

Abstract
As a major component of the extracellular matrix, proteoglycans influence the mechanical properties of connective tissue and play an important role in cell-cell and cell-matrix interactions. Genetic defects of proteoglycan biosynthesis lead to multi-system disorders, often most prominently affecting the skeletal system and skin. Specific deficiencies in the enzymes involved in the biosynthesis of the linkage region between the core of the proteoglycan protein and its glycosaminoglycan side chains are known as linkeropathies. We report on a patient from a second family with a homozygous c.830 G>A (p.Arg277Gln) mutation in the B3GAT3 gene. The clinical features expand the previously reported phenotype of B3GAT3 mutations and of linkeropathies in general. This patient has short stature, facial dysmorphisms, skeletal findings, joint laxity, and cardiac manifestations similar to those previously associated with B3GAT3 mutations. However, he also has developmental delay, a visual refractory defect, dental defects, pectus carinatum, and skin abnormalities that have only been associated with linkeropathies caused by mutations in B4GALT6 and B4GALT7. He has bilateral inguinal hernias and atlanto-axial as well as atlanto-occipital instability that have not been previously associated with B3GAT3 mutations. We provide a detailed clinical report and a comparative overview of the phenotypic features of the linkeropathies caused by mutations in B3GAT3, B4GALT6 and B4GALT7.
Keywords: B3GAT3, proteoglycan disorder, linkeropathy, Larsen-like syndrome
INTRODUCTION
Proteoglycans are cell surface molecules and major components of the extracellular matrix. They are major factors in determining the mechanical properties of connective tissue, and they play an important role in the regulation of cell-cell and cell-matrix interaction. They interact with major signaling pathways, influencing developmental processes and regulating cell adhesion, motility, proliferation, differentiation and morphogenesis [Perrimon and Bernfield, 2000; Häcker et al., 2005; Bernfield et al., 1999; Bishop et al., 2007; Sugahara, 2003; Haltiwanger and Lowe, 2004]. Defects in proteoglycan biosynthesis lead to multi-systemic disorders, often most prominently affecting the skeletal system and skin [Bishop et al., 2007; Mizumoto et al., 2013; Sugahara and Mikami, 2007].
The basic structure of each proteoglycan consists of a core protein and specific glycosaminoglycan (GAG) side chains that attach to a serine residue via a tetrasaccharide linkage region. This linkage region is common to all GAGs and usually consists of one xylose, two galactose and one glucuronic acid (GlcUA-Gal-Gal-Xyl-). The remainder GAG polysaccharide chain contains variable numbers of disaccharide repeats: Uronic acid (GlcUA) is paired with N-acetylgalatcosamine (GalNac) or with N-acetylglucosamine (GlcNac) to form Chondroitin/Dermatan or Heparan, respectively. Sulfatation and epimerization of uronic acid residues further modify and diversify the properties of the GAGs (Häcker et al., 2005).
The specific enzymes that catalyze each of the steps in GAG biosynthesis have been well characterized, including the enzymes synthesizing the linkage region and their respective genes: xylosyltransferases 1 and 2 (XYLT1, XYLT2), galactosyltransferase I (B4GALT7) and II (B3GALT6), and glucuronosyltransferase I (B3GAT3). Defects in these linkage enzyme genes, also known as linkeropathies [Nakajima et al., 2013], have so far been described for all but XYLT1 and XYLT2. Four patients from three families have been confirmed to have a mutation in B4GALT7 (OMIM# 130070) [Kresse et al., 1987; Quentin et al., 1990; Faiyaz-Ul-Haque et al., 2004; Guo et al., 2013]. Only recently, ten new pathogenic mutations in the B3GALT6 (OMIM# 615291) were described in 16 patients from multiple families [Malfait et al., 2013; Nakajima et al., 2013]. Thus far, there has only been a single case series of B3GAT3 deficiency in 5 siblings from a consanguineous family in the United Arab Emirates with a Larsen-like syndrome (OMIM# 245600) [Baasanjav et al., 2011].
