

Abstract
Previous studies have shown that (i) Dal81p and Dal82p are required for allophanate-induced gene expression in Saccharomyces cerevisiae; (ii) the cis-acting element mediating the induced transcriptional response to allophanate is a dodecanucleotide, UISALL; and (iii) Dal82p binds specifically to UISALL. Here we show that Dal82p is localized to the nucleus and parallels movement of the DNA through the cell cycle. Deletion analysis of DAL82 identified and localized three functional domains. Electrophoretic mobility shift assays identified a peptide (consisting of Dal82p amino acids 1– 85) that is sufficient to bind a DNA fragment containing UISALL. LexA-tethering experiments demonstrated that Dal82p is capable of mediating transcriptional activation. The activation domain consists of two parts: (i) an absolutely required core region (amino acids 66 –99) and (ii) less well defined regions flanking residues 66 –99 that are required for full wild-type levels of activation. The Dal82p C terminus contains a predicted coiled-coil motif that down-regulates Dal82p-mediated transcriptional activation.
The Saccharomyces cerevisiae allantoin degradative pathway genes can be classified according to one of three hierarchical control mechanisms. The first and least complex of these mechanisms is exemplified by the allantoin permease gene (DAL5), which is subject only to nitrogen catabolite repression (NCR)1 (1-5). NCR is the physiological process through which the expression of genes encoding proteins needed for the transport and catabolism of poor nitrogen sources is maintained at low levels when a good nitrogen source is available (6, 7). This process is the preeminent and most well characterized mode of regulation for nitrogen catabolic genes (8, 9). NCR is implemented by the regulation of Gln3p and Gat1p, the transcriptional activators responsible for the expression of NCR-sensitive genes (5, 10-18). Both Gln3p and Gat1p are predicted to contain C4 zinc-finger motifs and are members of the GATA family of DNA-binding proteins, and Gln3p binds to GATAcontaining UASNTR sequences situated upstream of NCR-sensitive genes (12, 16, 19-21). Similar binding experiments have not so far been performed with Gat1p due to its toxicity in Escherichia coli.
The second control mechanism is exemplified by the DAL3 gene, whose expression, like that of DAL5, is NCR-sensitive and dependent upon Gln3p (19, 20). In addition, however, this gene is down-regulated by Dal80p, i.e. DAL3 expression increases Δ20-fold when DAL80 is deleted (16, 20). Dal80p and its homologue, Deh1p, are also GATA family DNA-binding proteins that bind to the GATA sequences of the genes whose expression they regulate and that contain leucine zipper motifs at their C termini (20-23). The Dal80p-binding site differs from that of Gln3p in that Dal80p binding requires two specifically oriented GATA sequences, whereas for Gln3p binding, only one GATA sequence is necessary (16, 24). The demonstration that Gln3p and Dal80p are capable of binding to the same DAL3 GATA sequences led to the proposal that Dal80p regulates Gln3p- and Gat1p-dependent gene expression by competing with these transcriptional activators for binding to GATA sequences in the promoters of Dal80p-regulated genes (20, 25). Consistent with this proposal, we have shown that the level of nitrogen catabolic gene expression and NCR sensitivity is related to the amounts of Gat1p and Dal80p produced in the cell.2
The third and most complex allantoin pathway control mechanism is exemplified by DAL7, encoding malate synthase (26-28). DAL7 expression is NCR-sensitive; Gln3p- and Gat1p-dependent, like that of DAL5; and Dal80p-regulated, like that of DAL3 (17, 26). Superimposed on these two forms of regulation is a third layer of control, which occurs in response to the allantoin pathway inducer, allophanate, or its analogue, oxalurate (26, 27). Inducer-responsive DAL7 expression depends upon a cis-acting element, the allophanate-responsive up-stream induction sequence, UISALL (26). One or more copies of this dodecanucleotide element are found in the promoters of all allophanate-responsive genes, both in the allantoin pathway as well as in others such as CAR2 (29). Saturation mutagenesis of the 3' most DAL7 UISALL element demonstrated the sequence of UISALL to be 5'-(G/C)AAA(A/T)NTGCG(T/C)T (30). A single copy of this element, although sufficient for inducer responsiveness, will not alone support heterologous gene expression; multiple copies will, however, support very low level expression (26).
Two transcription factors are required for the UISALL-mediated gene expression, Dal81p/DurMp and Dal82p/DurLp/ Uga35p (31, 32). DAL81 and DAL82 were first identified genetically as loci required for growth with allantoin pathway nitrogen sources (31, 33). Null mutations in either of the loci result in loss of allophanate-dependent transcription (34, 35). Mutations in dal81 or dal82 also result in a significant decrease in basal level expression of the genes they regulate (35-37).
Initially, the best candidate to function as the UISALL-binding protein was the 109-kDa Dal81p because it possesses a Cys6(Zn+2)2 binuclear cluster motif (36). Enthusiasm for this candidacy quickly diminished, however, when Dal81p was found to function beyond the bounds of the allantoin pathway, e.g. inducer-dependent expression of the γ-aminobutyrate In addition, deletion of the Dal81p zinc-finger motif does not affect allophanate-responsive gene expression (36). These results focused our attention on the much smaller 29-kDa Dal82p as the UISALL-binding transcription factor. Using electrophoretic mobility shift assays (EMSAs), Dorrington and Cooper (38) demonstrated that Dal82p specifically binds to UISALL elements from all of the allophanate/oxalurate-responsive allantoin pathway genes. It has also recently been shown to bind to the UISALL element upstream of CAR2 (29).
