

Abstract
Inhibition of fatty acid amide hydrolase (FAAH) activity is under investigation as a valuable strategy for the treatment of several disorders, including pain and drug addiction. A number of potent FAAH inhibitors belonging to different chemical classes have been disclosed. O-aryl carbamates are one of the most representative families. In the search for novel FAAH inhibitors, we synthesized a series of O-(1,2,3-triazol-4-yl)methyl carbamate derivatives exploiting the copper-catalyzed [3 + 2] cycloaddition reaction between azides and alkynes (click chemistry). We explored structure-activity relationships within this new class of compounds and identified potent inhibitors of both rat and human FAAH with IC50 values in the single-digit nanomolar range.
Keywords: O-(Triazolyl)methyl carbamates, FAAH, inhibitors, stability, structure-activity relationship
Introduction
Fatty acid amide hydrolase (FAAH)[1] is a membrane-bound serine hydrolase that catalyses the hydrolytic cleavage of endogenous biologically active fatty acid ethanolamides (FAEs), such as anandamide (AEA), an agonist of cannabinoid receptors,[2] and palmitoylethanolamide (PEA)[3] and oleoylethanolamide (OEA),[4] which are agonists of type-α peroxisome proliferator-activated receptors (PPAR-α).[5] These natural FAAH substrates may play important roles in the central nervous system (CNS) and in peripheral tissues, where they are involved in a variety of physiological processes.[6]
Substantial efforts have been dedicated to the discovery of potent and selective FAAH inhibitors, with the objective of developing therapeutic approaches for pathologic conditions such as pain, drug addiction, anxiety, and depression.[6-7] Different classes of molecules are known to increase intracellular FAE levels through FAAH inhibition, including carbamates[8] and piperidine/piperazine ureas[9] that covalently bind to FAAH,[10] and α-keto heterocycles-based inhibitors,[11] which inhibit FAAH by reversible hemiketal formation with the catalytic serine of the enzyme.[12] Among them, the O-arylcarbamate series, exemplified by URB524[8a, 8b, 13] (1a, Figure 1), URB597[8a, 8b, 13] (1b, Figure 1) and URB694[8c, 14] (1c, Figure 1), has been extensively investigated.[15] In particular, it was shown that compound 1b exerts a combination of anxiolytic-like, antidepressant-like, and analgesic effects, because of its ability to inhibit FAAH activity in the CNS and peripheral tissues.[16]
Figure 1.
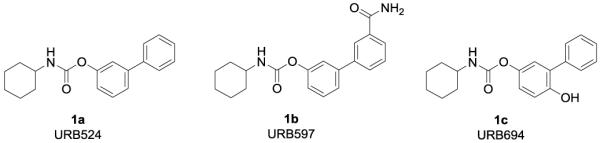
O-arylcarbamates FAAH inhibitors.
The active site of FAAH is characterized by an atypical catalytic triad, consisting of Ser241-Ser217-Lys142, which is capable of hydrolyzing amide and ester bonds at similar rates.[17] Several studies, including computational modeling,[18] supported by the resolution of the crystal structure of humanized rat FAAH in complex with 1b,[19] indicate that O-arylcarbamates bind covalently to FAAH and cause its irreversible inhibition. In particular, it has been proposed that this class of molecules is attacked at the carbonyl group by Ser 241, leading to the formation of carbamoylated, catalytically inactive FAAH and releasing the O-biphenyl moiety as the leaving group.
O-arylcarbamates such as 1b are selective for FAAH, but can also interact with select liver carboxylesterases, at least at high concentrations, and have limited plasma stability.[9a] Recently, however, highly potent O-arylcarbamates with markedly improved selectivity for FAAH were identified.[8c] The insertion of an electron-donating substituent, such as a hydroxy or amino group, in the para position of the proximal phenyl ring of 1a did not significantly affect inhibitory potency in vitro, but caused a marked increase in the stability of the compounds in plasma, in comparison to other molecules in the series. The compound URB694 (1c) was identified as a potent FAAH inhibitor with improved plasma stability, prolonged half-life in vivo, and decreased activity towards liver carboxylesterases in comparison to 1b.
In the search for better FAAH inhibitors with improved stability, we designed a novel class of carbamates where the O-aryl moiety is replaced by an O-(triazol-4-yl)alkyl group (Figure 2). We expected those compounds to be more stable than their O-aryl analogues, since the aliphatic alcohol resulting from the nucleophilic attack on the carbamate is a worse leaving group than a phenol. Although carbamate-based FAAH inhibitors containing an O-(heteroaryl)alkyl moiety have been reported in the patent literature,[12] no O-(triazol-4-yl)alkyl carbamate derivatives have been described to date.
Figure 2.
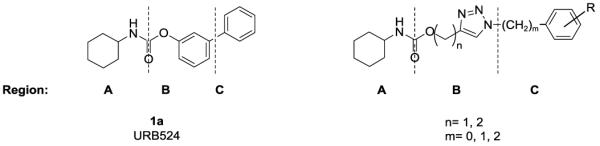
Structures of URB524 (1a) and O-(1,2,3-triazol-4-yl)alkyl carbamate derivatives
We report here the synthesis and characterization of a series of O-(1,2,3-triazol-4-yl)alkyl carbamates, prepared by copper-catalyzed [3 + 2] cycloaddition reaction between azides and alkynes.[20] The fast and versatile synthesis via click chemistry allowed us to prepare a number of analogues in a quick and reliable manner, and rapidly explore the SAR within this new class of FAAH inhibitors.
Results and Discussion
Chemistry
The (3-phenylphenyl)methyl N-cyclohexylcarbamate (4) was prepared from commercially available 3-phenylbenzoic acid (2) by lithium aluminum hydride reduction to 3-phenylbenzyl alcohol (3) followed by reaction with commercial cyclohexyl isocyanate (Scheme 1).
Scheme 1.
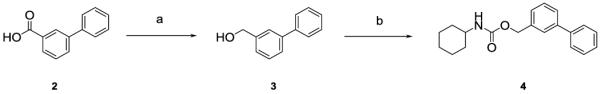
Synthesis of (3-phenylphenyl)methyl N-cyclohexylcarbamate (4). Reagents and conditions: a) LiAlH4 (2N solution in THF), dry THF, 0 °C to rt, 2h; (b) cyclohexyl isocyanate, DMAP, dry CH3CN, 80 °C, 6h.
