

Abstract
Inferring which protein species have been detected in bottom-up proteomics experiments has been a challenging problem for which solutions have been maturing over the past decade. While many inference approaches now function well in isolation, comparing and reconciling the results generated across different tools remains difficult. It presently stands as one of the greatest barriers in collaborative efforts such as the Human Proteome Project and public repositories like the PRoteomics IDEntifications (PRIDE) database. Here we present a framework for reporting protein identifications that seeks to improve capabilities for comparing results generated by different inference tools. This framework standardizes the terminology for describing protein identification results, associated with the HUPO-Proteomics Standards Initiative (PSI) mzIdentML standard, while still allowing for differing methodologies to reach that final state. It is proposed that developers of software for reporting identification results will adopt this terminology in their outputs. While the new terminology does not require any changes to the core mzIdentML model, it represents a significant change in practice, and, as such, the rules will be released via a new version of the mzIdentML specification (version 1.2) so that consumers of files are able to determine whether the new guidelines have been adopted by export software.
Keywords: protein identification, software, data standards, Proteomics Standards Initiative, XML
Introduction
In many proteomics workflows (so called bottom-up), proteins within the sample(s) are digested into peptides prior to analysis. This causes a loss of the link from an identified peptide to its parent protein, as many peptide sequences can be assigned to more than one protein. In such cases it is not possible to determine definitively which protein (or proteins) the peptide originated from and thus which proteins were present in the sample. Two proteins sharing one or more peptides may arise from the same gene but differ due to SNPs, post-translational cleavage (e.g. removal of a signal peptide), or alternative splicing; if arising from different genes they may be homologues (paralogues in a single species-derived database, or paralogues and orthologues if the search database contains proteins from multiple species), or unrelated but sharing a short sequence of amino acids. The concept of proteoform has been coined to describe the unit of protein as present in the cell and carrying a given sequence and a specific set of post-translational modifications (PTMs) [1]. It should be noted that PTMs can also introduce ambiguity in assignment of a parent protein, for example deamidation of asparagine is physically indistinguishable from aspartic acid, and as such different peptide sequences (from different proteins) could equally “explain” the same mass spectrum. Up to roughly the middle of the last decade it was common for investigators to report all protein sequences matching any putatively identified peptides, leading to highly inflated protein counts.
The so-called “protein inference” problem in proteomics aims to determine how many protein species have actually been detected and convey the remaining ambiguity in an optimal way, and has been tackled by many different groups [2-9]. Protein counting inflation has been brought under control in the last few years, driven by advances in protein inference algorithms and perhaps more importantly, increased awareness of their importance driven by journal publication guidelines [10-12]. It is now generally expected by journals that rules of parsimony are applied in producing the list of proteins identified [13]; i.e. the shortest list of proteins that can adequately explain all of the data is submitted for publication. While this pressure has forced the numbers of detected proteins reported by different methods to converge to some extent, there remains greater heterogeneity in the second major concern of protein inference – conveying the ambiguity.
Whether a result of the output of an algorithm or a subsequent choice made by a user, the way that ambiguity is conveyed in a protein identification result can have a major effect on how that result can be compared to other results. Even if multiple results use the same protein identifier system and are derived from the same database searched (problems not directly addressed here), insufficient description of ambiguity in protein groups can cause failure to recognize common protein detections between results, causing falsely low apparent intersections. Additionally, different protein inference tools describe ambiguity in different ways with different terminology. While individual publications may no longer report inflated protein lists, because of the missing information about ambiguity and how this was handled by the software employed, it is presently not possible to compare or combine findings from multiple laboratories adequately, when a broad range of different tools is used.
The challenges of comparing protein identification results were highlighted by the ABRF (Association of Biomolecular Resource Facilities) Proteome Informatics Research Group (iPRG) in 2008 [14, 15] where the committee, entirely comprised of creators of protein inference tools, attempted to analyze a common dataset and determine a consensus protein identification result, each using their respective software. The committee agreed a common terminology for describing identification results: protein accession - one entry in a database searched; protein group - a set of protein accessions that have some independent evidence in common (evidence distinguishing them from all other proteins) – generally considered to be a single unit of (protein-level) identification in proteomics; and a protein cluster – a set of protein groups that share some evidence in common (e.g. some peptides/spectra shared between groups), but within which different groups also have evidence independent from each other (e.g. some peptides/spectra uniquely assigned to some groups only). The minimal list of proteins “identified” from a study should be the count of the number of protein groups, for example passing a given threshold (Figure 1A-C). In earlier work by Nesvizhskii and Aebersold at providing a consistent nomenclature [13], concepts of “protein”, “protein group” and “protein family” were defined – which are broadly consistent with the three main concepts in the iPRG nomenclature. In [13], further classifications of “distinct proteins”, “differentiable proteins”, “indistinguishable proteins”, “subset proteins” and “subsumable proteins” were also described. In this work, the (simpler) iPRG concepts are used throughout, as they were derived by a consensus of protein inference tool creators, including the lead author of [13].