Experiments using cis-acting elements cloned into heterologous expression vector assay systems demonstrated that both Dal82p and Dal81p are required for the UIS element to mediate inducer-dependent transcription (34, 35). However, Dal82p possesses capabilities that depend upon neither inducer nor genes (32, 36). However, Dal82p possesses capabilities that depend upon neither inducer nor Dal81p. For example, if a UISALL element is placed adjacent to a mutated GATA sequence, it will efficiently suppress the phenotype of that cis-acting mutation (30). Suppression requires only a functional UISALL element and Dal82p, not inducer or Dal81p (30). In another situation, UISALL and Dal82p act together with Rap1p to constitute a strong upstream activation sequence in the CAR2 promoter (29). Again, neither Dal81p nor allophanate/oxalurate are required for this function. The pertinence of these two examples is their highlighting that Dal82p can bind to DNA and perform some functions in the absence of inducer or Dal81p.
Although much is known about the participation of Dal82p in transcription, little is known about how the functions of this small protein are carried out. Unfortunately, homology analyses of the deduced Dal82p amino acid sequence have not provided insights or hints in this regard. The only clearly recognizable motif is a predicted coiled-coil at the C terminus of Dal82p. DNA-binding motifs similar to those reported for other transcription factors are not apparent in Dal82p. In fact, the transcriptional activation ability of Dal82p has not been tested. The objective of this work has been to identify the component functions of Dal82p and the portions of the protein required to fulfill them.
MATERIALS AND METHODS
Strains and Media
The strains used in this work are listed in Table I. The media were standard formulations with the pertinent details described below and in the figure legends.
Table I.
S. cerevisiae and E. coli strains used in this work
|
S. cerevisiae
strains |
|
| GYC86 | MATa his3 leu2 trp1 ura3/Matα his3 leu2 trp1 ura3 |
| EGY48 | MATα 3lexAop::LEU2 ura3 trp1 his3 |
| TCY31 | MATa lys5 ura3 trp1Δ::hisG |
| STCY32 | MATa lys5 ura3 trp1Δ::hisG his3Δ::hisG |
| W303–1a | MATa leu2–3 trp1–1 can1–100 ura3–l ade2 |
| PB100 |
MATa leu2–3 trp1–1 canl–100 ura3–l
ade2dal81Δ::HIS3 |
| SS200 | MATa lys5 ura3 trp1Δ::hisG dal82::hisG |
| SS400 | MATa lys5 ura3 dal82Δ::TRP1 |
|
E. coli
strain |
|
| BL21(DE3) | F− ompT hsdSB (rB− rB+) dcm gal(DE3) plyS Cmr |
Construction of GAL1,10-GFP pNVS2
To construct GAL1,10-GFP pNVS2, the NdeI site in pRS316 (39) was blunt-ended to yield pAB52. A GAL1,10 XbaI-XhoI PCR fragment (containing promoter and ATG) was cloned into pAB52 to yield pAB53. Primers were 5'-GCTCTAGACCTTCTCTTTGGAACTTTCAGTAA-3' and 5'-CCGGCCCTCGAGCATTATAGTTTTTTCTCC-3', and the template was pEGKG (40). PCR-generated GFP (5'-ATGCTCGAGGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTCCCC-3' and 5'-GGGGTACCATCGATAAGCTTCTG-CAGGTCGACGGATCCCCCGGGGAATTCATTCAGTCATATGCTTG-TACAGCTTGTCCATGCCGAGAGTGATCCCGGA-3' primers and pJ22 as template (41)) was cloned into pAB53. A clone that fluoresced in a galactose-dependent manner, pNVS2, was isolated, verified, and used.
GFP-DAL82 Plasmids
The EcoRI-SalI fragment from pSS82BTM (N-terminal segment of Dal82p) was cloned into pNVS2. A pMO8 (34) SalI-HindIII fragment (remainder of Dal82p) was cloned into the resulting plasmid to yield pNVS82. HIS3-based ADH1-GFP p423 was constructed by replacing the XbaI-XhoI fragment (encoding the GAL1,10 promoter) from pNVS2 with an ADH1 promoter XbaI-XhoI PCR fragment (5'-GCGTCTAGAAAGAAATGATGGTAAATGAAATAG-G-3' and 5'-GCGCTCGAGCATAGTTGATTGTATGCTTGGTATAGCTT-3' primers and pEG202 (45, 46) as template). The SacI-EcoRI fragment from the resulting plasmid was then cloned into pRS423 (43) to yield p423. Next, the EcoRI-ClaI fragment from pNVS82 was cloned into p423, and finally, the 0.5-kilobase pair EcoRI fragment from pNVS82 was cloned into the resulting plasmid to yield ADH1-GFPDAL82 pSS423-82. To construct positive control pSS20 for dal82 complementation (urea amidolyase) assays, the pMO8 (34) EagI-ClaI fragment (full-length DAL82 and its promoter) was cloned into YCp50.
