH, Suh PG. Inositol 5′-phosphatase, SHIP1 interacts with phospholipase C-γ1 and modulates EGF-induced PLC activity. Exp Mol Med. 2005;37:161–168. doi: 10.1038/emm.2005.22. [DOI] [PubMed] [Google Scholar]
- 42.Ott VL, Tamir I, Niki M, Pandolfi PP, Cambier JC. Downstream of kinase, p62(dok), is a mediator of FcγIIB inhibition of FcεRI signaling. J Immunol. 2002;168:4430–4439. doi: 10.4049/jimmunol.168.9.4430. [DOI] [PubMed] [Google Scholar]
- 43.Isnardi I, Lesourne R, Bruhns P, Fridman WH, Cambier JC, Daëron M. Two distinct tyrosine-based motifs enable the inhibitory receptor FcgammaRIIB to cooperatively recruit the inositol phosphatases SHIP1/2 and the adapters Grb2/Grap. J Biol Chem. 2004;279:51931–51938. doi: 10.1074/jbc.M410261200. [DOI] [PubMed] [Google Scholar]
- 44.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, et al. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hazeki K, Nigorikawa K, Hazeki O. Role of phosphoinositide 3-kinase in innate immunity. Biol Pharm Bull. 2007;30:1617–1623. doi: 10.1248/bpb.30.1617. [DOI] [PubMed] [Google Scholar]
- 46.Hayakawa M, Kawaguchi K, Kaizawa H, Koizumi T, Ohishi T, Yamano M, Okada M, Ohta M, Tsukamoto S, Raynaud FI, et al. Synthesis and biological evaluation of sulfonylhydrazone-substituted imidazo[1,2-a]pyridines as novel PI3 kinase p110α inhibitors. Bioorg Med Chem. 2007;15:5837–5844. doi: 10.1016/j.bmc.2007.05.070. [DOI] [PubMed] [Google Scholar]
- 47.Ma P, Mali RS, Munugalavadla V, Krishnan S, Ramdas B, Sims E, Martin H, Ghosh J, Li S, Chan RJ, et al. The PI3K pathway drives the maturation of mast cells via microphthalmia transcription factor. Blood. 2011;118:3459–3469. doi: 10.1182/blood-2011-04-351809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakano T, Sonoda T, Hayashi C, Yamatodani A, Kanayama Y, Yamamura T, Asai H, Yonezawa T, Kitamura Y, Galli SJ. Fate of bone marrow-derived cultured mast cells after intracutaneous, intraperitoneal, and intravenous transfer into genetically mast cell-deficient W/Wv mice: evidence that cultured mast cells can give rise to both connective tissue type and mucosal mast cells. J Exp Med. 1985;162:1025–1043. doi: 10.1084/jem.162.3.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mrabet-Dahbi S, Metz M, Dudeck A, Zuberbier T, Maurer M. Murine mast cells secrete a unique profile of cytokines and prostaglandins in response to distinct TLR2 ligands. Exp Dermatol. 2009;18:437–444. doi: 10.1111/j.1600-0625.2009.00878.x. [DOI] [PubMed] [Google Scholar]
- 50.Takenaka H, Ushio H, Niyonsaba F, Jayawardana ST, Hajime S, Ikeda S, Ogawa H, Okumura K. Synergistic augmentation of inflammatory cytokine productions from murine mast cells by monomeric IgE and Toll-like receptor ligands. Biochem Biophys Res Commun. 2010;391:471–476. doi: 10.1016/j.bbrc.2009.11.082. [DOI] [PubMed] [Google Scholar]
- 51.Inami M, Inokuchi K, Yamaguchi H, Nakayama K, Watanabe A, Uchida N, Tanosaki S, Dan K. Oral administration of imatinib to P230 BCR/ABL-expressing transgenic mice changes clones with high BCR/ABL complementary DNA expression into those with low expression. Int J Hematol. 2006;84:346–353. doi: 10.1532/IJH97.05186. [DOI] [PubMed] [Google Scholar]
- 52.Kasakura K, Takahashi K, Aizawa T, Hosono A, Kaminogawa S. A TLR2 ligand suppresses allergic inflammatory reactions by acting directly on mast cells. Int Arch Allergy Immunol. 2009;150:359–369. doi: 10.1159/000226237. [DOI] [PubMed] [Google Scholar]