We report on a patient from a second consanguineous family from Sharjah, United Arab Emirates, with the same homozygous mutation in the B3GAT3 gene that was previously reported. Our clinical report broadens the phenotype associated with this gene defect. We also provide an overview of the overlapping and distinctive features of each of the linkeropathies that has been described, outlining similarities and differences.
CLINICAL REPORT
The patient was a 5-year-old boy who was born at full term to consanguineous parents from Sharjah, United Arab Emirates. The pregnancy was uncomplicated, and he was delivered by cesarean secondary to fetal distress. Birth weight was 2.7 kg (7th centile); birth length was unknown, but parents recalled that he was ‘short’. Shortly after birth, dysmorphic physical features were noted, along with bilateral hip and elbow dislocations which were treated conservatively. A right inguinal hernia was surgically corrected. During early childhood, failure to thrive and short stature were observed. Development was mildly delayed. He sat independently at age 9 months, walked at 2–3 years, spoke his first words between ages 2–3 years and was speaking in 3–4 word sentences at age 4 years. Visual correction was provided for hyperopia, esotropia, and right sided amblyopia. An echocardiogram obtained at age 9 months was reported as normal. A brain MRI obtained at age 22 months showed generalized cortical atrophy with a partially empty sella, but the infundibulum was present.
Family history was notable for the parents being first cousins through their mothers and second cousins through their fathers. The patient’s mother had had a previous male stillbirth at 6 months’ gestation due to preeclampsia and a spontaneous miscarriage of a male fetus at 17 weeks’ gestation. She also had a question of a thyroid disorder. The patient’s father and 14-month-old brother were healthy.
On physical examination, height was 92.5 cm (−3.43 SDS), weight was 13.3 kg, (−2.91 SDS), body mass index was 15.5 kg/m2 (0.06 SDS), and head circumference was 49.5 cm (approximately 10th centile [Rollins et al., 2010]). His lower segment was 40 cm with an upper to lower segment ratio of 1:1.3. We could not measure the arm span given a limited range of motion of the elbows. Father’s height was 177 cm, Mother’s height was 160cm, and mid-parental height was 175 cm (25–50th centile). He was brachycephalic with a flat occiput. He had a broad and relatively tall forehead, a flat face, mild bilateral proptosis, and a short nose with a depressed nasal bridge and a broad nasal tip (Fig 1A and B). His mouth was slightly small. His ears were low-set and the crus helix and inferior crus of the antihelix of his left ear were underdeveloped (Fig 1A and B, and Supplementary Figures 1H and I – see supporting information online). He had a short neck and low posterior hairline (Supplementary Figure 1G – see supporting information online). There were extensive cavities on his small-appearing primary teeth. Chest examination revealed significant pectus carinatum with asymmetry and protrusion of the sternum toward the right (Fig 1F). No scoliosis or kyphosis was observed on clinical examination. The cardiovascular, respiratory and abdominal examinations were normal. The genitourinary exam revealed prepubertal, circumcised genitalia with normal phallus and descended testicles. A visible bulge in the left inguinal canal was consistent with an easily reducible left inguinal hernia. The elasticity of the skin was normal, and there were no bruises or scars. However, there was excessive skin wrinkling on both palms and both soles. There was bilateral dislocation of the elbows, and the forearms appeared slightly shortened (Fig 1E). He had genu valgum, pes planus, and varus deformity of the feet bilaterally (Fig 1F). The tips of his fingers and toes were broad and the nails were short (Fig 1C and D). There was a sandal gap bilaterally (Fig 1C). His metacarpophalangeal and interphalangeal joints were markedly hyperextensible, as were his knees.
Figure 1.