dal82 Mutant Strains
The DAL82 EagI-HindIII fragment (from pMO8) was cloned into pBluescript KS+ (Stratagene). The BamHIEcoRI fragment from this plasmid was replaced with the TRP1,Knr BamHI-EcoRI fragment from pJA52 (42). Strain TCY31 was transformed with the resulting plasmid (Asp718Iand ClaI-digested). A Trp+ transformant (SS400) was isolated and verified by Southern analysis. A dal82 disruption (SS200) was constructed by inserting the HisG:URA3: HisG pNKY51 fragment (44) into the DAL82 BamHI site of pSS20. Strain SS200 was 5-FOA-selected after TCY31 cells were transformed with the linearized plasmid (Asp718I and SphI) and verified by Southern analysis.
dal82 Deletion Plasmids
DAL82 deletions were constructed by cloning WT DAL82 into pSelect (Promega). For each deletion, oligonucleotides were prepared such that they encompassed 20 base pairs preceding and following the designated deletion. After annealing and transformation into E. coli, several colonies were screened by sequence analysis. Clones containing the desired deletions were used to replace WT sequences in pRD41 (Fig. 1) (23). These plasmids (Fig. 2) were used to prepare E. coli extracts for EMSAs. pRD4 contains the NdeI-BamHI fragment (9-amino acid influenza hemagglutinin (HA) epitope) and Factor Xa recognition sequence fused to Dal82p amino acids 1– 6; pRD41 contains full-length DAL82 in place of amino acids 1– 6 (Fig. 1) (38). pRD403 carries the same HA-DAL82 as pRD41, except that two NdeI sites (5' of the AUG start codon of the HA epitope and 3' of DAL82) were destroyed, leaving one NdeI site within DAL82. pRD42, pRD43, and pRD44 were created from pRD41 by deleting the StyI-HindIII, XhoI-HindIII, and EcoRI-HindIII fragments, respectively (Fig. 1); sequence extensions were filled in and religated. pRD46 was constructed by replacing the WT BamHI-EcoRI fragment from pRD403 with a PCR product (pRD41 template and primers RD21 and RD22) (Table II). pRD47 and pRD48 were similarly constructed with PCR primers T7-7/ RD24 and T7-7/RD26, respectively. pRD476 is pRD403 with 1) the HindIII site at the 3'-end of DAL82 destroyed; 2) a G-to-T substitution in codon L78 (CTG to CTT), creating a unique HindIII site; 3) a G-to-A substitution (G116-R116), creating a unique StuI site; and 4) deletion of residues 81–114. pRD477, pRD478, pRD479, pRD480, pRD481, and pRD482 were constructed by cloning oligonucleotides RD39, RD42, RD38, RD41, RD40, and RD37 (Table II), respectively, into the HindIII-StuI site of pRD476. For pRD488 and pRD489, the WT BamHI-NdeI fragment from pRD403 was replaced with oligonucleotides RD57 and RD58 (Table II). pRD402 is a pRD403 derivative in which the StyI-HindIII fragment is deleted; sequence extensions were filled in and relegated. For pRD490, the WT NdeI-SalI fragment was replaced with oligonucleotide RD59.
FIG. 1. Plasmids used in this work: GFP pNVS2, GFP-DAL82 (inset below pNVS82), HA-DAL82 pRD41 (used to construct dal82.ii in EMSAs), and lexA-DAL82 activation measurements.

ORI, origin; Kb, kilobase pairs; ADH prom., alcohol dehydrogenase promoter; EGFP, enhanced GFP.
FIG. 2. Summary of DNA binding results.

Shown are maps of deletion plasmids derived from pRD41.
Table II.
