

Crotonaldehyde semicarbazone and crotonaldehyde thiosemicarbazone show the same E conformation around the imine C=N bond. Each molecule has an intramolecular N—H⋯N hydrogen bond, which generates an S(5) ring. Intermolecular N—H⋯O hydrogen bonds in the semicarbazone link the molecules into layers parallel to the bc plane, while weak intermolecular N—H⋯S hydrogen bonds in the thiosemicarbazone link the molecules into chains propagating in [110].
Keywords: crystal structure, crotonaldehyde, semicarbazone, thiosemicarbazone, powder X-ray diffraction, supramolecular structure, hydrogen bond, one-dimensional chain, two-dimensional networks
Abstract
Crotonaldehyde semicarbazone {systematic name: (E)-2-[(E)-but-2-en-1-ylidene]hydrazinecarboxamide}, C5H9N3O, (I), and crotonaldehyde thiosemicarbazone {systematic name: (E)-2-[(E)-but-2-en-1-yldene]hydrazinecarbothioamide}, C5H9N3S, (II), show the same E conformation around the imine C=N bond. Compounds (I) and (II) were obtained by the condensation of crotonaldehyde with semicarbazide hydrochloride and thiosemicarbazide, respectively. Each molecule has an intramolecular N—H⋯N hydrogen bond, which generates an S(5) ring. In (I), the crotonaldehyde fragment is twisted by 2.59 (5)° from the semicarbazide mean plane, while in (II) the corresponding angle (with the thiosemicarbazide mean plane) is 9.12 (5)°. The crystal packing is different in the two compounds: in (I) intermolecular N—H⋯O hydrogen bonds link the molecules into layers parallel to the bc plane, while weak intermolecular N—H⋯S hydrogen bonds in (II) link the molecules into chains propagating in [110].
Chemical context
The chemistry of semicarbazones and thiosemicarbazones is especially interesting due to their special role in biological applications such as anti-proliferative, anti-tumoral, anti-convulsant, anti-trypanosomal, herbicidal and biocidal activities (Beraldo et al., 2002 ▸; Kasuga et al., 2003 ▸; Teixeira et al., 2003 ▸; Beraldo & Gambino, 2004 ▸; Mikhaleva et al., 2008 ▸; de Oliveira et al., 2008 ▸; Alomar et al., 2012 ▸; Gan et al., 2014 ▸). They are also important intermediates in organic synthesis, mainly for obtaining heterocyclic rings, such as thiazolidones, oxadiazoles, pyrazolidones, and thiadiazoles (Greenbaum et al., 2004 ▸; Küçükgüzel et al., 2006 ▸). Semicarbazones and thiosemicarbazones have received considerable attention in view of their simplicity of preparation, various complexing abilities and coordination behavior that can be used in analytical applications (Garg & Jain, 1988 ▸; Casas et al., 2000 ▸). They are of interest from a supramolecular point of view since they can be functionalized to give different supramolecular arrays.
Structural commentary
Compounds (I) and (II) crystallize in centrosymmetric space groups P21/c and P
 , respectively, with one molecule in the asymmetric unit. Each molecule has an intramolecular N—H⋯N hydrogen bond (Tables 1 ▸ and 2 ▸), which forms an S(5) ring. The semicarbazone and thiosemicarbazone fragments in the compounds show an E conformation around the imine C=N bond. The molecules (Fig. 1 ▸) are approximately planar, with a dihedral angle of 2.59 (5)° between the C1/C2/C3 crotonaldehyde plane and the mean plane of the C4/N1/N2/C5/O1/N3 semicarbazone fragment for (I), and of 9.12 (5)° between the C1/C2/C3 crotonaldehyde plane and the mean plane of the C4/N1/N2/C5/S1/N3 thiosemicarbazone fragment for (II). All bond lengths and angles in (I) and (II) are normal and correspond well to those observed in the crystal structures of related semi- and thiosemicarbazone derivatives, viz. acetone semicarbazone and benzaldehydesemicarbazone (Naik & Palenik, 1974 ▸), 3,4- methylenedioxybenzaldehydesemicarbazone (Wang et al., 2004 ▸), isatin 3-semicarbazone and 1-methylisatin 3-semicarbazone (Pelosi et al., 2005 ▸), 4- (methylsulfanyl)benzaldehydethiosemicarbazone (Yathirajan et al., 2006 ▸), 4-(methylsulfanyl)benzaldehydesemicarbazone (Sarojini et al., 2007 ▸), 5-hydroxy-2-nitrobenzaldehyde thiosemicarbazone (Reddy et al., 2014 ▸) and 1-(4-formylbenzylidene) thiosemicarbazone (Carballo et al., 2014 ▸).