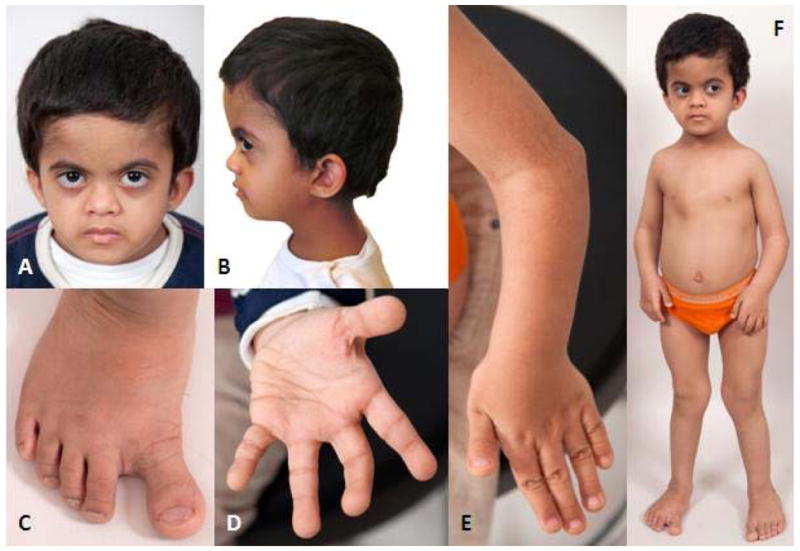
A. Face: Broad and tall forehead, flat face, mild bilateral proptosis, short nose with depressed nasal bridge and broad nasal tip, small mouth.
B. Head Profile: Brachycephaly, flat occiput and low-set ears.
C. Right foot: Pes planus and varus deformity, sandal gap, broad distal phalanges.
D. Left hand: Excessive wrinkling of the palm, broad finger tips.
E. Left elbow and hand: Elbow dislocation and shortened forearm, broad finger tips.
F. Full body: Protrusion of the sternum towards the right, genu valgum.
Imaging studies obtained at age 3 years 10 months showed flared metaphyses and epiphyses. Upper extremity X-rays confirmed dislocated elbows bilaterally with dysplastic radial heads, bowing of radius and ulna bilaterally, and proximal radio-ulnar dysplasia with functionally fused radio-ulnar joint (Fig 2A). The distal humerus was dysplastic, and shoulder imaging showed proximal humeral flattening, overgrowth of the acromion process and subluxation of the left humeral head, with a small and dysplastic ossification center, and a dysplastic left glenoid (Fig 2A). Feet X-rays showed broad toes. Detailed imaging of the hip at age 5 years 1 month showed flat acetabular angles, broad and somewhat flattened iliac wings which had a lacy trabecular pattern (Fig 2E and Supplementary Figures G–I – see supporting information online). Both femoral heads were flat with mild lateral subluxation. The femoral necks were wide. CT scan of the hips confirmed bilateral hip dysplasia with dislocation and dysmorphic appearance of the femoral heads. Spine imaging at ages 3 years 10 months and 5 years 1 month showed no segmentation abnormality and no platyspondylia in the thoracic spine. There was a gibbus deformity at the L1 level, and the ribs seemed gracile (Fig 2D). Cervical spine imaging showed significant atlantoaxial and atlantooccipital instability with flexion and extension (Fig 2B and C). Osteopenia was not observed on any of the imaging studies. An echocardiogram at age 5 years revealed mild dilation of the aortic root and main pulmonary artery (Fig 2F).
Figure 2.

A. Left shoulder and elbow: Proximal humeral flattening, subluxation of the humeral head, dysplastic glenoid, dysplastic distal humerus with dislocated elbow, dysplastic radial head, bowing of radius and ulna, and functionally fused radio-ulnar joint.
B., C. C-spine upright flexion (B) and extension (C): Atlantoaxial and atlantooccipital instability.
D. Spine Xray lateral: Gibbus deformity at the L1 level, and gracile ribs.
E. Hip: Flat acetabular angles, broad and flattened iliac wings, and a lacy trabecular pattern.
F. Echocardiogram: Mild dilation of the aortic root and main pulmonary artery.
Laboratory investigations showed mild vitamin D deficiency (19.7 ng/ml, normal range 30–80), but normal levels for electrolytes including calcium, phosphorus and magnesium, normal urine calcium to creatinine ratio, and normal alkaline phosphatase levels. A comprehensive endocrine and metabolic workup was unrevealing.