Oligonucleotide primers and DNA fragments used for dal82Δ constructs
| Primer | Sequence |
|---|---|
| T7–7 | 5'–GGG AGA CCA CAA CGG TTT CCC–3′ (T7 promoter) |
| RD21 | 5′–GGG CAT TGT GTG AAG AGT AGG–3′ |
| RD22 | 5′–G GGG GAT CCC TGG CAA AGG GTG CTC GAT GAG–3′ |
| RD24 | 5′–G GGG AAT TCG CCC CGG ATG ACT TTT AAT–3′ |
| RD25 | 5′–G GGG AAT TCC TCC CAT CAG CTT CAA CTG–3′ |
| RD37 |
AG CTT CAT CCA AGA CCA AGA ACT AAA TTC GGA AAT GAA TCA TCT TCA TCC A GTA GGT TCT GGT TCT TGA TTT AAG CCT TTA CTT AGT AGA AGT AGG TTA TCA TCA TCT TCT TTC ATT AAA AGT CAT CCG GGG AAT AGT AGT AGA AGA AAG TAA TTT TCA GTA GGC CCC |
| RD38 |
AG CTT TTC GGA AAT GAA TCA TCT TCA TCC TTA TCA TCA TCT TCT TTC ATT AAA A AAG CCT TTA CTT AGT AGA AGT AGG AAT AGT AGT AGA AGA AAG TAA TTT AGT CAT CCG GGG TCA GTA GGC CCC |
| RD39 |
AG CTT ATG GGA GCA CTC TTG GAC GAA GCA CCA GAA CAT CCA AGA CCA AGA ACT A TAC CCT CGT GAG AAC CTC CTT CGT GGT CTT GTA GGT TCT GGT TCT TGA AAA TTC GGA AAT GAA TCA TCT TCA TCC TTA TCA TCA TCT TCT TTT AAG CCT TTA CTT AGT AGA AGT AGG AAT AGT AGT AGA AGA |
| RD40 |
AG CTT ATG GGA GCA CTC TTG GAC GAA GCA CCA GAA CAT CCA AGA CCA AGA ACT A TAC CCT CGT GAG AAC CTC CTT CGT GGT CTT GTA GGT TCT GGT TCT TGA AAA TTC GGA AAT GAA ATT AAA AGT CAT CCG GGG TTT AAG CCT TTA CTT TAA TTT TCA GTA GGC CCC |
| RD41 |
AG CTT ATG GGA GCA CTC TTG GAC GAA GCA CCA GAA TTC GGA AAT GAA TCA TCT A TAC CCT CGT GAG AAC CTC CTT CGT GGT CTT AAG CCT TTA CTT AGT AGA TCA TCC TTA TCA TCA TCT TCT TTC ATT AAA AGT CAT CCG GGG AGT AGG AAT AGT AGT AGA AGA AAG TAA TTT TCA GTA GGC CCC |
| RD42 | AG CTT ATG GGA GCA CTC TTG GAC TTC ATT AAA AGT CAT CCG GGG A TAC CCT CGT GAG AAC CTC AAG TAA TTT TCA GTA GGC CCC |
| RD57 | GAT CCT GTG GAG CTG CTT CTA CGA CTA CTG ATA CCT GA CAC CTC GAC GAA GAT GCT GAT GAC TAT GGA AT |
| RD58 | GAT CCT CGG CAC AAA CCT CAT CTG AAA CCT GA GCC GTG TTT GGA GTA GAC TTT GGA AT |
| RD59 | TAC GCC TAC AGA CAA GAT AGC CG G CGG ATG TCT GTT CTA TCG GCA GCT |
lexA-DAL82 Plasmids
lexA-DAL82 p8202 was constructed by cloning the 1.5-kilobase pair pMO8 (34) BamHI-BamHI fragment into lexA pEG202 (45, 46) containing an EcoRI-BamHI adaptor (5'-GATCCACCGATTCATCCATG-3' and 5'-AATTCATGGATGAATCGGTG-3'). Deletions (dal82) were constructed by replacing WT sequences between BamHI and XhoI of pVS827 with desired deletions from the pRDΔ and pSSΔ series (Figs. 1 and 2). The SphI-SphI fragment from p8202 (ADH1-lexA-DAL82) was cloned into pBTM116 (2µ-based) to yield lexA- DAL82 pSS82BTM, containing TRP1. pSH18-34 and pEG202 were used for transcriptional activation assays (45, 46). All plasmids were verified by restriction and DNA sequence analyses.
Western Analysis, Enzymes, and EMSAs
HA-dal82Δ (constructed by pSelect) replaced the WT gene in pRD41 (38), and the resulting plasmids were expressed in E. coli strain BL21(DE3) (47). Western transfer procedures were essentially as described by Towbin et al. (48). HA-tagged antibody with horseradish peroxidase-conjugated secondary antibody (goat anti-mouse IgG; Bio-Rad) were used. EMSAs were performed using 10 µg of calf thymus + 50 ng of radiolabeled DNA fragment and 1–5 µg of protein (crude cell extract) (49). Urea amidolyase activity was determined in nystatin-permeabilized cells/transformants (4, 50, 51).
Fluorescence Microscopy
GYC86 cells transformed with pNVS2 or pNVS82 (grown overnight in medium containing 2% raffinose, 0.1% ammonia, and yeast nitrogen base (+casamino acids)) was induced for 3 h (4% galactose) and viewed with epifluorescent and/or weak white light using a Zeiss Axiophot microscope equipped with a fluorescein or UV filter and an Optronics analogue camera. 4,6-Diamidino-2-phenylindole staining followed standard protocols except that the cells were not fixed. Images were imported into Photoshop Version 4.0. To evaluate co-localization, the 4,6-diamidino-2-phenylindole-positive staining was pseudo-colored red, and the entire capture frame was merged with the GFP frame.
RESULTS
Localization of Dal82p
Complementation of dal82Δ with GFP-DAL82 pNVS82 demonstrated that the fusion protein retained native biological function (Table III). That only 2-fold induction occurred in WT STCY32 was not disturbing because this parameter is both highly strain- and gene-specific; for DAL7, induction ranges from 2- to 20-fold (26, 29). GFP pNVS2-transformed cells exhibited uniform cytoplasmic fluorescence (Fig. 3N). GFP-DAL82 pNVS82 transformants fluoresced (Fig. 3, A–F, H, and L) in a galactose-dependent manner (data not shown). Superimposition of GFP and 4,6-diamidino2-phenylindole-positive images indicated co-localization (Fig. 3, G–M), supporting that GFP-Dal82p is specifically nuclear. GFP-Dal82p localization was the same in WT and dal81 strains whether or not inducer was present. Decreasing the level of GAL1,10-GFP-DAL82 expression to the lower limit of fluorescence detection by omitting galactose or shortening induction to 1 h did not alter the results (data not shown).