, respectively, with one molecule in the asymmetric unit. Each molecule has an intramolecular N—H⋯N hydrogen bond (Tables 1 ▸ and 2 ▸), which forms an S(5) ring. The semicarbazone and thiosemicarbazone fragments in the compounds show an E conformation around the imine C=N bond. The molecules (Fig. 1 ▸) are approximately planar, with a dihedral angle of 2.59 (5)° between the C1/C2/C3 crotonaldehyde plane and the mean plane of the C4/N1/N2/C5/O1/N3 semicarbazone fragment for (I), and of 9.12 (5)° between the C1/C2/C3 crotonaldehyde plane and the mean plane of the C4/N1/N2/C5/S1/N3 thiosemicarbazone fragment for (II). All bond lengths and angles in (I) and (II) are normal and correspond well to those observed in the crystal structures of related semi- and thiosemicarbazone derivatives, viz. acetone semicarbazone and benzaldehydesemicarbazone (Naik & Palenik, 1974 ▸), 3,4- methylenedioxybenzaldehydesemicarbazone (Wang et al., 2004 ▸), isatin 3-semicarbazone and 1-methylisatin 3-semicarbazone (Pelosi et al., 2005 ▸), 4- (methylsulfanyl)benzaldehydethiosemicarbazone (Yathirajan et al., 2006 ▸), 4-(methylsulfanyl)benzaldehydesemicarbazone (Sarojini et al., 2007 ▸), 5-hydroxy-2-nitrobenzaldehyde thiosemicarbazone (Reddy et al., 2014 ▸) and 1-(4-formylbenzylidene) thiosemicarbazone (Carballo et al., 2014 ▸).
Table 1. Hydrogen-bond geometry (, ) for (I) .
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N3H2N3N1 | 0.87 | 2.33 | 2.629(19) | 100 |
| N2H1N2O1i | 0.88 | 2.07 | 2.910(11) | 158 |
| N3H1N3O1ii | 0.91 | 2.04 | 2.914(18) | 162 |
Symmetry codes: (i) 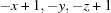 ; (ii)
; (ii)  .
.
Table 2. Hydrogen-bond geometry (, ) for (II) .
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N3H2N3N1 | 0.89 | 2.17 | 2.641(14) | 112 |
| N2H1N2S1i | 0.86 | 2.83 | 3.473(11) | 133 |
| N3H1N3S1ii | 0.87 | 2.77 | 3.398(11) | 130 |
Symmetry codes: (i)  ; (ii)
; (ii)  .
.
Figure 1.

The molecular structures of (a) (I) and (b) (II), showing the atom-labelling schemes. Displacement spheres (and the ellipsoid for S1) are drawn at the 50% probability level.
Supramolecular features
As a result of the presence of potential hydrogen-donor sites in molecules (I) and (II), supramolecular hydrogen-bonding interactions are observed in both compounds (Tables 1 ▸ and 2 ▸). In the crystal of (I), molecules are linked by pairs of N—H⋯O hydrogen bonds, forming inversion dimers with R22(8) ring motifs (Fig. 2 ▸ a). The resulting dimers are connected through N—H⋯O hydrogen bonds, forming layers parallel to bc plane. In the crystal of (II), molecules are linked by weak N—H⋯S hydrogen bonds, forming chains propagating in [110] (Fig. 2 ▸ b).