MOLECULAR GENETIC ANALYSES
After careful clinical evaluation, our patient’s features were suggestive of Larsen-Like Syndrome caused by B3GAT3 mutations. A clinical whole genome customized combined SNP and oligonucleotide chromosomal microarray was performed. Over 18% of the patient’s genome had regions of homozygosity, consistent with the high degree of consanguinity. There were no clinically relevant copy number variants identified. There was a 46.5 Mb block of homozygosity spanning chromosome 11 (coordinates: 32455853–78947645 (GRCh37)) which included the B3GAT3 gene, suggesting a possible recessive mutation in this gene. As the patient was from the Unites Arab Emirates, which is where the initially reported 5 siblings with a B3GAT3 mutation were identified [Baasanjav et al., 2011], we hypothesized that the patient may share the same rare pathogenic variant. We therefore performed Sanger sequencing of the exon containing the previously identified pathogenic variant, using the primer sequences published by Baasanjav et al.[Baasanjav et al., 2011], and we found that our patient was homozygous for the same variant c.830G>A (p.Arg277Gln) (Supplementary Figure 3 – see supporting information online).
We also performed whole exome sequencing (WES) through a clinical diagnostic laboratory to determine whether he might have another co-morbid genetic disorder since the high degree of parental consanguinity puts him at risk for having another autosomal recessive condition. The only pathogenic finding on WES was the same homozygous mutation that had been identified through Sanger sequencing as indicated above.
DISCUSSION
In this report, we describe the second family with a confirmed case of B3GAT3 mutation. Our patient carries the same c.830 G>A mutation that has previously been reported [Baasanjav et al., 2011], and similarly to the first family, our patient’s parents are from the United Arab Emirates and have a high degree of consanguinity.
While our patient presents many of the previously described characteristics of a defect in glucuronosyltransferase I [Baasanjav et al., 2011], he displays additional clinical features that expand the phenotype of B3GAT3 mutation. His developmental delay, pectus carinatum, dental abnormalities, refractive error and skin wrinkling were not reported in the five affected siblings from the previous family, but are features that have been reported in linkeropathies due to B3GALT6 [Malfait et al., 2013; Nakajima et al., 2013] and B4GALT7 [Kresse et al., 1987; Faiyaz-Ul-Haque et al., 2004; Guo et al., 2013] mutations (Table I). His atlanto-axial and atlanto-occipital instability, as well as his bilateral inguinal hernias could be incidental findings. However, the fact that both stem from connective tissue defects suggests that they are part of his proteoglycan synthesis disorder. To our knowledge, neither has previously been associated with any of the described linkeropathies.
Table 1.
Comparison of clinical features in patients with mutations in B4GALT7, B3GALT6, B3GAT3, and our patient
| B4GALT7 (n=4) [Kresse et al., 1987; Faiyaz-Ul-Haque et al., 2004; Guo et al., 2013] | B3GALT6 (n=16) [Nakajima et al., 2013; Malfait et al., 2013] | B3GAT3 (n=5) [Baasanjav et al., 2011] | Our patient | |
|---|---|---|---|---|
| Skeletal Abnormalities | ||||
| Short stature | + (−5.1 SDS*; range −3.6 to −6.3) | + (−5.4 SDS**; range −1 to −9.1) | + (P3 or less) | + (−3.43 SDS) |
| Elbow joint abnormality# | + (4/4) | + (10/15) | + (5/5) | + |
| Dislocated joints | − | + (6/13) shoulder, hip | + (5/5) elbow, shoulder | + elbow, shoulder, hip |
| Joint laxity | + (4/4) fingers, large joints | + (11/14) fingers, large joints¶ | + (5/5) fingers only | + |