Table III.
GFP-DAL82 complementation of dal82Δ
| Strain | Plasmid description |
β- Galactosidase activity |
|
|---|---|---|---|
| Proa | Pro + OXLU |
||
| Miller units | |||
| STCY32 (WT) | 1946 | 4684 | |
| SS200 (dal82Δ) | 201 | 139 | |
| SS200 (dal82Δ) | Vector (YCp50) | 233 | 152 |
| SS200 (dal82Δ) | GFP vector (p423) | 231 | 167 |
| SS400 (dal82Δ) | Full-length Dal82p (pSS20) | 1874 | 4834 |
| SS200 (dal82Δ) | GFP-Dal82p (pSS423–82) | 2839 | 5646 |
Pro ; OXLU, oxalurate.
WT STCY32 and dal82 SS200 were transformed with DAL7-lacZ reporter pHY43–1 (26); pSS20 (DAL82 under its own promoter), YCp50, or pSS423–82 was used for a second transformation. β-Galactosidase was assayed in transformants grown (split culture) in medium containing 2% minimal glucose, 0.1% proline, and yeast nitrogen base without amino acids or NH4 ± inducer, oxalurate (66 mg/liter). Note that the vector used for microscopy (GAL1,10) and complementation (ADH1) differed by the promoter driving GFP-DAL82 because the strains used for the experiments were of necessity different; dal82 mutant SS400 does not use galactose as sole carbon source.
FIG. 3.

A–F, GFP-DAL82 expressed in WT GYC86 cells. Co-localization of 4,6-diamidino-2-phenylindole-positive staining (I and K, blue; and J and M, pseudo-colored red) and GFP (H and L, green) is indicated in yellow (J and M). G, visible light only; N, GFP vector expressed in the wild type.
DNA-binding Domain of Dal82p
Dal82p was not homologous to any known DNA-binding motifs. However, since Dal82p specifically binds to UISALL (38), we prepared crude extracts from E. coli BL21(DE3) transformants of HA-tagged dal82Δ plasmids (Fig. 2) and investigated the stability of the mutant proteins. All extracts, except those from pT7-7 (vector), pRD4, and pSS495, contained proteins that could be easily detected by Western blot analysis (Fig. 4); pRD41 and pRD4 were the positive and negative controls (38). The extracts were also used in EMSAs with DAL7 fragment JD72 (−254 to −199), containing a well characterized UISALL, as probe (38) (see Fig. 5 for data and Fig. 2 for a summary). A Dal82p-specific band ran close to the free DNA (Fig. 5, lanes 2 and 5). This complex contains a stable degradation product with the same binding specificity as full-length Dal82p (38); it is clearer when less protein was assayed (Fig. 5, lanes 7 and 8). These data argue that the first 85 Dal82p residues are sufficient for DNA binding.
FIG. 4. Western blot analyses of extracts from E. coli BL21(DE3) pLysS cells transformed with HA-tagged Dal82p deletion derivatives.

See Fig. 2 for maps.
FIG. 5. EMSAs of truncated Dal82p.

See Fig. 2. The probe was the synthetic DNA fragment JD72 (38) containing a DAL7 UISALL element. -EXT, extract omitted. Lane 8 contains 10 time more protein than lane 7. Data are summarized in Fig. 2.
Transcriptional Activation Domain of Dal82p
If Dal82p is the UISALL-binding protein, then Dal81p might complex to it and function as a transcriptional activator. By this reasoning, Dal82p might not contain a transcriptional activation motif (53). Therefore, we transformed WT EGY48 cells with lexAlacZ transcriptional activation reporter pSH18-34, pSH18-34 + vector pBTM116 (45, 46), or pSH18-34 + lexA-DAL82 pSS82BTM (15, 16). High level activation occurred only in lexA-DAL82 pSS82BTM transformants (Fig. 6), indicating the ability of Dal82p to support transcriptional activation. The only easily recognizable motif in Dal82p was a C-terminal coiledcoil. When it was deleted (pSH18-34 + pSS43), Δ-galactosidase activity increased 5-fold (Fig. 6).
FIG. 6. Gene activation supported by lexA pBTM116, WT lexA-DAL82 pSS82BTM, or lexA-dal82.ii pSS43 in strain M1682-19b containing reporter pSH18-34.

Transformants were grown in minimal glucose/proline medium + oxalurate (66 mg/liter).
Two considerations prompted us to use the dal82 coiled-coil deletion to investigate the activation domain. (i) It removes one variable from the analysis; and (ii) increased activation permits greater resolution of the genetic dissection. lacZ expression was measured in EGY48 cells transformed with reengineered lexA-dal82Δ plasmids (Fig. 7). Two regions were required for activation: (i) an absolutely required core (residues 66 –99) and (ii) residues flanking it required only for full activity.