Figure 2.

(a) A portion of the crystal packing of (I) viewed down the b axis (parallel to the hydrogen-bonded layer). (b) A portion of the crystal packing of (II), showing the hydrogen-bonded chain of the molecules. Thin dotted lines denote intermolecular hydrogen bonds.
Synthesis and crystallization
All reactions and manipulations were carried out in air with reagent grade solvents. The IR spectra were recorded on a Jasco FT–IR 300E instrument. 1H and 13C{1H} NMR spectra were recorded on a Bruker Bio spin 400 spectrometer. Microanalysis was performed using EURO EA. Powder X-ray diffraction data were collected with Stoe Transmission diffractometer (Stadi P).
For the synthesis of (I), a mixture of semicarbazide hydrochloride (CH5N3O·HCl; 0.5 g, 4.5 mmol) and sodium acetate (CH3COONa; 0.75 g, 9.1 mmol) in 10 ml water was agitated well and crotonaldehyde (0.5 g, 7.1 mmol) was added slowly with stirring. On completion of the addition, the reaction mixture was agitated for 24 h at room temperature. The solid product which formed was separated by filtration and washed with water and finally recrystallized from absolute ethanol to produce the product (I) (white powder; m.p. 481–482 K) in 55.5% yield.
IR (KBr, ν, cm−1): 3456, 3281, 3192 (NH2), (1668–1638) (C=O); 1H NMR (400 MHz, CD3OD) δ p.p.m. 1.76 (d, J = 4.42 Hz, 3H, –CH3), 6.43–5.46 (m, 2H, –HC=CH–), 7.39 (d, J = 7.19 Hz, 1H, HC=N–).13C NMR (100 MHz, CD3OD) δ p.p.m. 18.52 (CH3), 130.01 (–HC=CH–), 137.62 (–HC=CH–), 145.64 (N=C), 160.19 (C=O). Analysis calculated for (I): C, 47.23; H, 7.13; N, 33.05, 12.58 O%. Found: C, 46.43; H, 6.08; N, 34.69%
For the synthesis of (II), crotonaldehyde (0.5 g, 7.1 mmol) was added to thiosemicarbazide (CH5N3S; 0.65 g, 7.1 mmol) in 5 ml water and the mixture was stirred at room temperature for 24 h. The product was separated by filtration and recrystallized from absolute ethanol to produce the product (II) (white powder; m.p. 435–436 K) in 72.5% yield.
IR (KBr, ν, cm−1): 3323, 3244, 3164 (NH2), 1650(C=S). 1H NMR (400 MHz, CDCl3) δ p.p.m. 1.90 (d, J = 5.86 Hz, 3H, –CH3), 6.07–6.27 (m, 2H, –HC=CH–), 6.49 (sb, 1H), 7.10 (sb, 1H) 7.60 (d, J = 8.57 Hz, 1H, HC=N–), 10.10 (sb, 2H). 13C NMR (100.6 MHz, CDCl3) 18.73 (CH3), 127.70 (–HC=CH–), 140.58 (–HC=CH–), 146.21 (N=C), 177.95 (C=S). Analysis calculated for (II): C, 41.93; H, 6.33; N, 29.34.05, 22.39 S%. Found: C, 41.89; H, 6.25; N, 31.88%.