The preparation of 1,4-disubstituted-1,2,3-triazoles was accomplished by [3 + 2] cycloaddition reaction between azides and alkynes, in the presence of copper (I) salts (click chemistry).[20]
We designed a versatile synthetic strategy that allowed us to generate a first set of molecules, bearing an O-(1,2,3-triazol-4-yl)methyl moiety. The desired compounds (17-30, 32, 33) were synthesized as shown in Scheme 2.
Scheme 2.
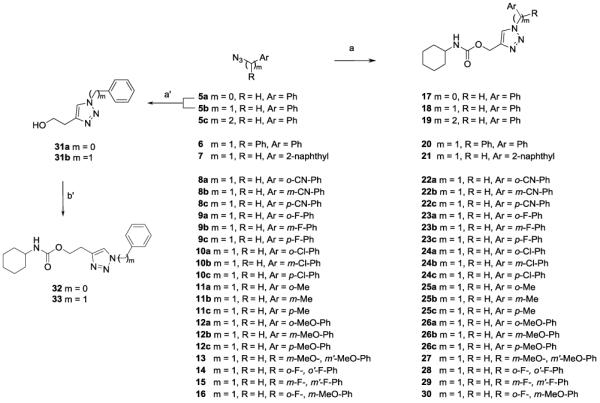
Synthesis of O-(triazol-4-yl)alkyl carbamates 17-30, 32-33. Reagents and conditions: a) prop-2-yn-1-yl N-cyclohexylcarbamate, sodium L-ascorbate, copper (II) sulfate pentahydrate, H2O-tert-BuOH 1:1, rt, 3h; a’) but-3-yn-1-ol, sodium L-ascorbate, copper (II) sulfate pentahydrate, H2O-tert-BuOH 1:1, rt, 3h; b’) cyclohexyl isocyanate, DMAP, dry MeCN, 80 °C, 5h.
The aromatic azide 5a was prepared from aniline by a diazotation-azidation protocol,[21] while 5b, 5c, and 6-16 were obtained in good to excellent yields by reacting the corresponding halides with sodium azide.[22] Final compounds 17, 18, and 19 were prepared via click chemistry, starting from prop-2-yn-1-yl N-cyclohexylcarbamate,[23] prepared by reaction of cyclohexylamine with the commercially available prop-2-ynyl chloroformate, and the azides 5a, 5b, and 5c, respectively (Scheme 2). Then, copper catalyzed [3 + 2] cycloaddition reaction between azides 5a and 5b with the commercially available but-3-yn-1-ol, allowed us to obtain compounds 31a and 31b in acceptable yields.[24] Finally, compounds 32 and 33 were prepared by coupling alcohols 31a and 31b, respectively, with commercial cyclohexyl isocyanate (Scheme 2).
The O-(1,2,3-triazol-4-yl)methyl carbamate derivatives 20-30 (Scheme 2) were prepared by reaction of the azides 6-16 with prop-2-yn-1-yl N-cyclohexylcarbamate, under click chemistry conditions.
A second set of analogues was synthesized, as reported in Scheme 3, in order to explore region A (Figure 2). Prop-2-ynyl-N-prop-2-ynyl carbamate,[25] obtained by reaction of the commercially available propargyl amine with prop-2-ynyl chloroformate, was reacted with aromatic azides 12a and 12b thus affording compounds 34a and 34b, which bear the same substituent on both aromatic rings (Scheme 3).
Scheme 3.
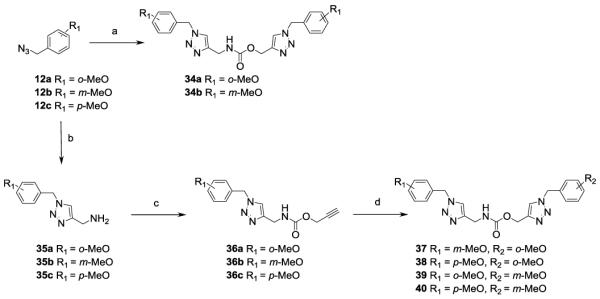
Syntheses of compounds 34a, b and 37-40. Reagents and conditions: a) 12a or 12b, prop-2-yn-1-yl N-(prop-2-yn-1-yl)carbamate, sodium L-ascorbate, copper (II) sulfate pentahydrate, H2O-tert-BuOH 1:1, rt, 3h; b) prop-2-yn-1-amine, sodium L-ascorbate, copper (II) sulfate pentahydrate, H2O-tert-BuOH 1:1, rt, 3h; c) prop-2-ynyl chloroformate, Et3N, dry DCM, 0 °C, 30 min; d) 12a or 12b, sodium L-ascorbate, copper (II) sulfate pentahydrate, H2O-tert-BuOH 1:1, rt, 3h.
Bis-(triazol-4-yl)methyl carbamates with different substitution pattern on the aromatic rings were synthesized according to Scheme 3. Aromatic azides 12a-c were reacted with propargyl amine, under click chemistry conditions, to give the corresponding aminomethyl triazoles 35a-c, which were subsequently coupled with prop-2-ynyl chloroformate to afford N-(triazol-4-yl)methyl-O-propargyl carbamates 36a-c. The latter compounds were reacted with aromatic azides 12a-b, under click chemistry conditions, to afford N-(triazol-4-yl)methyl-O-(triazol-4-yl)methyl carbamates 37-40.
The compounds bearing a (1,2,4-triazol-3-yl-)methyl moiety, 43 and 46, were synthesized as reported in Scheme 4. Compound 43 was prepared by lithium aluminum hydride reduction of the commercially available 1-phenyl-1,2,4-triazole-3-carboxylic acid 41 followed by reaction with cyclohexyl isocyanate. Then, the commercially available methyl 1H-1,2,4-triazole-3-carboxylate 44 was reacted with benzyl bromide in presence of potassium carbonate to obtain compound 45. Reduction by lithium aluminum hydride followed by reaction with cyclohexyl isocyanate afforded compound 46.
Scheme 4.
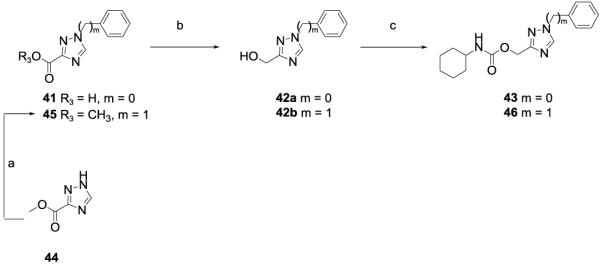
Syntheses of (1,2,4-triazol-3-yl)methyl N-cyclohexylcarbamates 43and 46. Reagents and conditions: a) BnBr, K2CO3, dry DMF, 80 °C, 16h; b) LiAlH4 (2N solution in THF), dry THF, 0 °C to rt, 2h; c) cyclohexyl isocyanate, DMAP, dry CH3CN, 80 °C, 6h.