FIG. 7. Reporter gene activation supported by truncated Dal82p.

dal82 mutants replaced WT sequences in pVS827 (pEG202-derived). Transformants were in strain EGY48 containing pSH18-34 grown as described in the legend to Fig. 6.
Only Full-length Dal82p Is Able to Complement dal82Δ in Vivo
To determine whether the above constructs could complement dal82Δ, SS400 cells were transformed with representative plasmids, and urea amidolyase was measured. Urea amidolyase was highly inducible only in a transformant containing WT pSS20 (Table IV). The lack of complementation with pSS43 is significant because the protein it encoded tested positive for DNA binding and transcriptional activation. Measuring complementation with DAL7-lacZ yielded similar results (Table IV); DUR1,2 (encoding urea amidolyase) is always far more inducible than DAL7 (8, 26, 37, 55).
Table IV.
Complementation of dal82Δ by portions of DAL82
| Plasmid | Residues deleted |
Dal82p |
UALase activity |
β-Galactosidase activity |
|||
|---|---|---|---|---|---|---|---|
| Binding | Activation | −OXLU | +OXLU | −OXLU | +OXLU | ||
| pSS20 | None | + | + | 4 | 211 | 2413 | 5789 |
| YCp50 | NA | NA | 6 | 3 | 25 | 13 | |
| pSS482 | 79–88 | − | − | 4 | 12 | 51 | 43 |
| pSS480 | 89–95 | + | − | 2 | 5 | 47 | 25 |
| pSS489 | 8–16 | − | + | 6 | 8 | 21 | 20 |
| pSS488 | 17–23 | − | + | 6 | 5 | 19 | 37 |
| pSS43 | 225–255 | + | + | 3 | 2 | 26 | 41 |
a UALase, urea amidolyase; OXLU, oxalurate.
dal82Δ SS400 was the transformation recipient. The deletion constructs replaced WT sequences in pSS20 and thus are under the native DAL82 promoter. Culture conditions were as described in the legend to Table III. Induction was for one generation (30–60 Klett units). Urea amidolyase activity was assayed at a culture density of 60 Klett units. DAL7-lacZ (pHY43–1) was the reporter.
DISCUSSION
Our experiments argue that Dal82p is localized to the nucleus, positively correlating with the in vitro demonstration that it binds to DNA. We were unable to obtain evidence that Dal82p localization is influenced by Dal81p or inducer. This possibility was interesting in light of recent reports that Msn2p and Msn4p are strongly regulated by controlling their entry into and exit from the nucleus (54). Whether our lack of success in demonstrating an effect was due to the overproduction of the protein required for us to see it microscopically could not be determined because without some overproduction, the fluorescent signal was too faint to see. Reducing expression to the limit of detection, however, did not affect the results.
Three important regions were identified in Dal82p (Fig. 8). The DNA-binding domain consists of residues 1– 85. There is modest ambiguity in delineating the N terminus of the domain because it was defined by pRD489 encoding a protein lacking amino acids 8 –16. It is unclear which of the first 16 residues are necessary for function. The C terminus is tightly defined by pSS493 and pRD478 (Fig. 2). Although the Dal82p DNA-binding domain appears devoid of periodic structures and exhibits little similarity to known DNA-binding motifs, amino acids 1– 85 do share limited homology with insect homeobox proteins HMEN (engrailed) and H17.
FIG. 8. Schematic diagram of Dal82p indicating the putative functional domains.
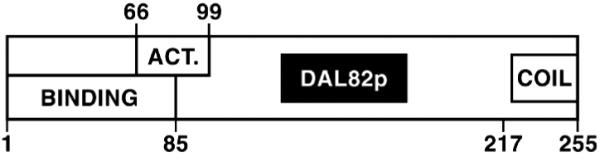
ACT., activation domain.
The Dal82p domain required for transcriptional activation appears to be more complex, consisting of residues 66 –99 with the boundaries being rather tightly defined. Flanking this region were residues whose presence was not absolutely required, but they were nonetheless needed to achieve high level activation. Without residues 32– 66 and those in the vicinity of positions 99 –152, transcriptional activation reached only onethird to one-fifth of the parental (pVS827) activity. When residues 1–31 were removed, reporter gene expression decreased 20 – 40%.
The Dal82p DNA-binding and transcriptional activation domains overlap. Since DNA binding involves a DNA-protein complex, whereas activation probably involves a protein-protein interaction, this overlap might at first seem a bit paradoxical. However, such data would be expected if the N-terminal region of the protein is part of a domain whose integrity is required for both DNA binding and transcriptional activation. By this explanation, removing the non-overlapping portions of the domain damages, but does not destroy, its ability to carry out one of its functions. On the other hand, removing residues common to both DNA binding and transcriptional activation distorts the putatively shared domain to a point of losing all function. Consistent with this speculation, most Dal82p preparations, including those in the above experiments, are contaminated with a proteolytic product that more or less comigrates with truncated Dal82p 85 amino acids long (Ref. 38 and this work).