Refinement details
Crystal data, data collection and structure refinement details are summarized in Table 3 ▸. Compounds (I) and (II) crystallized in the form of a very fine white powder. Since no single crystals of sufficient size and quality could be obtained, the crystal structures of both compounds were determined from X-ray powder diffraction patterns. The powder samples of (I) and (II) were lightly ground in a mortar, loaded into two Mylar foils and fixed onto the sample holder with a mask of suitable internal diameter (8.0 mm). The powder X-ray diffraction data were collected at room temperature with a STOE transmission STADI-P diffractometer using monochromatic Cu Ka
1 radiation (λ= 1.54060 Å) selected with an incident beam curved-crystal germanium Ge(111) monochromator with a linear position-sensitive detector (PSD). The patterns were scanned over the angular range 5.0–80.0° (2θ). For pattern indexing, the extraction of the peak positions was carried out with the program WinPLOTR (Roisnel & Rodríguez-Carvajal, 2000 ▸). Pattern indexing was performed with the program DICVOL4.0 (Boultif & Louër, 2004 ▸). The first 20 lines of the powder pattern were indexed completely on the basis of a monoclinic cell for (I) and a triclinic cell for (II). The figures of merit (de Wolff et al., 1968 ▸; Smith & Snyder, 1979 ▸) are sufficiently acceptable to support the obtained indexing results [M(20) = 50.5, F(20) = 71.9 (0.0034, 83)] for (I) and [M(20) = 61.8, F(20) = 96.0 (0.0051, 41)] for (II). The best estimated space groups were P21/c in the monoclinic system for (I) and P
in the triclinic system for (II).
Table 3. Experimental details.
| (I) | (II) | |
|---|---|---|
| Crystal data | ||
| Chemical formula | C5H9N3O | C5H9N3S |
| M r | 127.15 | 143.21 |
| Crystal system, space group | Monoclinic, P21/c | Triclinic, P
|
| Temperature (K) | 298 | 298 |
| a, b, c () | 11.1646(3), 5.13891(9), 13.0301(2) | 5.86650(17), 8.0313(2), 9.0795(4) |
| , , () | 90, 112.3496(11), 90 | 104.1407(18), 101.0403(19), 106.3511(17) |
| V (3) | 691.43(3) | 382.15(2) |
| Z | 4 | 2 |
| Radiation type | Cu K 1, = 1.5406 | Cu K 1, = 1.5406 |
| (mm1) | 0.74 | 3.11 |
| Specimen shape, size (mm) | Flat sheet, 8 8 | Flat sheet, 8 8 |
| Data collection | ||
| Diffractometer | Stoe transmission Stadi-P | Stoe transmission Stadi-P |
| Specimen mounting | Powder loaded into two Mylar foils | Powder loaded into two Mylar foils |
| Data collection mode | Transmission | Transmission |
| Scan method | Step | Step |
| 2 values () | 2min = 5 2max = 80 2step = 0.02 | 2min = 4.980 2max = 79.960 2step = 0.02 |
| Refinement | ||
| R factors and goodness of fit | R p = 0.027, R wp = 0.036, R exp = 0.029, R(F 2) = 0.02795, 2 = 1.613 | R p = 0.033, R wp = 0.043, R exp = 0.034, R(F 2) = 0.02670, 2 = 1.664 |
| No. of data points | 3750 | 3750 |
| No. of parameters | 121 | 114 |
| No. of restraints | 0 | 1 |
| H-atom treatment | H-atom parameters not refined | H-atom parameters not refined |
The whole powder diffraction patterns from 5 to 80° (2θ) for the two compounds (I) and (II) were subsequently refined with cell and resolution constraints (Le Bail et al., 1988 ▸) using the profile-matching option of the program FULLPROF (Rodríguez-Carvajal, 2001 ▸). The number of molecules per unit cell was estimated to be Z = 4 for (I) and Z = 2 for (II). The initial crystal structures for (I) and (II) were determined by direct methods using the program EXPO2014 (Altomare et al., 2013 ▸). The models found by this program were introduced into the program GSAS (Larson & Von Dreele, 2004 ▸) implemented in EXPGUI (Toby, 2001 ▸) for Rietveld refinement. During the Rietveld refinements, the background was refined using a shifted Chebyshev polynomial with 20 coefficients. The effect of asymmetry of low-order peaks was corrected using a pseudo-Voigt description of the peak shape (Thompson et al., 1987 ▸), which allows for angle-dependent asymmetry with axial divergence (Finger et al., 1994 ▸) and microstrain broadening, as described by Stephens (1999 ▸). The two asymmetry parameters of this function, S/L and D/L, were both fixed at 0.022 during this refinement. Intensities were corrected from absorption effects with a function for a plate sample in transmission geometry with a μ·d value of 0.15 for (I) and 0.72 for (II) (μ is the absorption coefficient and d is the sample thickness). These μ·d values were determined experimentally.