Figure 1.
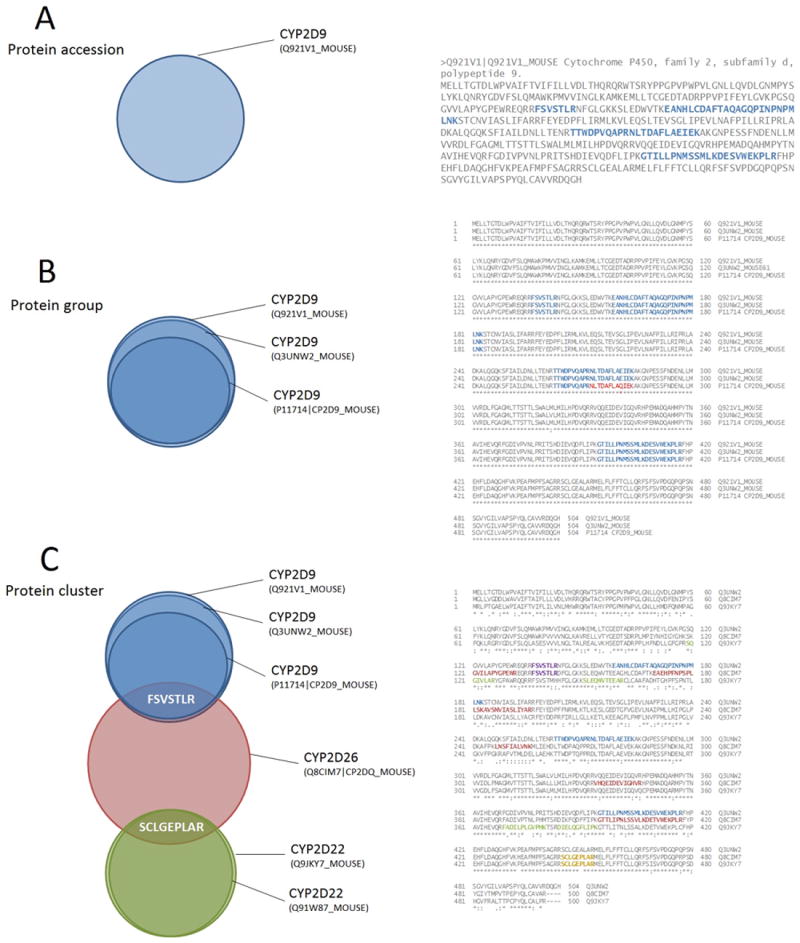
Terminology defined by the iPRG2008 working group, for a protein accession (A), protein group (B) and protein cluster (C), with a multiple sequence alignment displaying the peptides shared between the different proteins.
Even with the exceptional advantage of having direct input from each tool’s creator, the synthesis exercise performed in iPRG2008 still proved time-consuming, requiring much manual intervention. For repositories and collaborative studies to function, this same kind of synthesis needs to be accomplished not only without the benefit of direct interaction with software creators, but via automated computation. Additional standardization is needed to achieve this, which is the aim of this work.
The Proteomics Standards Initiative (PSI) is an entirely open collaboration of academic research groups, instrument and software vendors and journal representatives, which has been developing resources to facilitate data sharing and public deposition for over ten years [16]. Each PSI workgroup develops broadly three types of output: minimum reporting guidelines, standard data formats, and controlled vocabularies sets. The Minimum Information About a Proteomics Experiment (MIAPE) specifications are a set of modules that provide minimum reporting guidelines for specific experimental techniques or approaches [17]. Data format standards seek to improve data exchange between software packages and databases; efforts to date include mzML for MS data [18], mzIdentML for peptide and protein identification data [19] and new formats for quantitation results – mzQuantML [20] and mzTab [21]. Established controlled vocabularies (CVs) containing well-defined terminology to use within the data formats enable concepts to be unambiguously interpreted. Examples include the PSI-MOD [22] and Unimod [23] nomenclatures for describing peptide modifications and the PSI-MS CV [24] used in a variety of PSI standards.