| Hip dysplasia/dislocation | NR | + (16/16) dislocation, short ilia | + (number of subjects NR) | + |
| Leg deformity | + (1/4) varus | NR | − | + valgus |
| Broad fingertips/toes | + (4/4) | + (11/16) | + (5/5) | + |
| Foot deformity± | + (4/4) | + (7/12) | + (5/5) | + |
| Kyphoscoliosis | − | + (10/10) before age 2y | − | − |
| Pectus deformity | + (2/2) carinatum | + (4/12) carinatum, excavatum | − | + carinatum |
| Frequent fractures | NR | + (3/3) | − | − |
| Decreased bone density | + (3/4) | + (3/3) | +(number of subjects NR) | − |
| X-ray abnormalities | ||||
| Vertebral abnormality | NR | + (14/14) platyspondyly | + (?/?) | + L1 gibbus deformity |
| Metaphyseal abnormality | + (3/4) | + (13/13) | NR | + |
| Other bony features | Clavicle exostosis | Overtubulatio, trabecular pattern abnormal, delayed bone age | Delayed bone age | Atlantoaxial and atlantooccipital instability; lacy trabecular pattern |
| Cranio-facial features | ||||
| Brachycephaly | NR | NR | + (5/5) | + |
| Prominent forehead | + (4/4) | + (9/10) | − | + (mild) |
| Prominent eyes/proptosis | + (3/4) | + (8/11) | + (5/5) | + |
| Refractive error | + (2/4) esotropia, hyperopia | + (3/4) myopia, retinal detachment | NR | + hyperopia, esotropia, amblyopia |
| Blue sclerae | NR | + (11/14) | NR | − |
| Ear abnormality | + (2/4) | + (2/2) | + (3/5) | + |
| Small mouth | + (3/4) | + (5/10) | + (3/5) micrognathia | + |
| Short/webbed neck | NR | NR | + (5/5) | + |
| Dental abnormalities | + (3/3) | + (3/4) | NR | + |
| Skin/hair findings | ||||
| Hyperextensible skin | + (4/4) | + (9/14) | NR | − |
| Excessive wrinkling | − (2/2) | + (2/4) | NR | + |
| Atrophic scarring | + (3/3) | +(2/2) | NR | − |
| Other | ||||
| Hypotonia | + (4/4) | + (9/16) | NR | − |
| Developmental delay | + (3/4) | + (6/16) | − (0/5) | + mild |
| Cardiovascular abnormality | NR | + (1/16) mitral regurgitation | + (5/5) various | + aortic root/pulm. artery dilation |
mean of 4 patients;
mean of 15 patients (1 patient data missing);
comprises: dislocation, radio-ulnar synostosis, radial/ulnar bowing, subluxed radial head;
one 34 year-old patient had joint contractures that were progressive with age;
talipes equinovarus, metatarsus varus, pes planus
A comparison of the clinical features that have been observed in patients with a documented linkeropathy reveals remarkable similarities as well as some distinguishing differences between the three different enzyme defects (Table I). Short stature is ubiquitous, but possibly more pronounced in the B3GALT6 and B4GALT7 mutations. Shared skeletal abnormalities of variable severity and expression include foot deformities, hip dysplasia, and other joint and bony abnormalities, whereas elbow joint abnormalities, finger joint laxity and broad fingertips and toes are almost universal to all patients. Large joint laxity is described in patients with B3GALT6 and B4GALT7 mutations but not in any of the six patients with B3GAT3 mutation. Kyphoscoliosis is very prominent in patients with B3GALT6 mutation, but not in those with mutations in B4GALT7 or B3GAT3. Common craniofacial features are a prominent forehead, prominent eyes, ear deformities and refractory errors, whereas webbed neck and low hairline has only been reported in patients with a B3GAT3 mutation. On the other hand, blue sclera, skin hyperextensibility, easy bruising or thin skin is characteristically absent in patients with a mutation in B3GAT3. Hypotonia seems ubiquitous, whereas developmental delay is only present in some patients. Thus far, only patients with B3GAT3 mutations have been reported to have cardiovascular abnormalities, although not all authors reported echocardiograms on their patients.