Before the final refinement, all H atoms were introduced in geometrically calculated positions. The coordinates of these H atoms were refined with strict restraints on bond lengths and angles until a suitable geometry was obtained, after that they were fixed in the final stage of the refinement. No soft restraints were imposed for (I), while for (II) the CH3—CH bond was clearly stretched (close to 1.6 Å), therefore a single soft restraint was carried out to obtain a normal value (1.49 Å). The final refinement cycles were performed using isotropic atomic displacement parameters for the C, N and O atoms, an anisotropic atomic displacement parameter for S atom in (II) and a fixed global isotropic atomic displacement parameter for the H atoms. The preferred orientation was modelled with 12 coefficients using a spherical harmonics correction (Von Dreele, 1997 ▸) of intensities in the final refinement. The use of the preferred orientation correction leads to a better molecular geometry with better agreement factors. The final Rietveld plots of the X-ray diffraction patterns for both (I) and (II) are given in Fig. 3 ▸.
Figure 3.

The final Rietveld plots for (a) (I) and (b) (II). Experimental intensities are indicated by dots and the best-fit profile (upper trace) and difference pattern (lower trace) are shown as solid lines. The vertical bars indicate the calculated positions of the Bragg peaks.
Supplementary Material
Crystal structure: contains datablock(s) CROTON-CZ_Publ, I, II. DOI: 10.1107/S2056989015000663/cv5481sup1.cif
Rietveld powder data: contains datablock(s) I. DOI: 10.1107/S2056989015000663/cv5481Isup2.rtv
Rietveld powder data: contains datablock(s) II. DOI: 10.1107/S2056989015000663/cv5481IIsup3.rtv
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors thank Professor I. Othman, Director General, and Professor Z. Ajji, Head of the Chemistry Department, for their support and encouragement during this work. We also thank Miss D. Naima for her kind assistance.
supplementary crystallographic information
Crystal data
| C5H9N3S | V = 382.15 (2) Å3 |
| Mr = 143.21 | Z = 2 |
| Triclinic, P1 | F(000) = 152 |
| Hall symbol: -P 1 | Dx = 1.245 Mg m−3 |
| a = 5.86650 (17) Å | Cu Kα1 radiation, λ = 1.5406 Å |
| b = 8.0313 (2) Å | µ = 3.11 mm−1 |
| c = 9.0795 (4) Å | T = 298 K |
| α = 104.1407 (18)° | Particle morphology: fine powder |
| β = 101.0403 (19)° | white |
| γ = 106.3511 (17)° | flat sheet, 8 × 8 mm |
Data collection