The initial stable version of mzIdentML (version 1.1 [19]) has now become a well-established standard for capturing the outputs of proteomic search engines, particularly the scores and statistical values associated with peptide-spectrum matches (PSMs). The mzIdentML version 1.1 specifications enabled protein identification results to be reported as grouped accessions (where shared peptide evidence exists) in a relatively flexible structure, with the intention that more robust guidelines could be developed later. In this work, we have now developed guidelines for reporting (grouped) protein identification results in a format that can be consumed and interpreted in an unambiguous manner, and supporting the majority of known approaches for inferring protein identifications. The guidelines do not require an update to the core mzIdentML structure (the XML Schema), but do represent a change in practice in how protein-level results should be encoded, and, as such, we are now releasing a new version of mzIdentML (version 1.2), so that consuming software is able to differentiate those following the new guidelines on protein reporting.
Methods
The guidelines reported here have been developed through an open consultation process at PSI meetings [16] and teleconferences. The guidelines have been formally captured in the mzIdentML specification document: http://code.google.com/p/psi-pi/source/browse/trunk/specification_document/specdoc1_2/, supporting examples files (http://code.google.com/p/psi-pi/source/browse/trunk/examples/) updates to the PSI-MS controlled vocabulary [24], a new mapping file indicating how the CV terms should be used within the format (http://code.google.com/p/psi-pi/source/browse/trunk/cv/) and updates to the mzIdentML validator [25].
mzIdentML overview
The mzIdentML standard has been designed to capture the outputs of peptide/protein identification software, such as sequence database search engines and search result verification/post-processing software. The format captures the software used, the sequence database searched, software parameters (including modifications) and output results – one or more lists of peptide-spectrum matches (PSMs) and the set of proteins inferred from those PSMs. Each PSM or protein identification can be reported with one or more scores or statistical measures, such as e-values or p-values (encoded using standard terminology from the PSI-MS CV), which allow subsequent manual or automated assessment of the quality of individual results. An up-to-date listing of software implementations for mzIdentML can be found here: http://www.psidev.info/tools-implementing-mzidentml.
Each mzIdentML file has a <SequenceCollection> containing elements called <DBSequence> (Figure 2A). <DBSequence> is a reusable (referenced from several elements in an mzIdentML file) representation of a single database entry, capturing the accession in the source database and optionally the protein sequence, description, taxonomy and so on. One or more <DBSequence> elements is referenced from every PSM (not shown on Figure 2), capturing all possible parent proteins for every peptide prior to protein inference. An mzIdentML file could in theory encode an entire search database (for example from a FASTA formatted file) in the <SequenceCollection>, although generally the <SequenceCollection> contains only the listing of all possible proteins mapped from PSMs, which is typically a superset of the protein accessions identified following protein inference.
Figure 2.

A and B. Graphical representation of how the concepts defined in Figure 1 map onto an mzIdentML file, following the recommendations presented in this manuscript. Each <ProteinDetectionHypothesis> has references back to all peptide spectrum matches (PSMs) on which the protein identifications are based (not shown – consult [19] for more details).
The <ProteinDetectionList> contains a hierarchical structure in which the protein identifications are represented (Figure 2B). Each <ProteinDetectionList> contains <ProteinAmbiguityGroup> elements (here referred as PAG), each capturing a single identified protein or a group of proteins where there is some ambiguity in exactly which protein has been identified. Each protein within a group is recorded as an element called <ProteinDetectionHypothesis> (here referred as PDH). Each PDH references exactly one <DBSequence> element, indicating the database entry that has been potentially identified. Each PDH also references the set of PSMs on which it is based, completing the evidence trail for its identification (not shown on Figure 2). Also, each PDH has a mandatory true/false attribute called passThreshold, indicating whether the protein identification is deemed to have passed a threshold reported elsewhere within the file. This attribute was included in mzIdentML 1.1 (and earlier releases) to allow the data producer to export identifications both above and below the threshold. However, no such attribute was also present on the protein group (PAG) level.