- 53.Supajatura V, Ushio H, Nakao A, Okumura K, Ra C, Ogawa H. Protective roles of mast cells against enterobacterial infection are mediated by Toll-like receptor 4. J Immunol. 2001;167:2250–2256. doi: 10.4049/jimmunol.167.4.2250. [DOI] [PubMed] [Google Scholar]
- 54.Chiba N, Masuda A, Yoshikai Y, Matsuguchi T. Ceramide inhibits LPS-induced production of IL-5, IL-10, and IL-13 from mast cells. J Cell Physiol. 2007;213:126–136. doi: 10.1002/jcp.21101. [DOI] [PubMed] [Google Scholar]
- 55.Keck S, Freudenberg M, Huber M. Activation of murine macrophages via TLR2 and TLR4 is negatively regulated by a Lyn/PI3K module and promoted by SHIP1. J Immunol. 2010;184:5809–5818. doi: 10.4049/jimmunol.0901423. [DOI] [PubMed] [Google Scholar]
- 56.Luyendyk JP, Schabbauer GA, Tencati M, Holscher T, Pawlinski R, Mackman N. Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J Immunol. 2008;180:4218–4226. doi: 10.4049/jimmunol.180.6.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuo CC, Lin WT, Liang CM, Liang SM. Class I and III phosphatidylinositol 3′-kinase play distinct roles in TLR signaling pathway. J Immunol. 2006;176:5943–5949. doi: 10.4049/jimmunol.176.10.5943. [DOI] [PubMed] [Google Scholar]
- 58.Wiener Z, Pocza P, Racz M, Nagy G, Tolgyesi G, Molnar V, Jaeger J, Buzas E, Gorbe E, Papp Z, et al. IL-18 induces a marked gene expression profile change and increased Ccl1 (I-309) production in mouse mucosal mast cell homologs. Int Immunol. 2008;20:1565–1573. doi: 10.1093/intimm/dxn115. [DOI] [PubMed] [Google Scholar]
- 59.Kataoka TR, Nishizawa Y. Stat4 suppresses the proliferation of connective tissue-type mast cells. Lab Invest. 2008;88:856–864. doi: 10.1038/labinvest.2008.51. [DOI] [PubMed] [Google Scholar]
- 60.Ekoff M, Strasser A, Nilsson G. FcepsilonRI aggregation promotes survival of connective tissue-like mast cells but not mucosal-like mast cells. J Immunol. 2007;178:4177–4183. doi: 10.4049/jimmunol.178.7.4177. [DOI] [PubMed] [Google Scholar]
- 61.Haddon DJ, Antignano F, Hughes MR, Blanchet MR, Zbytnuik L, Krystal G, McNagny KM. SHIP1 is a repressor of mast cell hyperplasia, cytokine production, and allergic inflammation in vivo. J Immunol. 2009;183:228–236. doi: 10.4049/jimmunol.0900427. [DOI] [PubMed] [Google Scholar]
- 62.Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM, Borowski A, Jirik F, Krystal G, Humphries RK. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V, Huxham L, Minchinton AI, Mui A, Krystal G. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23:361–374. doi: 10.1016/j.immuni.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 64.Oh SY, Zheng T, Bailey ML, Barber DL, Schroeder JT, Kim YK, Zhu Z. Src homology 2 domain-containing inositol 5-phosphatase 1 deficiency leads to a spontaneous allergic inflammation in the murine lung. J Allergy Clin Immunol. 2007;119:123–131. doi: 10.1016/j.jaci.2006.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Malaviya R, Ikeda T, Ross E, Abraham SN. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-α. Nature. 1996;381:77–80. doi: 10.1038/381077a0. [DOI] [PubMed] [Google Scholar]
- 66.Sutherland RE, Olsen JS, McKinstry A, Villalta SA, Wolters PJ. Mast cell IL-6 improves survival from Klebsiella pneumonia and sepsis by enhancing neutrophil killing. J Immunol. 2008;181:5598–5605. doi: 10.4049/jimmunol.181.8.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.