| Stoe transmission Stadi-P diffractometer | Data collection mode: transmission |
| Radiation source: sealed X-ray tube | Scan method: step |
| Ge 111 monochromator | 2θmin = 4.980°, 2θmax = 79.960°, 2θstep = 0.02° |
| Specimen mounting: Powder loaded into two Mylar foils |
Refinement
| Least-squares matrix: full | Profile function: CW Profile function number 4 with 21 terms Pseudovoigt profile coefficients as parameterized in (Thompson et al., 1987) Asymmetry correction of Finger et al., 1994. #1(GU) = 0.000 #2(GV) = 0.000 #3(GW) = 2.793 #4(GP) = 0.000 #5(LX) = 5.477 #6(ptec) = 2.45 #7(trns) = 0.00 #8(shft) = 0.0000 #9(sfec) = 0.00 #10(S/L) = 0.0220 #11(H/L) = 0.0220 #12(eta) = 0.6000 Peak tails are ignored where the intensity is below 0.0010 times the peak Aniso. broadening axis 0.0 0.0 1.0 |
| Rp = 0.033 | 114 parameters |
| Rwp = 0.043 | 1 restraint |
| Rexp = 0.034 | H-atom parameters not refined |
| R(F2) = 0.02670 | (Δ/σ)max = 0.03 |
| χ2 = 1.664 | Background function: GSAS Background function number 1 with 20 terms. Shifted Chebyshev function of 1st kind 1: 590.360 2: -469.557 3: 198.126 4: -45.2586 5: -2.75624 6: 13.8508 7: 4.35563 8: -5.95029 9: -12.8815 10: 35.6051 11: -12.9276 12: -11.1488 13: 8.85293 14: -2.01034 15: -0.496121 16: 8.39616 17: -2.33367 18: -5.14527 19: 10.5079 20: -3.85249 |
| 3750 data points |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.184 (2) | 0.841 (2) | 0.515 (2) | 0.103 (6)* | |
| H1A | 0.15342 | 0.79934 | 0.39748 | 0.12* | |
| H1B | 0.2323 | 0.97016 | 0.55142 | 0.12* | |
| H1C | 0.02574 | 0.78525 | 0.53491 | 0.12* | |
| C2 | 0.370 (2) | 0.7688 (17) | 0.5865 (18) | 0.054 (5)* | |
| H2 | 0.53963 | 0.83335 | 0.59455 | 0.055* | |
| C3 | 0.325 (2) | 0.6393 (16) | 0.6524 (15) | 0.034 (5)* | |
| H3 | 0.14582 | 0.56255 | 0.63049 | 0.055* | |
| C4 | 0.487 (3) | 0.5747 (19) | 0.7264 (19) | 0.039 (5)* | |
| H4 | 0.66632 | 0.65671 | 0.74816 | 0.055* | |
| N1 | 0.4514 (17) | 0.4461 (12) | 0.7878 (15) | 0.035 (4)* | |
| N2 | 0.6486 (16) | 0.4005 (12) | 0.8462 (13) | 0.021 (4)* | |
| H1n2 | 0.79218 | 0.45838 | 0.841 | 0.05* | |
| C5 | 0.611 (3) | 0.2572 (16) | 0.907 (2) | 0.034 (4)* | |
| N3 | 0.3681 (15) | 0.1560 (12) | 0.8849 (13) | 0.017 (4)* | |
| H1n3 | 0.34725 | 0.13246 | 0.97116 | 0.05* | |
| H2n3 | 0.26401 | 0.20773 | 0.84645 | 0.05* | |
| S1 | 0.8446 (6) | 0.1980 (5) | 0.9772 (6) | 0.04081 |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.032 (4) | 0.039 (5) | 0.082 (8) | 0.026 (4) | 0.043 (5) | 0.035 (5) |