When the mzIdentML standard was completed as a stable release (version 1.1), a set of CV terms was added to the PSI-MS CV allowing basic annotations as to the role that each protein (PDH) played within its group (PAG) – intended to capture same-set, subset and subsumable relationships between PDHs. However, the original mzIdentML specification document did not enforce the use of these CV terms and provided little guidance on how more general grouping relationships should be captured. The result is that software reading mzIdentML files containing protein identification results would have difficulty comparing the results exported from different packages. Specific problems that have been identified are as follows. Most critically, the specifications did not contain a clear statement in terms of how the concepts represented in Figure 1A-C should be mapped onto mzIdentML – e.g. different exporters could choose to map onto a PAG either a “cluster” or a “protein group” or, for some software packages that define sets/groups of proteins at yet further levels of granularity, something else. Second, when reading an mzIdentML file, the answer to a simple question “how many proteins were being reported as identified” could not be decisively determined, and different users or software could arrive at different answers (for example one might count PAGs or count PDHs). Third, the specification documentation was not clear on how the passThreshold attribute on PDH should be interpreted – potentially implying that this protein was determined as identified and representative of a group or only that it had statistically significant PSMs. In this manuscript, we describe work undertaken to formalize how protein inference should be reported in mzIdentML (although the guidelines could be adapted for other methods of reporting proteomics identification results), including the definition of a new set of CV terms that have been added to the PSI-MS CV.
Results and Discussion
The primary result reported here is a standardized set of rules for mapping the concepts represented in Figure 1 onto mzIdentML, as shown graphically in Figure 2 and as actual mzIdentML XML code in Figure 3. The following mappings and rules have now been established in this work - capitalized MUST, SHOULD and MAY have a formal interpretation by validation software:
Figure 3.

A snippet of mzIdentML showing a <ProteinAmbiguityGroup> (lines 5038 to 5095) containing four <ProteinDetectionHypothesis> elements (two minimised on lines 5068 and 5080). In this example, the first PDH (lines 5039-5055) has been flagged as both a “leading protein” and “group representative” (lines 5051 and 5052). The second PDH (lines 5056-5067) has been assigned as a “non-leading protein” (line 5066) and a “sequence sub-set protein” (line 5065). CV terms assigned to the PAG-level are on lines 5092-5094, including the mandatory term “protein group passes threshold” (line 5092).
-
As in mzIdentML version 1.1, a single protein accession that has been cited by software (Figure 1A) is captured in mzIdentML in <ProteinDetectionHypothesis> (PDH).
A PDH MAY contain scores or statistical values produced by the export software, encoded as CV terms.
-
A “protein group” (Figure 1B), representing a “biological entity” for which the software claims independent evidence is present, MUST be mapped onto <ProteinAmbiguityGroup> (PAG).
A PAG MAY have additional scores produced by the export software, encoded as CV terms.
-
The reporting of protein identification thresholds is now mapped onto PAGs. There is no desire to change the core XML Schema Document (XSD) for mzIdentML and as such, a new CV term “protein group passes threshold” value= “xsd:boolean” MUST be present on every PAG (MS:1002415). If no thresholding has been done by the software, all protein groups MUST be annotated as “protein group passes threshold” value= “true”.
The attribute passThreshold = “true∣false” remains present on PDH and MAY be used if software packages wish to report a two-level hierarchy of thresholds applied, however, it is not expected that consuming software will use this attribute to determine which proteins have been reported as identified.
As in mzIdentML 1.1, the threshold applied to protein-level results MUST be present in the <ProteinDetectionProtocol>. However, the mzIdentML 1.1 specifications implied that the threshold value present here was used to determine the passThreshold attribute on all PDH elements. In mzIdentML 1.2, the threshold value reported here corresponds with PAG-level thresholding applied – either to a score reported specifically on each PAG itself, or a score on the PDH flagged as a “group representative”. In approaches that do not use the “group representative” CV term, there is an expectation that thresholds SHOULD be applied to scores reported at the PAG-level. The <ProteinDetectionProtocol> MUST contain either the “no threshold” term or a suitable score/value pair sourced from the PSI-MS CV – such as p-value, FDR, e-value and so on, determined by any type of statistical analysis (i.e. not limited to target-decoy approaches). The file reader can then determine the error rate that has been estimated by the software in determining those PAGs that pass the reported threshold.
The <ProteinDetectionList> MUST contain the CV term “count of identified proteins” value= “xsd:integer” (MS:1002404). The value MUST be derived from the count of PAGs passing the threshold reported in the file and will be checked by validation software. Optional CV terms for alternative methods for counting protein identifications or providing ranges can be requested from the working group.