Structure–Activity Relationship (SAR) and Stability Studies
The compounds were tested for their ability to inhibit the hydrolysis of [3H]anandamide by FAAH prepared from rat brain homogenates. Median inhibitory concentration (IC50) values are reported in Tables 1-4.
Table 1.
FAAH inhibitory activity and rat plasma stability of 1a, 4, and O-[1-substituted-(1,2,3-triazol-4-yl)]alkyl carbamates
| Compounds | Structure | IC50
[nM]a |
Plasma half-life (min.) |
% remaining after 7 hb |
|---|---|---|---|---|
| 1a |
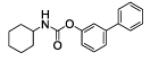
|
5.7 ± 1.2 |
62 ± 19 | 0 |
| 4 |
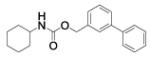
|
65% @ 100 μM |
- | - |
| 17 |
|
380.6 ± 62.9 |
- | 90 ± 9 |
| 18 |
|
26.2 ± 3.8 |
- | 92 ± 8 |
| 19 |
|
833 ± 129 |
- | - |
| 32 |
|
1278 ± 84 |
- | - |
| 33 |
|
2535 ± 106 |
- | - |
| 43 |
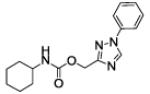
|
70% @ 100 μM |
- | - |
| 46 |
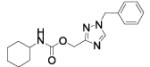
|
51500 ± 1125 |
- | - |
IC50 values are reported as the mean ± SEM (n= 3);
Percent of parent compound remaining after 7 h incubation.
Table 4.
Comparison of r-FAAH vs. h-FAAH-1 inhibitory activity of selected compounds.
| Compounds | r-FAAH IC50 [nM]a | h-FAAH IC50 [nM]a |
|---|---|---|
| 1a | 5.7 ± 1.2 | 3.1 ± 1.3 |
| 23a | 9.9 ± 1.9 | 619 ± 138 |
| 23b | 12.1 ± 0.8 | 180 ± 6 |
| 24a | 6.2 ± 1.3 | 1068 ± 144 |
| 24b | 13.2 ± 2.8 | 190 ± 8. |
| 26a | 1.4 ± 0.2 | 337 ± 101 |
| 26b | 11.8 ± 3.7 | 158 ± 12 |
| 27 | 10.5 ± 2.9 | 3.6 ± 0.9 |
| 28 | 11.9 ± 1.0 | 812 ± 105 |
| 29 | 10.4 ± 2.6 | 216 ± 6 |
| 34b | 3.9 ± 0.7 | 4.2 ± 1.0 |
| 37 | 3.2 ± 1.2 | 30.4 ± 3.7 |
| 38 | 7.6 ± 0.6 | 30.7 ± 6.2 |
| 39 | 9.8 ± 0.4 | 39.1 ± 0.6 |
| 40 | 5.8 ± 0.6 | 9.4 ± 2.0 |
IC50 values are reported as the mean ± SEM (n= 3).
In a first attempt to improve the stability of O-biphenyl carbamate FAAH inhibitors, we replaced the O-(3-phenylphenyl) residue of 1a with an O-(3-phenylphenyl)methyl group, as in compound 4. This change caused an almost complete loss of activity, as 4 showed only 65% inhibition of FAAH activity at 100 µM (Table 1). Interestingly, the substitution of the O-(3-phenylphenyl)methyl group with an O-(1-phenyl-1,2,3-triazol-4-yl)methyl residue, as in compound 17 (IC50 = 381 nM), allowed to recover the FAAH inhibitory activity. Encouraged by this result, we synthesized a set of close analogues of 17, compounds 18-19 and 32-33, to identify the best substituents at position 1 and 4 of the triazole ring for FAAH inhibition. The results are reported in Table 1.
The replacement of the phenyl group at position 1 of the triazole with a benzyl residue, compound 18 (IC50 = 26 nM), led to a ca. 15-fold increase in potency. It is interesting to note that this compound was only 4.6-fold less potent than 1a (URB524). When the methylene linker at position 1 or 4 of the triazole ring was substituted by an ethylene moiety, as in compounds 19 and 33, respectively, a drop in potency with respect to 18 was observed. The replacement of the 4-methylene residue in compound 17 with a 4-ethylene one, leading to compound 32 (IC50 = 1278 nM), resulted in a ca. 3-fold decrease in potency.
We then asked whether 1-phenyl- or 1-benzyl-(1,2,4-triazol-3yl)methyl moieties worked as a replacement of the O-(3-phenylphenyl) residue of 1a and whether they were interchangeable with the isomeric 1-phenyl- and 1-benzyl-(1,2,3-triazol-4-yl)methyl residues. Compounds 43 and 46 showed a dramatic decrease in potency with respect to 1a and, surprisingly, were much less potent than 17 and 18. We speculate that the loss in potency might be the ascribed to the different electronic properties of the 1,2,4-triazole ring with respect to the isomeric 1,2,3-triazole counterpart, leading to an unfavourable interaction with the active site of the enzyme.
To test our hypothesis that O-(triazol-4-yl)alkyl carbamate derivatives are more stable than O-aryl carbamates, we compared the rat plasma stability of compounds 1a, 17 and 18. The results are reported in Table 1. Consistent with our expectation, the O-(1,2,3-triazol-4-yl)methyl carbamates 17 and 18 showed a significantly higher plasma stability than did 1a. The latter compound displayed half-life of 62 min[26], but was no longer detectable after 7 hours incubation with rat plasma. In contrast, ca. 90% of the initial amount of compound 17 and 18 was still detectable after 7 hours.
The limited decrease in potency of compound 18 vs. 1a, coupled with its significantly higher plasma stability, prompted us to explore further this chemical class. We first investigated region C (Figure 2) by preparing a series of compounds bearing variously substituted benzyl residues at position 1 of the triazole ring. The results are summarized in Table 2.
Table 2.
FAAH inhibitory activity of O-(triazol-4-yl)methyl carbamates variously substituted at region C.