Geometric parameters (Å, º)
| C1—H1A | 0.999 | C4—N1 | 1.274 (12) |
| C1—H1B | 0.946 | N1—N2 | 1.361 (10) |
| C1—H1C | 0.983 | N2—H1n2 | 0.856 |
| C1—C2 | 1.49 (2) | N2—C5 | 1.377 (13) |
| C2—H2 | 0.963 | C5—N3 | 1.376 (13) |
| C2—C3 | 1.311 (13) | C5—S1 | 1.638 (13) |
| C3—H3 | 1.008 | N3—C5 | 1.376 (13) |
| C3—C4 | 1.352 (14) | N3—H1n3 | 0.872 |
| C4—H4 | 1.024 | N3—H2n3 | 0.894 |
| H1A—C1—H1B | 109.0 | C3—C4—N1 | 130.8 (16) |
| H1A—C1—H1C | 106.2 | H4—C4—N1 | 116.8 |
| H1A—C1—C2 | 108.5 | C4—N1—N2 | 118.6 (11) |
| H1B—C1—H1C | 110.2 | N1—N2—H1n2 | 119.6 |
| H1B—C1—C2 | 113.8 | N1—N2—C5 | 119.2 (10) |
| H1C—C1—C2 | 108.8 | H1n2—N2—C5 | 121.2 |
| C1—C2—H2 | 115.8 | N2—C5—N3 | 115.6 (12) |
| C1—C2—C3 | 125.6 (13) | N2—C5—S1 | 120.4 (11) |
| H2—C2—C3 | 118.2 | N3—C5—S1 | 123.5 (9) |
| C2—C3—H3 | 116.8 | C5—N3—H1n3 | 110.5 |
| C2—C3—C4 | 128.4 (15) | C5—N3—H2n3 | 112.2 |
| H3—C3—C4 | 113.9 | H1n3—N3—H2n3 | 113.2 |
| C3—C4—H4 | 111.8 | ||
| C4—N1—N2—C5 | −177.4 (14) | N1—N2—C5—N3 | 8.0 (19) |
| N2—N1—C4—C3 | 175.6 (15) | C1—C2—C3—C4 | −176.2 (15) |
| N1—N2—C5—S1 | 179.6 (11) | C2—C3—C4—N1 | −177.6 (16) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N3—H2N3···N1 | 0.89 | 2.17 | 2.641 (14) | 112 |
| N2—H1N2···S1i | 0.86 | 2.83 | 3.473 (11) | 133 |
| N3—H1N3···S1ii | 0.87 | 2.77 | 3.398 (11) | 130 |
Symmetry codes: (i) −x+2, −y+1, −z+2; (ii) −x+1, −y, −z+2.
References
- Alomar, K., Gaumet, V., Allain, M., Bouet, G. & Landreau, A. (2012). J. Inorg. Biochem. 115, 36–43. [DOI] [PubMed]
- Altomare, A., Cuocci, C., Giacovazzo, C., Moliterni, A., Rizzi, R., Corriero, N. & Falcicchio, A. (2013). J. Appl. Cryst. 46, 1231–1235.
- Beraldo, H. & Gambinob, D. (2004). Mini Rev. Med. Chem. 4, 31–39. [DOI] [PubMed]
- Beraldo, H., Sinisterra, R. D., Teixeira, L. R., Vieira, R. P. & Doretto, M. C. (2002). Biochem. Biophys. Res. Commun. 296, 241–246. [DOI] [PubMed]
- Boultif, A. & Louër, D. (2004). J. Appl. Cryst. 37, 724–731.
- Carballo, R., Pino-Cuevas, A. & Vázquez-López, E. M. (2014). Acta Cryst. E70, o970. [DOI] [PMC free article] [PubMed]
- Casas, J. S., Garc\?ía-Tasende, M. S. & Sordo, J. (2000). Coord. Chem. Rev. 209, 197–261.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Finger, L. W., Cox, D. E. & Jephcoat, A. P. (1994). J. Appl. Cryst. 27, 892–900.
- Gan, C., Cui, J., Su, S., Lin, Q., Jia, L., Fan, L. & Huang, Y. (2014). Steroids, 87, 99–107. [DOI] [PubMed]
- Garg, B. S. & Jain, V. K. (1988). Microchem. J. 38, 144–169.