Few software packages report “protein clusters” at present (Figure 1C), but for those packages that wish to report clusters, a CV term “cluster identifier” value = “xsd:integer” SHOULD be used (MS:1002407). The integer identifier MUST be shared by all PAGs belonging to the same cluster. If cluster identifiers are used, all PAGs MUST have a cluster identifier. An optional term “count of identified clusters” value = “xsd:integer” (MS:1002406) MAY be annotated on the <ProteinDetectionList>.
-
Every PDH MUST be annotated as either a “leading protein” (MS:1002401) or a “non-leading protein” (MS:1002402), as defined in Table 1, within a PAG. This recommendation thus makes it explicit for consuming software whether one or more proteins have stronger evidence than others in the group (see Table 2 for examples).
An additional term, “group representative” (MS:1002403) MAY be used to annotate one PDH, which is also flagged as a “leading protein”, if the export software wishes to enforce that only one of potential several “leading proteins” will be interpreted by the consuming software as the representative of the group, for example acting as a tiebreaker.
If the export software does not explicitly flag one protein as the “group representative”, it is assumed that if consuming software requires a single accession to represent the group, an arbitrary choice will be made (among “leading proteins” only if these exist).
-
Any PDHs MAY be annotated with terms present in the CV for spectrum/sequence same-set, spectrum/sequence subset, spectrum/sequence subsumable, marginally distinguished and so on (Table 1).
A PDH MAY be annotated with more than one of these terms if appropriate to describe the complex set relationships that exist within a group.
Developers of software packages MAY propose additional terms for describing group membership of PDHs, which will be incorporated into the CV.
The associated value for these CV terms MAY be used to annotate which PDH(s) are the super/same-set of the annotated PDH.
There is no expectation that consuming software should be aware of these terms, but they may be useful in internal pipeline or visualization software packages that are specifically designed to work with this terminology set.
Some PDHs could be mapped to more than one PAG, for example where proteins are multiply subsumed. To capture these cases, multiple PDHs in different PAGs MAY reference the same <DBSequence>.
Table 1.
New CV terms for reporting protein set (group) relationships and global statistics about the protein identification results. The semantic validation software for mzIdentML (v.1.2) reports an error (MUST), a warning (SHOULD) or an informational message (MAY) if these terms are not reported within the file.
| mzIdentML context | CV term | Values | Requirement level | Description |
|---|---|---|---|---|
| ProteinDetectionList | count of identified proteins | xsd:integer | MUST | The value reported MUST equal the number of PAGs with “protein group passes threshold” value = “true” |
| ProteinDetectionList | count of identified clusters | xsd:integer | MAY | If protein clusters have been reported in the file, the exporter may choose to annotate the ProteinDetectionList with the number identified above threshold. |
| ProteinAmbiguity-Group | number of distinct protein sequences | xsd:integer | MAY | The number of distinct protein sequences among the PDHs in the group. For example, if there are two PDHs with different identifiers that have identical full length sequences, the value would be 1. |
| ProteinAmbiguity-Group | cluster identifier | xsd:integer | MAY | An identifier applied to protein groups to indicate that they are linked by shared peptides. |
| ProteinDetection-Hypothesis | leading protein OR non-leading protein |
- | MUST OR MUST |
Every PDH in each PAG MUST be flagged as a leading protein or a non-leading protein and each PAG MUST contain at least one leading protein, but MAY contain more than one. A “leading protein” is defined as a protein that has the strongest or near strongest (further explained in Table 2) set of evidence for being present in the sample studied, amongst the grouped protein accessions. A “non-leading protein” is defined as a protein that has (substantially) less evidence than other proteins within the same group, and is thus less likely to have been present in the sample studied. |
| ProteinDetection-Hypothesis | group representative | - | MAY | Each PAG MAY contain zero or one PDH flagged as the group representative, if the software wishes to flag a preference (often arbitrary or for example based on alphabetical ordering) amongst the leading proteins. The group representative term can thus be viewed a “tiebreaker” if the export software wishes to make this distinction. |
| ProteinDetection-Hypothesis | Sequence Same-Set Protein | xsd: “list_of_strings” space separated list of PDH IDs that are same-set. | MAY | A protein that is indistinguishable or equivalent to another protein in the group, having matches to an identical set of peptide sequences. |
| ProteinDetection-Hypothesis | Spectrum Same-Set Protein | xsd: “list_of_strings” space separated list of PDH IDs that are same-set. | MAY | A protein that is indistinguishable or equivalent to another protein in the group, having PSMs derived from the same set of spectra. |
| ProteinDetection-Hypothesis | Sequence Subset Protein | xsd: “list_of_strings” space separated list of PDH IDs that are super-set. | MAY | A protein for which the matched peptide sequences are a subset of the matched peptide sequences for another protein in the group. |