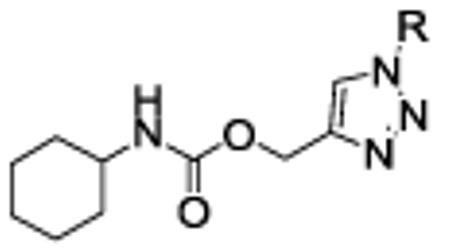
| |||||
|---|---|---|---|---|---|
|
| |||||
| Cpds | R | IC50
[nM]a |
Cpds | R | IC50
[nM]a |
| 20 |
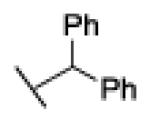
|
73% @ 100 μM |
25a |
|
21.5 ± 1.2 |
| 21 |
|
2003 ± 636 |
25b |
|
24.5 ± 6.2 |
| 22a |
|
19.5 ± 4.3 |
25c |
|
500 ± 44 |
| 22b |
|
21.5 ± 7.9 |
26a |
|
1.4 ± 0.2 |
| 22c |
|
2282 ± 585 |
26b |
|
11.8 ± 3.7 |
| 23a |
|
9.9 ± 1.9 |
26c |
|
627 ± 173 |
| 23b |
|
12.1 ± 0.8 |
27 |
|
10.5 ± 2.9 |
| 23c |
|
95.5 ± 22.2 |
28 |
|
11.9 ± 1.0 |
| 24a |
|
6.2 ± 1.3 |
29 |
|
10.4 ± 2.6 |
| 24b |
|
13.2 ± 2.8 |
30 |
|
44.6 ± 10.3 |
| 24c |
|
215 ± 31 |
|||
IC50 values are reported as the mean ± SEM (n= 3).
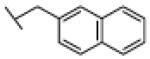
Replacement of the benzyl residue in 18 with a benzhydryl moiety, compound 20 (73% inhibition at 100 µM), or a 2-naphthylmethyl group, compound 21 (IC50 = 2.0 µM), led to a significant decrease in FAAH inhibitory potency, indicating that bulky arylmethyl groups linked to the triazole were not tolerated, most likely because of steric clash at the active site of the enzyme.
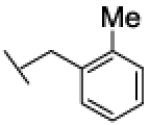
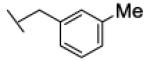
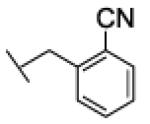
Interestingly, the nature of the substituent on the phenyl ring appeared to have a limited effect on the potency of the compounds as FAAH inhibitors. In fact, benzyl residues bearing both electron-withdrawing (CN, F, Cl) and electron-donating (Me, OMe) substituents at ortho or meta position all led to low-nanomolar inhibitors. Among them, the ortho-methoxybenzyl derivative 26a showed the highest potency (IC50 = 1.4 nM). All the para-substituted derivatives were less potent than the corresponding ortho- or meta-substituted analogues, irrespective of the electronic properties of the susbtituent. In particular, compound 22c (IC50 = 2282 nM), bearing a para-cyano group, showed the highest loss in potency within this sub-set of analogues. As for the naphthylmethyl compound 21, we interpret this finding as the result of an unfavorable steric interaction between the para-substituted phenyl ring and the active site of the enzyme. Together, from this small series of derivatives the rank order of potency ortho- > meta- > para-substituted compounds clearly emerged.
The excellent potency of benzyl derivatives bearing a fluorine or a methoxyl group at position ortho and meta led us to synthesize the di-substituted compounds 27-30 to verify whether any additive effect on potency was observed. With the exception of the 2-fluoro-3-methoxy-derivative 30 (IC50 = 44.6 nM), all the compounds retained an excellent potency, with IC50 in the range 10.4 - 11.9 nM, but none of them improved significantly over the corresponding mono-substituted analogue.
The most potent compound, 26a, was effective at inhibiting FAAH activity ex vivo. One hour after systemic administration of 26a (3 mg/kg, intraperitoneally) to CD1 mice, FAAH activity measured ex vivo in brain tissue was reduced by 78% (n=3) with respect to control.
As the next step in the investigation of the SAR of this new class of FAAH inhibitors, we conducted a preliminary exploration of region A (Figure 2). Previous studies on O-arylcarbamates showed that replacement of the cyclohexyl group of 1a with an arylalkyl moiety led to inhibitors of greater potency.[15a] Exploiting the click chemistry approach, we replaced the cyclohexyl group with a [1-(methoxybenzyl)triazol-4-yl]methyl residue, as in compounds 34a and 34b, and 37-40. The results are reported in Table 3.
Table 3.
FAAH inhibitory activity of N-(triazol-4-yl)methyl O-(triazol-4-yl)methyl carbamates 34a-40.
| Compounds | Structure | −IC50 [nM]a |
|---|---|---|
| 34a |
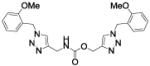
|
154 ± 14 |
| 34b |
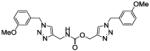
|
3.9 ± 0.7 |
| 37 |
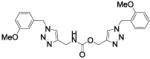
|
3.2 ± 1.2 |
| 38 |
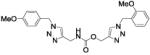
|
7.6 ± 0.6 |
| 39 |
|
9.8 ± 0.1 |
| 40 |
|
5.8 ± 0.6 |
IC50 values are reported as the mean ± SEM (n= 3).
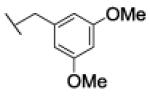
Compound 34a, bearing an ortho-methoxybenzyl residue on both triazolyl rings turned out to be the least potent derivative (IC50 = 154 nM). Moving the methoxyl group to the meta position of the N-(1-benzyltriazol-4-yl)methyl moiety led to derivative 37 (IC50 = 3.2 nM), which showed a 48-fold increase in potency vs. 34a and confirmed as the most potent compound of this small series. Consistent with the previous finding, replacement of the ortho-methoxybenzyl residue at position 1 of the triazole ring in region C with a meta-methoxybenzyl group, as in compound 39 (IC50 = 9.8 nM), was accompanied by a ca. 15-fold increase in potency with respect to 34a. Introduction of a meta-methoxybenzyl group at position 1 on both triazolyl rings led to the potent inhibitor 34b (IC50 = 3.9 nM). Interestingly, a para-methoxybenzyl group at position 1 of the (triazol-4-yl)methyl moiety in region A was not detrimental for potency, as compounds 38 and 40 inhibited FAAH activity with IC50 of 7.6 and 5.8 nM, respectively.