It appears that the N-terminal DNA-binding and activation domains are able to function independently of the coiled-coil. We were, however, surprised by the apparent negative role that the predicted coiled-coil domain appears to play. Previously reported genetic results concerning DAL82 led to the conclusion that it is a positive regulator. However, removing the coiled-coil resulted in a 5-fold increase in transcriptional activation ability, arguing in favor of it potentially participating in a negative function as well. What is not clear at this point is whether the coiled-coil interacts with another domain in Dal82p or an unknown “repressor” protein to perform this function.
Acknowledgments
We thank Dr. Harry Jarrett for the GFP plasmid we used and the University of Tennessee Yeast Group, who suggested improvements for the manuscript. Tim Higgins prepared the figures.
Footnotes
The work was supported by United States Public Health Service Grant GM-35642. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- NCR
- nitrogen catabolite repression
- EMSAs
- electrophoretic mobility shift assays
- PCR
- polymerase chain reaction
- GFP
- green fluorescent protein
- WT
- wild-type
- HA
- hemagglutinin
T. S. Cunningham, R. Anhare, and T. G. Cooper, manuscript in preparation.
REFERENCES
- 1.Bysani N, Daugherty JR, Cooper TG. J. Bacteriol. 1991;173:4977–4982. doi: 10.1128/jb.173.16.4977-4982.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rai R, Genbauffe FS, Cooper TG. J. Bacteriol. 1987;170:266–271. doi: 10.1128/jb.170.1.266-271.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rai R, Genbauffe FS, Cooper TG. J. Bacteriol. 1987;169:3521–3524. doi: 10.1128/jb.169.8.3521-3524.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rai R, Genbauffe FS, Sumrada RA, Cooper TG. Mol. Cell. Biol. 1989;9:602–608. doi: 10.1128/mcb.9.2.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper TG, Rai R, Yoo HS. Mol. Cell. Biol. 1989;9:5440–5444. doi: 10.1128/mcb.9.12.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper TG. In: The Molecular Biology of the Yeast Saccharomyces: Metabolism and Gene Expression. Strathern JN, Jones EW, Broach J, editors. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1982. pp. 39–99. [Google Scholar]
- 7.Wiame J-M, Grenson M, Arst H. Adv. Microb. Physiol. 1985;26:1–87. doi: 10.1016/s0065-2911(08)60394-x. [DOI] [PubMed] [Google Scholar]
- 8.Cooper TG. In: Mycota. Marzluf G, Bambrl R, editors. III. Springer-Verlag; Berlin: 1994. pp. 139–169. [Google Scholar]
- 9.Cooper TG, Sumrada RA. J. Bacteriol. 1983;155:623–627. doi: 10.1128/jb.155.2.623-627.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooper TG, Ferguson D, Rai R, Bysani N. J. Bacteriol. 1990;172:1014–1018. doi: 10.1128/jb.172.2.1014-1018.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minehart PL, Magasanik B. Mol. Cell. Biol. 1991;11:6216–6228. doi: 10.1128/mcb.11.12.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blinder D, Magasanik B. J. Bacteriol. 1995;177:4190–4193. doi: 10.1128/jb.177.14.4190-4193.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanbrough M, Rowen DW, Magasanik B. Proc. Natl. Acad. Sci. U. S. A. 1995;92:9450–9454. doi: 10.1073/pnas.92.21.9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coffman JA, Rai R, Cooper TG. J. Bacteriol. 1995;177:6910–6918. doi: 10.1128/jb.177.23.6910-6918.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coffman JA, Rai R, Cunningham TS, Svetlov V, Cooper TG. Mol. Cell. Biol. 1996;16:847–858. doi: 10.1128/mcb.16.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cunningham TS, Svetlov V, Rai R, Cooper TG. J. Bacteriol. 1995;178:3470–3479. doi: 10.1128/jb.178.12.3470-3479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daugherty JR, Rai R, ElBerry HM, Cooper TG. J. Bacteriol. 1993;175:64–73. doi: 10.1128/jb.175.1.64-73.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minehart PL, Magasanik B. J. Bacteriol. 1992;174:1828–1836. doi: 10.1128/jb.174.6.1828-1836.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chisholm G, Cooper TG. Mol. Cell. Biol. 1982;2:1088–1095. doi: 10.1128/mcb.2.9.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coffman JA, Rai R, Loprete DM, Cunningham T, Svetlov V, Cooper TG. J. Bacteriol. 1997;179:3416–3429. doi: 10.1128/jb.179.11.3416-3429.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cunningham TS, Dorrington RA, Cooper TG. J. Bacteriol. 1994;176:4718–4725. doi: 10.1128/jb.176.15.4718-4725.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galibert F. EMBO J. 1996;15:2031–2049. [PMC free article] [PubMed] [Google Scholar]