| ProteinDetection-Hypothesis | Spectrum Subset Protein | xsd: “list_of_strings” space separated list of PDH IDs that are super-set. | MAY | A protein for which the matched spectra are a subset of the matched spectra for another protein in the group. |
| ProteinDetection-Hypothesis | Sequence Multiply Subsumable Protein | xsd: “list_of_strings” space separated list of PDH IDs that subsume this PDH. | MAY | A protein for which the matched peptide sequences are the same, or a subset of, the matched peptide sequences for two or more other proteins combined. These other proteins need not all be in the same group. |
| ProteinDetection-Hypothesis | Spectrum Multiply Subsumable Protein | xsd: “list_of_strings” space separated list of PDH IDs that subsume this PDH. | MAY | A protein for which the matched spectra are the same, or a subset of, the matched spectra for two or more other proteins combined. These other proteins need not all be in the same group. |
| ProteinDetection-Hypothesis | Marginally distinguished protein | - | MAY | Assigned to a non-leading PDH that has some independent evidence to support its presence relative to the leading protein(s) e.g. the PDH may have a unique peptide but not sufficient to be promoted as, for example, a leading protein of another a PAG. |
Table 2.
A summary of grouping options and recommendation for CV term annotations, assuming a group of four related proteins A-D.
| Scenario | Software preference | Encoding |
|---|---|---|
| Software scores A and B as same-set, C and D as subset. | Software wishes to make A the group representative (arbitrary) |
|
| As above | Software does not wish to choose which is the group representative |
|
| Software scores A as best protein, B, C and D are all subset or subsumed | N/A |
|
| Software scores all four proteins as same-set or more generally as having equal evidence | Software wishes to make A the group representative (arbitrary) |
|
| As above | Software does not wish to choose which is the group representative |
|
| Software scores A as having slightly more evidence than B. B has additional weak independent evidence relative to A. C and D have less evidence than either A or B. | Software wishes to assign A as the leading protein and the independent evidence for B is not sufficient for it to form a new PAG. |
|
| As above | Software does not wish to choose which is the leading protein out of A and B or group representative |
|
| As above | Software does not wish to choose which is the leading protein but does select a group representative |
|
These guidelines have been developed as a consensus of opinion from the creators of protein inference tools, and we believe they can accommodate all currently known approaches – including those that are spectrum or peptide-based, statistical and/or set-based, those that include only confidently identified PSMs or those that take evidence from weakly identified PSMs.
The CV terms and mapping into mzIdentML described have been added to the mzIdentML specification document (version 1.2 candidate) – standardization process described in [26]. The semantic validation software has been updated to encode these rules and report errors (“MUST” rule), warnings (“SHOULD” rule) or informational messages (“MAY” rule) [27]. We have also started collecting information describing how concepts from a number of different protein inference packages map onto the terminology described here (http://www.psidev.info/mzidentml#mzid12 – link “Rosetta”). A set of example files is available from the project website. The example files can be visualized using the ProteoIDViewer software [25], which has been updated to support the new specifications (available from: http://code.google.com/p/mzidentml-viewer/). Several examples have been generated by different protein inference tools from the same artificially constructed set of spectra, known to produce grouping and clustering scenarios when searched against databases containing more or less redundancy, thus ensuring we have standardized example files that test the full range of biological conditions that might exist and different software approaches.
A number of other issues have been identified since the release of the stable mzIdentML 1.1 in 2011, which will also be resolved in the release of mzIdentML 1.2. These include explicit support for approaches using multiple database search engines; and approaches where multiple MS analyses originating from separation of the same sample (e.g. fractionation) are combined in a single database search or the protein inference stage. We have also improved support for capturing peptide identifications from de novo sequencing approaches and for experiments where statistical analysis was performed at the peptide-level, removing redundant PSMs reporting on the same peptide unit. Details are available in the new specification document: http://code.google.com/p/psi-pi/source/browse/trunk/specification_document/specdoc1_2/ and will be described fully in a separate publication.
The PSI will continue to support mzIdentML 1.1 for the foreseeable future (for example the mzIdentML 1.1 validator will remain in general use), and it is expected that both mzIdentML 1.1 and 1.2 should be supported by importing software and databases. New export software will only be expected to create mzIdentML 1.2 however, and over time we expect software packages exporting mzIdentML currently to move over to the new guidelines.