The most interesting compounds identified from the SAR exploration were tested for their inhibitory activity against human FAAH-1 (h-FAAH-1). A comparison between the inhibitory potency of selected compounds on r-FAAH vs. h-FAAH-1 is reported in Table 4.
The series of substituted O-(1-benzyltriazol-4-yl)methyl N-cyclohexylcarbamate derivatives resulted to be generally less active at inhibiting h-FAAH-1 than r-FAAH, displaying a 14- to 240-fold drop in potency. The only exception was represented by 27 (IC50 = 3.6 nM), which showed 3-fold higher potency on h-FAAH-1 with respect to r-FAAH. Moreover, the ortho-substituted analogues 23a, 24a, and 26a, which displayed single-digit nanomolar r-FAAH inhibition, and the 2,6-dibustituted derivative 28, suffered the most marked loss of potency: 62, 172, 240, and 73-fold respectively. The meta-substituted analogues 23b, 24b, and 26b, and the 3,5-disubstituted derivative 29 were more potent inhibitors than their ortho-substituted analogues, thus reversing the preference for ortho-substituted benzyl residues observed with r-FAAH.
By contrast, compounds bearing substituted (1-benzyltriazol-4-yl)methyl residues at both region A and C (Figure 2) displayed a minor decrease in activity on h-FAAH-1 compared to the N-cyclohexylcarbamate derivatives. Indeed, compounds 37, 38, and 39 showed 4 to 10-fold lower potency, but retained double-digit nanomolar potency on h-FAAH-1. The most promising molecules in terms of potency were 34b and 40 that possessed IC50 for h-FAAH-1 inhibition of 4.2 nM and 9.4 nM respectively. Interestingly, both of them are characterized by a meta-methoxybenzyl group on region C (Figure 2), suggesting that the binding site on h-FAAH-1 prefers meta-substituents on that benzyl group.
The most potent compounds against both rat and human FAAH, i.e. 27, 34b, and 40, were tested for their selectivity versus monoacylglycerol lipase (MGL), a serine hydrolase that inactivates the endocannabinoid 2-arachidonoylglycerol (2-AG).[27] None of the compounds inhibited MGL activity when tested at concentrations up to 100 μM (Figure S1). The selective inhibition of FAAH activity by compounds 27, 34b, and 40 is in agreement with previous observations with the O-arylcarbamates 1a and 1b.[28]
Finally, compounds 27, 34b, and 40 were further characterized by determining their rat plasma and mouse liver microsomal (MLM) stability, and their kinetic solubility in buffer. A comparison of the overall profile of O-arylcarbamate 1a with compounds 27, 34b, and 40 is reported in Table 5.
Table 5.
Characterization of selected compounds
| Cp d |
Structure |
r- FAA H IC50 [nM]a |
h
-FAA H IC50 [nM]a |
Plasm a Stab. (% at 7 h)b |
MLM Stab. half- life (min.) c |
Kineti c sol. (μM)d |
|---|---|---|---|---|---|---|
| 1a |
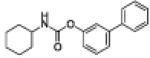
|
5.7 ± 1.2 |
3.1 ± 1.3 |
0 | <5 | 6 |
| 27 |
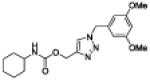
|
10.5 ± 2.9 |
3.6 ± 0.9 |
91 ± 3 | 5 | 18 |
| 34b |
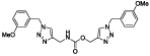
|
3.9 ± 0.7 |
4.2 ± 1.0 |
88 ± 11 |
6 | 174 |
| 40 |
|
5.8 ± 0.6 |
9.4 ± 2.0 |
89 ± 9 | 8 | 152 |
IC50 values for FAAH inhibition are reported as the mean ± SEM (n= 3);
Data are the mean ± SEM of three determinations;
Data are the mean of three determinations;
Phosphate buffered saline, pH 7.4, single determination.
The selected O-[(1-benzyltriazol-4-yl)methyl]carbamate derivatives 27, 34b, and 40 showed FAAH inhibitory activity and MLM stability comparable to those of the O-arylcarbamate 1a. However, they displayed much higher rat plasma stability than 1a, as ca. 90% of the initial amount of the compounds was still present after 7 hours incubation with rat plasma. Moreover, with the exception of compound 27, the kinetic solubility in buffer also improved significantly.
Conclusion
In the present study, we report the synthesis and characterization of O-(1,2,3-triazol-4-yl)alkyl carbamates as a novel class of FAAH inhibitors. In these compounds, an O-(triazol-4-yl)methyl group replaces the O-aryl moiety of known and potent FAAH inhibitors such as compound 1a (URB524) and 1b (URB597). A number of compounds were prepared by copper-catalyzed [3 + 2] cycloaddition reactions between azides and alkynes (click chemistry). Exploiting the same chemistry, we also synthesized carbamates bearing a substituted (1-benzyltriazol-4-yl)methyl moiety at both the O and N end. The click chemistry approach allowed us rapidly to explore the structure-activity relationships within the class. Several single-digit nanomolar inhibitors of rat FAAH were obtained, including the potent derivative 26a, which showed an IC50 value of 1.4 nM and inhibited brain FAAH activity in vivo. Some of these compounds potently inhibited human FAAH-1. In particular, compounds 34b and 40, bearing a [1-[(methoxyphenyl)methyl]triazol-4-yl]methyl group at both the O and N end of the carbamate function, displayed single-digit nanomolar IC50 values for both rat and human FAAH. In addition, they showed a remarkable improvement in rat plasma stability and kinetic solubility in buffer with respect to URB524.
The dramatic decrease in FAAH inhibitory activity of 1-phenyl- or 1-benzyl-substituted O-(1,2,4-triazol-3-yl)mehtyl carbamates 43 and 46 demonstrated that the 1-substituted-(1,2,3-triazol-4-yl)methyl core structure, easily accessible by click chemistry, was essential for obtaining potent inhibition of FAAH activity.
In conclusion, exploiting a click chemistry approach we prepared a novel series of potent and drug-like FAAH inhibitors containing an O-(1,2,3-triazol-4-yl)alkyl carbamate moiety. The compounds described in the present study represent a promising starting point for the development of new FAAH inhibitors with improved drug-like properties.