Nakajima et al. showed that the three linkage genes are expressed at slightly different levels in body tissues and show highest expression in cartilage, skeletal muscle, tendon and bone [Nakajima et al., 2013]. The authors suggested that qualitative differences between the three linkeropathies may be due to the varying levels of gene expression in affected tissues, whereas quantitative differences may be more likely influenced by the amount of enzyme deficiency caused by each mutation [Nakajima et al., 2013]. For example, B3GAT3 seems to have higher levels of expression in the heart compared to B3GALT6 and B4GALT7, possibly explaining the higher incidence of cardiovascular abnormalities in these patients. Another possible explanation for the overlapping clinical features is the idea that the enzymes of proteoglycan biosynthesis may be part of a larger enzyme complex rather than functioning completely independently. This idea was first introduced by Schwartz et al. in 1974 [Schwartz and Roden, 1974], and recently expanded to suggest the presence of a so-called GAGosome where the enzymes of proteoglycan biosynthesis may be gathered [Esko and Selleck, 2002; Presto et al., 2008]. Further collaborative research between clinicians and basic scientists is needed to elucidate the interplay of the linkage enzymes and their clinical correlates, and to confirm a relationship between gene expression in peripheral tissues and phenotype.
In summary, our report expands the phenotype of the B3GAT3 mutation and of the linkeropathies in general. Features in our patient that have not previously been associated with B3GAT3 mutation include developmental delay, dental anomalies, bilateral inguinal herniae, refractive error, pectus carinatum, and atlanto-axial as well as atlanto-occipital instability. Although there is a possibility that given the high degree of consanguinity in his parents, our patient may have an additional autosomal recessive disorder, his clinical features are highly suggestive of a proteoglycan disorder and the majority can be readily explained by the mutation in B3GAT3. It is interesting to note that there is some variability in the clinical presentation of B3GAT3 deficiency even in subjects with the same pathogenic variant. Additional clinical case descriptions will help refine the phenotype of linkeropathies and other proteoglycan disorders. Nevertheless, the clinician should have a high index of suspicion for a linkeropathy in a child presenting with short stature and specific skeletal findings, namely joint hypermobility particularly of the fingers, wrists and knees, foot deformity, elbow dislocation or contracture, and broad based distal phalanges.
Supplementary Material
Supplemental Figure 1. G. Posterior neck: Short neck and low posterior hairline.
H. Right ear: Low-set but normally formed.
I. Left ear: Underdeveloped crus helix and inferior crus of the antihelix.
Supplemental Figure 2. G.–I. Hip views von Rosen (G), standing (H), and frog leg (I): Flat acetabular angles, broad and flattened iliac wings.
Supplemental Figure 3
Acknowledgments
This work was conducted with support from Harvard Catalyst | The Harvard Clinical and Translational Science Center (NIH Award #UL1 RR 025758 and financial contributions from Harvard University and its affiliated academic health care centers). The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, the National Center for Research Resources, or the National Institutes of Health. This work was also supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development at the NIH grant #1K23HD073351 to AD.
Footnotes
The authors have no conflicts of interest to disclose.
References
- Baasanjav S, Al-Gazali L, Hashiguchi T, Mizumoto S, Fischer B, Horn D, Seelow D, Ali BR, Aziz SAA, Langer R, Saleh Aa H, Becker C, Nürnberg G, Cantagrel V, Gleeson JG, Gomez D, Michel J-B, Stricker S, Lindner TH, Nürnberg P, Sugahara K, Mundlos S, Hoffmann K. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am J Hum Genet. 2011;89:15–27. doi: 10.1016/j.ajhg.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernfield M, Götte M, Park P. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Faiyaz-Ul-Haque M, Zaidi SHE, Al-Ali M, Al-Mureikhi MS, Kennedy S, Al-Thani G, Tsui L-C, Teebi AS. A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and Ehlers-Danlos syndrome resembling the progeroid type. Am J Med Genet. 2004;128A:39–45. doi: 10.1002/ajmg.a.30005. [DOI] [PubMed] [Google Scholar]