Concluding remarks
In this work, we have described a standardized terminology for use with the mzIdentML data standard for reporting protein identification results in a standard way. The new guidelines are released as a new version (1.2) of the standard. We anticipate that the mechanism described here for reporting protein grouping results will improve capabilities for multi-site collaborations, comparisons between different approaches and consistent import of data into public repositories, such as PRIDE [28] and other members of the ProteomeXchange Consortium [29]
Acknowledgments
ARJ would like to acknowledge funding from BBSRC [BB/K01997X/1, BB/K004123/1] and ARJ/JAV [BB/I00095X/1, BB/K01997X/1]. ARJ, JAV and PAB would like to acknowledge funding from EU FP7 grant ProteomeXchange (grant no. 260558). JAV is also funded by the Wellcome Trust [grant numbers WT085949MA and WT101477MA]. PAB is part of the Swiss-Prot group, which activities are supported by the Swiss Federal Government through the Federal Office of Education and Science and by the National Institutes of Health (NIH) grant 1 U41 HG006104-01. JU and ME would like to acknowledge funding by PURE (Protein Unit for Research in Europe, Az.: 131/1.08-031), a project of North Rhine-Westphalia, Germany. RJC is supported by NIH NIGMS grant 8P41GM103481. TF is supported by federal funds from the National Institutes of Health-National Human Genome Research Institute (grant No. 1RC2-HG005805-01), The National Science Foundation MRI (grant No. 0923536) and the National Institute of General Medical Sciences (grant No. R01 GM087221).
Abbreviations
- ABRF
Association of Biomolecular Resource Facilities
- CV
Controlled Vocabulary
- iPRG
Proteome Informatics Research Group
- MIAPE
Minimum Information About a Proteomics Experiment
- PAG
Protein Ambiguity Group
- PDH
Protein Detection Hypothesis
- PRIDE
PRoteomics IDEntifications (database)
- PSI
Proteomics Standards Initiative
- PSM
Peptide Spectrum Match
- SNP
Single Nucleotide Polymorphism
- XSD
XML Schema Document
Footnotes
Conflict of interest statement
The authors have declared no conflict of interest.
References
- 1.Smith LM, Kelleher NL. roteoform: a single term describing protein complexity. Nat Meth. 2013;10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koskinen VR, Emery PA, Creasy DM, Cottrell JS. Hierarchical Clustering of Shotgun Proteomics Data. Molecular & Cellular Proteomics. 2011;10 doi: 10.1074/mcp.M110.003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 4.Searle BC. Scaffold: A bioinformatic tool for validating MS/MS-based proteomic studies. PROTEOMICS. 2010;10:1265–1269. doi: 10.1002/pmic.200900437. [DOI] [PubMed] [Google Scholar]
- 5.Slotta DJ, McFarland MA, Markey SP. MassSieve: Panning MS/MS peptide data for proteins. PROTEOMICS. 2010;10:3035–3039. doi: 10.1002/pmic.200900370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabb DL, McDonald WH, Yates JR. DTASelect and Contrast: Tools for Assembling and Comparing Protein Identifications from Shotgun Proteomics. Journal of Proteome Research. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang X, Dondeti V, Dezube R, Maynard DM, et al. DBParser: Web-Based Software for Shotgun Proteomic Data Analyses. Journal of Proteome Research. 2004;3:1002–1008. doi: 10.1021/pr049920x. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B, Chambers MC, Tabb DL. Proteomic Parsimony through Bipartite Graph Analysis Improves Accuracy and Transparency. Journal of Proteome Research. 2007;6:3549–3557. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qeli E, Ahrens CH. PeptideClassifier for protein inference and targeted quantitative proteomics. Nat Biotech. 2010;28:647–650. doi: 10.1038/nbt0710-647. [DOI] [PubMed] [Google Scholar]
- 10.Bradshaw RA, Burlingame AL, Carr S, Aebersold R. Reporting Protein Identification Data. Molecular & Cellular Proteomics. 2006;5:787–788. doi: 10.1074/mcp.E600005-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Carr S, Aebersold R, Baldwin M, Burlingame A, et al. The Need for Guidelines in Publication of Peptide and Protein Identification Data: Working Group On Publication Guidelines For Peptide And Protein Identification Data. Mol Cell Proteomics. 2004;3:531–533. doi: 10.1074/mcp.T400006-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Wilkins M, Appel R, Van Eyk J, Chung M, et al. Guidelines for the next 10 years of proteomics. PROTEOMICS. 2006;6:4–8. doi: 10.1002/pmic.200500856. [DOI] [PubMed] [Google Scholar]
- 13.Nesvizhskii AI, Aebersold R. Interpretation of Shotgun Proteomic Data: The Protein Inference Problem. Molecular & Cellular Proteomics. 2005;4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.iPRG2008 results poster. http://www.abrf.org/ResearchGroups/ProteomicsInformaticsResearchGroup/EPosters/iPRG2008_InitialResultsPoster.pdf.