Experimental Section
Chemistry
Chemicals, Materials and Methods. Solvents and reagents were obtained from commercial suppliers and were used without further purification. For simplicity, solvents and reagents were indicated as follows: acetonitrile (CH3CN), benzyl bromide (BnBr), cyclohexane (Cy), dichloromethane (DCM), diethyl ether (Et2O), 4-(dimethylamino)-pyridine (DMAP), ethanol (EtOH), ethyl acetate (EtOAc), hydrochloric acid (HCl), methanol (MeOH), N,N’-dimethylformamide (DMF), room temperature (rt), sodium sulfate (Na2SO4), sodium bicarbonate (NaHCO3), sulfuric acid (H2SO4), tert-butanol (t-BuOH), tetrahydrofuran (THF), triethylamine (Et3N).
Automated column chromatography purifications were performed by using a Teledyne ISCO apparatus (CombiFlash® Rf) with pre-packed silica gel columns of different sizes (from 4 g to 120 g). Mixtures of increasing polarity of Cy and EtOAc or DCM and MeOH were used as eluents. NMR experiments were run on a Bruker Avance III 400 system (400.13 MHz for 1H, and 100.62 MHz for 13C), equipped with a BBI probe and Z-gradients. Spectra were acquired at 300 K, using deuterated dimethylsulfoxide ([D6]DMSO) or deuterated chloroform (CDCl3) as solvents. Chemical shifts for 1H and 13C spectra were recorded in parts per million using the residual non-deuterated solvent as the internal standard (for CDCl3: 7.26 ppm, 1H and 77.16 ppm, 13C; for [D6]DMSO: 2.50 ppm, 1H; 39.52 ppm, 13C). UPLC/MS analyses were run on a Waters ACQUITY UPLC/MS system consisting of a Single Quadropole Detector (SQD) Mass Spectrometer (MS) equipped with an Electrospray Ionization (ESI) interface and a Photodiode Array (PDA) Detector. PDA range was 210-400 nm. ESI in positive and negative mode was applied. Mobile phases: (A) 10mM NH4OAc in H2O, pH 5; (B) 10mM NH4OAc in MeCN/H2O (95:5) pH 5. Analyses were performed either with method A or B. Method A: gradient 5 to 95% B over 3 min; flow rate 0.5 mL/min; temperature 40 °C. Pre column: Vanguard BEH C18 (1.7µm 2.1x5mm). Column: BEH C18 (1.7µm 2.1x50mm). Method B: gradient: 50 to 100% B over 3 min, flow rate 0.5 mL/min; temperature 40 °C. Pre column: Vanguard BEH C18 (1.7µm 2.1x5mm). Column: BEH C18 (1.7µm 2.1x50mm). Accurate mass measurement (HMRS) was performed on a Synapt G2 Quadrupole-Tof Instrument (Waters, USA), equipped with an ESI ion source.
All final compounds (4, 17-30, 32, 33, 34a-b, 37-40, 43 and 46) showed ≥ 95% purity by NMR and UPLC/MS analysis. The syntheses of reaction intermediates 3, 5a-c, 6-16, 31a-b, 35a-c, 36a-c, 41, 42a-b, and 45 are described in the Supporting Information.
General procedure (1) for the synthesis of triazoles (17-30, 37-40)
1 equiv. of the ethynyl derivatives and 1 equiv. of the azido compounds were suspended in a solution of water / t-BuOH 1:1. Sodium ascorbate (0.1 eq) of a freshly prepared 1 M solution in water was added, followed by the addition of copper (II) sulfate pentahydrate (0.01 eq). The resulting reaction vigorously stirred for 3 h at rt. The reaction mixture was then diluted with water, cooled on ice, and the precipitate was collected by filtration. When addition of water failed to precipitate the desired triazoles, evaporation of the solvent allowed the recovery of the crude products. Purifications were performed by column chromatography.
General procedure (2) for the synthesis of double triazoles carbamates (34a-b)
0.5 equiv. of the ethynyl derivatives and 1 equiv. of the azido compounds were suspended in a solution of water / t-BuOH 1:1. Sodium ascorbate (0.1 eq) of a freshly prepared 1 M solution in water was added, followed by the addition of copper (II) sulfate pentahydrate (0.01 eq). The resulting reaction vigorously stirred for 8 h rt. Afterwards, evaporation of the solvent allowed the recovery of the crude products. Purification were performed by column chromatography.
(3-phenylphenyl)methyl N-cyclohexylcarbamate (4)
(3-phenylphenyl)methanol (3, 0.125 g, 0.68 mmol) were dissolved in dry CH3CN (5 mL) while stirring at rt. Then, DMAP (0.08 g, 0.68 mmol) and cyclohexyl isocyanate (0.09 g, 0.75 mmol) were added and the reaction mixture was stirred at 80 °C for 3 h. Afterwards, the reaction mixture was diluted with EtOAc and washed once with 2N HCl, and once with brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by chromatography employing a gradient of MeOH in DCM from 0% to 2%, to afford compound 4 as a white powder (0.13 g; 61%): 1H NMR (400 MHz, [D6]DMSO): δ=7.69 – 7.61 (m, 3H), 7.60 (dt, J=7.9, 1.4 Hz, 1H), 7.47 (q, J=7.3 Hz, 3H), 7.42 – 7.31 (m, 2H), 7.19 (d, J=7.9 Hz, 1H), 5.07 (s, 2H), 3.31 – 3.22 (m, 1H), 1.76 (dd, J=12.6, 3.6 Hz, 2H), 1.67 (dq, J=12.5, 3.7 Hz, 2H), 1.53 (dq, J=11.5, 3.7 Hz, 1H), 1.31 – 1.00 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=155.2, 140.2, 139.9, 138.0, 128.9, 128.9, 127.5, 126.7, 126.6, 126.0 (2C), 65.0, 49.5, 32.6 (2C), 25.1, 24.6 ppm (2C); UPLC-MS: Method B, Rt 1.97, ionization: m/z 310 [M+H]+; HRMS–ESI: m/z [M+Na]+ calcd for C20H23NO2Na: 332.1626, found: 332.1622.