- Greenbaum, D. C., Mackey, Z., Hansell, E., Doyle, P. S., Gut, J., Caffrey, C. R., Lehrman, J., Rosenthal, P. J., McKerrow, J. H. & Chibale, K. (2004). J. Med. Chem. 47, 3212–3219. [DOI] [PubMed]
- Kasuga, N. C., Sekino, K., Ishikawa, M., Honda, A., Yokoyama, M., Nakano, S., Shimada, N., Koumo, C. & Nomiya, K. (2003). J. Inorg. Biochem. 96, 298–310. [DOI] [PubMed]
- Küçükgüzel, G., Kocatepe, A., De Clercq, E., Şahin, F. & Güllüce, M. (2006). Eur. J. Med. Chem. 41, 353–359. [DOI] [PubMed]
- Larson, A. C. & Von Dreele, R. B. (2004). GSAS. Report LAUR 86-748. Los Alamos National Laboratory, New Mexico, USA.
- Le Bail, A., Duroy, H. & Fourquet, J. L. (1988). Mater. Res. Bull. 23, 447–452.
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst. 39, 453–457.
- Mikhaleva, A. I., Ivanov, A. V., Vasil’tsov, A. M., Ushakov, I. A., Ma, J. S. & Yang, G. (2008). Chem. Heterocycl. Compd, 44, 1117–1122.
- Naik, D. V. & Palenik, G. J. (1974). Acta Cryst. B30, 2396–2401.
- Oliveira, R. B. de, de Souza-Fagundes, E. M., Soares, R. P. P., Andrade, A. A., Krettli, A. U. & Zani, C. L. (2008). Eur. J. Med. Chem. 43, 1983–1988. [DOI] [PubMed]
- Pelosi, G., Pelizzi, C., Belicchi Ferrari, M., Rodríguez-Argüelles, M. C., Vieito, C. & Sanmartín, J. (2005). Acta Cryst. C61, o589–o592. [DOI] [PubMed]
- Reddy, M. S., Sarala, Y., Jagadeesh, M., Das, S. K. & Ammireddy, V. R. (2014). Acta Cryst. E70, o846. [DOI] [PMC free article] [PubMed]
- Rodríguez-Carvajal, J. (2001). Commission on Powder Diffraction (IUCr) Newsletter, 26, 12–19.
- Roisnel, T. & Rodríguez-Carvajal, J. (2000). Mater. Sci. Forum, 378–381, 118–123.
- Sarojini, B. K., Narayana, B., Bindya, S., Yathirajan, H. S. & Bolte, M. (2007). Acta Cryst. E63, o2946.
- Smith, G. S. & Snyder, R. L. (1979). J. Appl. Cryst. 12, 60–65.
- Stephens, P. W. (1999). J. Appl. Cryst. 32, 281–289.
- Stoe & Cie (1999). WinXPOW. Stoe & Cie, Darmstadt, Germany.
- Teixeira, L. R., Sinisterra, R. D., Vieira, R. P., Doretto, M. C. & Beraldo, H. (2003). J. Incl. Phenom. Macrocycl. Chem. 47, 77–82.
- Thompson, P., Cox, D. E. & Hastings, J. B. (1987). J. Appl. Cryst. 20, 79–83.
- Toby, B. H. (2001). J. Appl. Cryst. 34, 210–213.
- Von Dreele, R. B. (1997). J. Appl. Cryst. 30, 517–525.
- Wang, J.-L., Jia, Y.-J. & Yu, M. (2004). Acta Cryst. E60, o662–o663.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Wolff, P. M. de (1968). J. Appl. Cryst. 1, 108–113.
- Yathirajan, H. S., Bindya, S., Narayana, B., Sarojini, B. K. & Bolte, M. (2006). Acta Cryst. E62, o5925–o5926.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) CROTON-CZ_Publ, I, II. DOI: 10.1107/S2056989015000663/cv5481sup1.cif
Rietveld powder data: contains datablock(s) I. DOI: 10.1107/S2056989015000663/cv5481Isup2.rtv
Rietveld powder data: contains datablock(s) II. DOI: 10.1107/S2056989015000663/cv5481IIsup3.rtv
Additional supporting information: crystallographic information; 3D view; checkCIF report