- 15.iPRG2008 Results presentation. http://www.abrf.org/ResearchGroups/ProteomicsInformaticsResearchGroup/EPosters/iPRG2008_InitialResultsOralPresentation.pdf.
- 16.Orchard S, Binz P-A, Jones AR, Vizcaino JA, et al. Preparing to Work with Big Data in Proteomics – A Report on the HUPO-PSI Spring Workshop. PROTEOMICS. 2013;13:2931–2937. doi: 10.1002/pmic.201370166. [DOI] [PubMed] [Google Scholar]
- 17.Taylor CF, Paton NW, Lilley KS, Binz P-A, et al. The minimum information about a proteomics experiment (MIAPE) Nat Biotech. 2007;25:887–893. doi: 10.1038/nbt1329. [DOI] [PubMed] [Google Scholar]
- 18.Martens L, Chambers M, Sturm M, Kessner D, et al. mzML—a Community Standard for Mass Spectrometry Data. Molecular & Cellular Proteomics. 2011;10:R110.000133. doi: 10.1074/mcp.R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones AR, Eisenacher M, Mayer G, Kohlbacher O, et al. The mzIdentML data standard for mass spectrometry-based proteomics results. Molecular & Cellular Proteomics. 2012;11:M111.014381. doi: 10.1074/mcp.M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walzer M, Qi D, Mayer G, Uszkoreit J, et al. The mzQuantML data standard for mass spectrometry-based quantitative studies in proteomics. Molecular & Cellular Proteomics. 2013 doi: 10.1074/mcp.O113.028506. mcp.O113.028506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griss J, Sachsenberg T, Walzer M, Gatto L, et al. The mzTab Data Exchange Format: communicating MS-based proteomics and metabolomics experimental results to a wider audience. Mol Cell Proteomics. 2014 doi: 10.1074/mcp.O113.036681. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montecchi-Palazzi L, Beavis R, Binz P-A, Chalkley RJ, et al. The PSI-MOD community standard for representation of protein modification data. Nat Biotech. 2008;26:864–866. doi: 10.1038/nbt0808-864. [DOI] [PubMed] [Google Scholar]
- 23.Creasy DM, Cottrell JS. Unimod: Protein modifications for mass spectrometry. PROTEOMICS. 2004;4:1534–1536. doi: 10.1002/pmic.200300744. [DOI] [PubMed] [Google Scholar]
- 24.Mayer G, Montecchi-Palazzi L, Ovelleiro D, Jones AR, et al. The HUPO proteomics standards initiative- mass spectrometry controlled vocabulary. Database 2013. 2013 doi: 10.1093/database/bat009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghali F, Krishna R, Lukasse P, Martínez-Bartolomé S, et al. Tools (Viewer, Library and Validator) that Facilitate Use of the Peptide and Protein Identification Standard Format, Termed mzIdentML. Molecular & Cellular Proteomics. 2013;12:3026–3035. doi: 10.1074/mcp.O113.029777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vizcaíno JA, Martens L, Hermjakob H, Julian RK, Paton NW. The PSI formal document process and its implementation on the PSI website. PROTEOMICS. 2007;7:2355–2357. doi: 10.1002/pmic.200700064. [DOI] [PubMed] [Google Scholar]
- 27.Montecchi-Palazzi L, Kerrien S, Reisinger F, Aranda B, et al. The PSI semantic validator: A framework to check MIAPE compliance of proteomics data. PROTEOMICS. 2009;9:5112–5119. doi: 10.1002/pmic.200900189. [DOI] [PubMed] [Google Scholar]
- 28.Vizcaíno JA, Côté R, Reisinger F, Barsnes H, et al. The Proteomics Identifications database: 2010 update. Nucleic acids research. 2010;38:D736–D742. doi: 10.1093/nar/gkp964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vizcaino JA, Deutsch EW, Wang R, Csordas A, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotech. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]