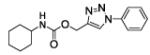
(1-phenyltriazol-4-yl)methyl N-cyclohexylcarbamate (17)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.23 g, 1.26 mmol) and azidobenzene (0.15 g, 1.26 mmol), sodium ascorbate (0.02, 0.12 mmol), and copper (II) sulfate pentahydrate (0.003 g, 0.01 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 17 as a white powder (0.22 g; 59%): 1H NMR (400 MHz, [D6]DMSO): δ=8.82 (s, 1H), 7.91 (m, 2H), 7.61 (m, 2H), 7.52 (m, 1H), 7.20 (d, J=8.0 Hz, 1H), 5.13 (s, 2H), 3.28 (m, 1H), 1.76 (m, 2H), 1.67 (dt, J=12.2, 3.8 Hz, 2H), 1.54 (m, 1H), 1.18 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=155.4, 144.3, 137.0, 130.3 (2-C), 129.2, 123.2, 120.6 (2-C), 57.0, 50.0, 33.1 (2-C), 25.6, 25.0 ppm (2-C); UPLC-MS: Method A, Rt 2.42, ionization: m/z 301 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C16H20N4O2: 301.1665, found: 301.1666.
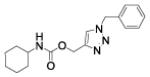
(1-benzyltriazol-4-yl)methyl N-cyclohexylcarbamate (18)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and azidomethylbenzene (0.11 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 50% EtOAc in Cy, to afford compound 18 as a white powder (0.18 g; 71%): 1H NMR (400 MHz, [D6]DMSO): δ=8.15 (s, 1H), 7.35 (m, 5H), 7.13 (d, J=7.9 Hz, 1H), 5.60 (s, 2H), 5.02 (s, 2H), 3.25 (m, 1H), 1.69 (m, 4H), 1.53 (dt, J=12.7, 3.8 Hz, 1H), 1.15 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=155.4, 143.5, 136.5, 129.2 (2-C), 128.6, 128.4 (2-C), 125.0, 57.2, 53.2, 49.9, 33.0 (2-C), 25.6, 25.0 ppm (2-C); UPLC-MS: Method A, Rt 2.37, ionization: m/z 315 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C17H22N4O2: 315.1821, found: 315.1826.
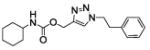
(1-phenethyltriazol-4-yl)methyl N-cyclohexylcarbamate (19)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 2-azidoethylbenzene (0.12 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 19 as a white powder (0.135 g; 50%): 1H NMR (400 MHz, [D6]DMSO): δ=8.03 (s, 1H), 7.29 (m, 2H), 7.21 (m, 3H), 7.13 (d, J=7.9 Hz, 1H), 4.99 (s, 2H), 4.61 (dd, J=7.9, 6.8 Hz, 2H), 3.24 (m, 1H), 3.16 (t, J=7.4 Hz, 2H), 1.69 (m, 4H), 1.54 (d, J=13.3 Hz, 1H), 1.18 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=154.6, 142.5, 137.5, 128.6 (2-C), 128.3 (2-C), 126.5, 124.4, 56.6, 50.3, 49.4, 35.6, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.44, ionization: m/z 329 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C18H24N4O2: 329.1978, found: 329.1982.
(1-benzhydryltriazol-4-yl)methyl N-cyclohexylcarbamate (20)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and [azido(phenyl)methyl]benzene (0.17 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 40% EtOAc in Cy, to afford compound 20 as a white powder (0.23 g; 71%): 1H NMR (400 MHz, [D6]DMSO): δ=8.08 (s, 1H), 7.39 (m, 6H), 7.31 (s, 1H), 7.21 (m, 4H), 7.14 (d, J=7.9 Hz, 1H), 5.02 (s, 2H), 3.22 (m, 1H), 1.67 (m, 4H), 1.52 (d, J=12.5 Hz, 1H), 1.12 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=155.4, 144.3, 138.6 (2-C), 128.7 (4-C), 128.2 (2-C), 127.9 (4-C), 66.5, 56.7, 49.4, 32.5 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.81, ionization: m/z 391 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C23H26N4O2: 391.2134, found: 391.2132.
[1-(2-naphthylmethyl)triazol-4-yl]methyl N-cyclohexylcarbamate (21)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 2-(azidomethyl)naphthalene (0.15 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 21 as a white powder (0.16 g; 54%): 1H NMR (400 MHz, [D6]DMSO): δ=8.19 (s, 1H), 7.92 (m, 3H), 7.86 (s, 1H), 7.54 (m, 2H), 7.44 (dd, J=8.5, 1.6 Hz, 1H), 7.10 (d, J=7.8 Hz, 1H), 5.76 (s, 2H), 5.01 (s, 2H), 3.22 (m, 1H), 1.66 (dd, J=26.8, 12.6 Hz, 4H), 1.51 (d, J=12.5 Hz, 1H), 1.11 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=154.9, 143.0, 133.4, 132.7, 132.4, 128.4, 127.7, 127.5, 126.9, 126.5, 126.4, 125.7, 124.6, 56.7, 52.9, 49.4, 32.5 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.65, ionization: m/z 365 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C21H24N4O2: 365.1978, found: 365.1975.
[1-[(2-cyanophenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (22a)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 2-(azidomethyl)benzonitrile (0.13 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 22a as a white powder (0.15 g; 53%): 1H NMR (400 MHz, [D6]DMSO): δ=8.19 (s, 1H), 7.92 (dd, J=7.7, 1.0 Hz, 1H), 7.72 (td, J=7.7, 1.2 Hz, 1H), 7.57 (td, J=7.7, 0.9 Hz, 1H), 7.36 (d, J=7.8 Hz, 1H), 7.14 (d, J=7.8 Hz, 1H), 5.81 (s, 2H), 5.03 (s, 2H), 3.25 (m, 1H), 1.68 (dd, J=27.0, 12.5 Hz, 4H), 1.53 (d, J=12.7 Hz, 1H), 1.13 ppm (dtd, J=31.1, 24.1, 12.1 Hz, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=154.9, 143.1, 138.7, 133.8, 133.3, 129.4, 129.2, 125.0, 116.9, 111.2, 56.6, 50.9, 49.4, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.25, ionization: m/z 340 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C18H21N5O2: 340.1773, found: 340.1779.
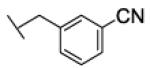
[1-[(3-cyanophenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (22b)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 3-(azidomethyl)benzonitrile (0.13 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 22b as a white powder (0.17 g; 62%): 1H NMR (400 MHz, [D6]DMSO): δ=8.21 (s, 1H), 7.82 (m, 2H), 7.62 (m, 2H), 7.12 (d, J=7.8 Hz, 1H), 5.67 (s, 2H), 5.01 (s, 2H), 3.24 (m, 1H), 1.68 (dd, J=25.4, 12.5 Hz, 4H), 1.52 (d, J=12.6 Hz, 1H), 1.13 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=154.9, 143.2, 137.5, 132.9, 131.9, 131.6, 130.0, 124.8, 118.3, 111.6, 56.6, 51.7, 49.4, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.25, ionization: m/z 340 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C18H21N5O2: 340.1773, found: 340.1781.