- 23.Soussi-Boudekou S, Vissers SS, Urrestarazu A, Jauniaux J-C, Andre B. Mol. Microbiol. 1997;23:1157–1168. doi: 10.1046/j.1365-2958.1997.3021665.x. [DOI] [PubMed] [Google Scholar]
- 24.Cunningham TS, Cooper TG. J. Bacteriol. 1993;175:5851–5861. doi: 10.1128/jb.175.18.5851-5861.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andre B, Talibi D, Boudekou SS, Hein C, Vissers S, Coornaert D. Nucleic Acids Res. 1995;23:558–564. doi: 10.1093/nar/23.4.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoo HS, Cooper TG. Mol. Cell. Biol. 1989;9:3231–3243. doi: 10.1128/mcb.9.8.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoo HS, Cooper TG. Gene (Amst.) 1991;104:55–62. doi: 10.1016/0378-1119(91)90464-m. [DOI] [PubMed] [Google Scholar]
- 28.Hartig A, Simon MM, Schuster T, Daugherty JR, Yoo HS, Cooper TG. Nucleic Acids Res. 1992;20:5677–5686. doi: 10.1093/nar/20.21.5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park HD, Scott S, Rai R, Dorrington R, Cooper TG. J. Bacteriol. 1999;181:7052–7064. doi: 10.1128/jb.181.22.7052-7064.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Vuuren HJJ, Daugherty JR, Rai R, Cooper TG. J. Bacteriol. 1991;173:7186–7195. doi: 10.1128/jb.173.22.7186-7195.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turoscy V, Cooper TG. J. Bacteriol. 1982;151:1237–1246. doi: 10.1128/jb.151.3.1237-1246.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vissers S, Andre B, Muyldermansm F, Grenson M. Eur. J. Biochem. 1989;181:357–361. doi: 10.1111/j.1432-1033.1989.tb14732.x. [DOI] [PubMed] [Google Scholar]
- 33.Lemoine Y, Dubois E, Wiame J-M. Mol. Gen. Genet. 1978;166:251–258. [PubMed] [Google Scholar]
- 34.Olive MJ, Daugherty JR, Cooper TG. J. Bacteriol. 1991;173:255–261. doi: 10.1128/jb.173.1.255-261.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bricmont PA, Cooper TG. Mol. Cell. Biol. 1989;9:3869–3877. doi: 10.1128/mcb.9.9.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bricmont PA, Daugherty JR, Cooper TG. Mol. Cell. Biol. 1991;11:1161–1166. doi: 10.1128/mcb.11.2.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Genbauffe FS, Cooper TG. Mol. Cell. Biol. 1986;6:3954–3964. doi: 10.1128/mcb.6.11.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dorrington RA, Cooper TG. Nucleic Acids Res. 1993;21:3777–3784. doi: 10.1093/nar/21.16.3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sikorski RS, Heiter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitchell DA, Marshall TK, Deschenes RJ. Yeast. 1993;9:715–722. doi: 10.1002/yea.320090705. [DOI] [PubMed] [Google Scholar]
- 41.Jarrett HW, Taylor WL. J. Chromatogr. 1998;803:131–139. doi: 10.1016/s0021-9673(97)01257-0. [DOI] [PubMed] [Google Scholar]
- 42.Allen JB, Elledge SJ. Yeast. 1994;10:1267–1272. doi: 10.1002/yea.320101003. [DOI] [PubMed] [Google Scholar]
- 43.Christianson TW, Sikorski RS, Dante M, Shero JH, Heiter P. Gene (Amst.) 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- 44.Alani E, Cao L, Kleckner N. Genetics. 1987;116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Golemis EA, Gyuris J, Brent R. In: Current Protocols in Molecular Biology. Ausabel FM, Brent R, Kingston R, Moore D, Seidman J, Struhl K, editors. John Wiley & Sons, Inc.; New York: 1996. [Google Scholar]
- 46.Bartel P, Chien C, Sternglanz R, Fields S. In: Cellular Interactions in Development: A Practical Approach. Hartley DA, editor. Oxford University Press; Oxford: 1993. pp. 153–179. [Google Scholar]
- 47.Dubendorff JW, Studier FW. J. Mol. Biol. 1992;219:45–59. doi: 10.1016/0022-2836(91)90856-2. [DOI] [PubMed] [Google Scholar]
- 48.Towbin H, Stachelin T, Gordon J. Biotechnology. 1992;24:145–149. [PubMed] [Google Scholar]
- 49.Luche RM, Sumrada R, Cooper TG. Mol. Cell. Biol. 1990;10:3884–3895. doi: 10.1128/mcb.10.8.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whitney PA, Cooper TG, Magasanik B. J. Biol. Chem. 1973;248:6203–6209. [PubMed] [Google Scholar]
- 51.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. p. 403. [Google Scholar]
- 52.Kunze I, Adler HG, Bernard J, Nilsson NB, Stoltenburg R, Kohlvein SD, Kunze G. Biochim. Biophys. Acta. 1999;410:287–298. doi: 10.1016/s0005-2728(99)00006-7. [DOI] [PubMed] [Google Scholar]
- 53.Svetlov V, Cooper TG. J. Bacteriol. 1997;179:7644–7652. doi: 10.1128/jb.179.24.7644-7652.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gorner W, Durchschlag E, Martinez-Pastor MT, Estruch F, Ammerer G, Hamilton B, Ruis H, Schuller T. Genes Dev. 1998;12:586–597. doi: 10.1101/gad.12.4.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roon RJ, Levenberg B. J. Biol. Chem. 1968;243:5213–5215. [PubMed] [Google Scholar]