- Guo MH, Stoler J, Lui J, Nilsson O, Bianchi DW, Hirschhorn JN, Dauber A. Redefining the progeroid form of ehlers-danlos syndrome: Report of the fourth patient with B4GALT7 deficiency and review of the literature. Am J Med Genet. 2013 doi: 10.1002/ajmg.a.36128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häcker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- Kresse H, Rosthøj S, Quentin E, Hollmann J, Glössl J, Okada S, Tønnesen T. Glycosaminoglycan-free small proteoglycan core protein is secreted by fibroblasts from a patient with a syndrome resembling progeroid. Am J Hum Genet. 1987;41:436–453. [PMC free article] [PubMed] [Google Scholar]
- Malfait F, Kariminejad A, Van Damme T, Gauche C, Syx D, Merhi-Soussi F, Gulberti S, Symoens S, Vanhauwaert S, Willaert A, Bozorgmehr B, Kariminejad MH, Ebrahimiadib N, Hausser I, Huysseune A, Fournel-Gigleux S, De Paepe A. Defective Initiation of Glycosaminoglycan Synthesis due to B3GALT6 Mutations Causes a Pleiotropic Ehlers-Danlos Syndrome-like Connective Tissue Disorder. Am J Hum Genet. 2013 doi: 10.1016/j.ajhg.20. pii: S0002–9297(13)00177–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizumoto S, Ikegawa S, Sugahara K. Human genetic disorders caused by mutations in genes encoding biosynthetic enzymes for sulfated glycosaminoglycans. J Biol Chem. 2013;288:10953–10961. doi: 10.1074/jbc.R112.437038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M, Mizumoto S, Miyake N, Kogawa R, Iida A, Ito H, Kitoh H, Hirayama A, Mitsubuchi H, Miyazaki O, Kosaki R, Horikawa R, Lai A, Mendoza-Londono R, Dupuis L, Chitayat D, Howard A, Leal GF, Cavalcanti D, Tsurusaki Y, Saitsu H, Watanabe S, Lausch E, Unger S, Bonafé L, Ohashi H, Superti-Furga A, Matsumoto N, Sugahara K, Nishimura G, Ikegawa S. Mutations in B3GALT6, which Encodes a Glycosaminoglycan Linker Region Enzyme, Cause a Spectrum of Skeletal and Connective Tissue Disorders. Am J Hum Genet. 2013 doi: 10.1016/j.ajhg.20. pii: S0002–9297(13)00164–X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrimon N, Bernfield M. Specificities of heparan sulphate proteoglycans in developmental processes. Nature. 2000:725–728. doi: 10.1038/35008000. [DOI] [PubMed] [Google Scholar]
- Presto J, Thuveson M, Carlsson P, Busse M, Wilén M, Eriksson I, Kusche-Gullberg M, Kjellén L. Heparan sulfate biosynthesis enzymes EXT1 and EXT2 affect NDST1 expression and heparan sulfate sulfation. Proc Natl Acad Sci U S A. 2008;105:4751–4756. doi: 10.1073/pnas.0705807105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quentin E, Gladen A, Roden L, Kresse H. A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc Natl Acad Sci U S A. 1990;87:1342–1346. doi: 10.1073/pnas.87.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins JD, Collins JS, Holden KR. United States head circumference growth reference charts: birth to 21 years. J Pediatr. 2010;156:907–913. doi: 10.1016/j.jpeds.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Schwartz NB, Roden L. Biosynthesis of Chondroitin Sulfate. Purification of UDP-D-Xylose: Core Protein 1-D-Xylotransferase by Affinity Chromatography. Carbohydr Res. 1974;56:717–724. doi: 10.1016/s0008-6215(00)87072-x. [DOI] [PubMed] [Google Scholar]
- Sugahara K. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr Opin Struct Biol. 2003;13:612–620. doi: 10.1016/j.sbi.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Sugahara K, Mikami T. Chondroitin/dermatan sulfate in the central nervous system. Curr Opin Struct Biol. 2007;17:536–545. doi: 10.1016/j.sbi.2007.08.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. G. Posterior neck: Short neck and low posterior hairline.
H. Right ear: Low-set but normally formed.
I. Left ear: Underdeveloped crus helix and inferior crus of the antihelix.
Supplemental Figure 2. G.–I. Hip views von Rosen (G), standing (H), and frog leg (I): Flat acetabular angles, broad and flattened iliac wings.
Supplemental Figure 3