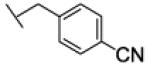
[1-[(4-cyanophenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (22c)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 4-(azidomethyl)benzonitrile (0.13 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 22c as a white powder (0.21 g; 77%): 1H NMR (400 MHz, [D6]DMSO): δ=8.21 (s, 1H), 7.86 (d, J=8.2 Hz, 2H), 7.45 (d, J=8.2 Hz, 2H), 7.14 (d, J=7.8 Hz, 1H), 5.72 (s, 2H), 5.02 (s, 2H), 3.24 (m, 1H), 1.69 (dd, J=25.1, 12.6 Hz, 4H), 1.53 (d, J=12.4 Hz, 1H), 1.14 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=154.9, 143.2, 141.4, 132.7 (2-C), 128.6 (2-C), 124.9, 118.4, 110.9, 56.6, 52.1, 49.4, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.22, ionization: m/z 340 [M+H]+; HRMS–ESI: m/z [M+Na]+ calcd for C18H21N5O2Na: 362.1593, found: 362.1594.
[1-[(2-fluorophenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (23a)
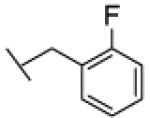
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-2-fluoro-benzene (0.12 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 23a as a white powder (0.17 g; 61%): 1H NMR (400 MHz, [D6]DMSO): δ=8.11 (s, 1H), 7.42 (m, 1H), 7.34 (td, J=7.6, 1.4 Hz, 1H), 7.23 (m, 2H), 7.13 (d, J=7.8 Hz, 1H), 5.66 (s, 2H), 5.00 (s, 2H), 3.24 (m, 1H), 1.67 (m, 4H), 1.52 (d, J=12.5 Hz, 1H), 1.12 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=160.0 (d, J=246.7 Hz), 155.0, 143.0, 130.7 (d, J=4.6 Hz), 130.7, 124.8 (d, J=3.4 Hz), 124.7, 122.8 (d, J=14.7 Hz), 115.6 (d, J=20.8 Hz), 56.7, 49.4, 46.8 (d, J=3.7 Hz), 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.37, ionization: m/z 333 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C17H21FN4O2: 333.1727, found: 333.1732.
[1-[(3-fluorophenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (23b)
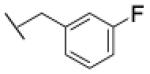
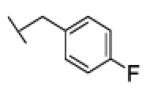
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-3-fluoro-benzene (0.12 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 50% EtOAc in Cy, to afford compound 23b as a white powder (0.17 g; 63%): White powder; yield 63%; 1H NMR (400 MHz, [D6]DMSO): δ=8.18 (s, 1H), 7.42 (m, 1H), 7.16 (m, 4H), 5.62 (s, 2H), 5.01 (s, 2H), 3.24 (m, 1H), 1.68 (dd, J=26.2, 12.5 Hz, 4H), 1.52 (d, J=12.5 Hz, 1H), 1.12 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=162.5 (d, J=244.3 Hz), 155.4, 143.6, 139.2, 131.3 (d, J=8.3 Hz), 125.2, 124.4 (d, J=2.7 Hz), 115.3 (m, 2-C), 57.1, 52.5, 49.9, 33.0 (2-C), 25.6, 25.0 ppm (2-C); UPLC-MS: Method A, Rt 2.40, ionization: m/z 333 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C17H21FN4O2: 333.1727, found: 333.1731.
[1-[(4-fluorophenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (23c)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-4-fluoro-benzene (0.12 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 23c as a white powder (0.13 g; 49%): 1H NMR (400 MHz, [D6]DMSO): δ=8.14 (s, 1H), 7.38 (ddd, J=8.4, 5.3, 2.5 Hz, 2H), 7.20 (m, 2H), 7.12 (d, J=7.5 Hz, 1H), 5.58 (s, 2H), 5.00 (s, 2H), 3.23 (m, 1H), 1.67 (m, 4H), 1.52 (d, J=12.5 Hz, 1H), 1.11 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=161.8 (d, J=244.5 Hz), 154.9, 143.0, 132.2, 130.2 (d, J=8.4 Hz), 124.5, 115.5 (d, J=21.6 Hz), 56.7, 51.9, 49.4, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.39, ionization: m/z 333 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C17H21FN4O2: 333.1727, found: 333.1731.
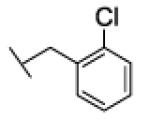
[1-[(2-chlorophenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (24a)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-2-chloro-benzene (0.14 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 24a as a white powder (0.19 g; 68%): 1H NMR (400 MHz, [D6]DMSO): δ=8.11 (s, 1H), 7.52 (dd, J=7.7, 1.5 Hz, 1H), 7.38 (m, 2H), 7.22 (dd, J=7.4, 1.7 Hz, 1H), 7.14 (d, J=7.7 Hz, 1H), 5.70 (s, 2H), 5.02 (s, 2H), 3.24 (m, 1H), 1.68 (dd, J=26.2, 12.5 Hz, 4H), 1.53 (m, 1H), 1.11 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=154.9, 142.9, 133.2, 132.6, 130.4, 130.2, 129.6, 127.6, 124.9, 56.7, 50.5, 49.4, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.49, ionization: m/z 349 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C17H21ClN4O2: 349.1431, found: 349.1435.
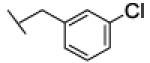
[1-[(3-chlorophenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (24b)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-3-chloro-benzene (0.14 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 50% EtOAc in Cy, to afford compound 24b as a white powder (0.24 g; 83%): 1H NMR (400 MHz, [D6]DMSO): δ=8.19 (s, 1H), 7.40 (dd, J=6.1, 2.3 Hz, 3H), 7.27 (dq, J=5.9, 2.8 Hz, 1H), 7.13 (d, J=7.9 Hz, 1H), 5.61 (s, 2H), 5.01 (s, 2H), 3.25 (m, 1H), 1.68 (dd, J=26.4, 12.4 Hz, 4H), 1.52 (d, J=12.5 Hz, 1H), 1.13 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=154.9, 143.1, 138.4, 133.2, 130.6, 128.1, 127.8, 126.6, 124.7, 56.6, 51.9, 49.4, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.55, ionization: m/z 349 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C17H21ClN4O2: 349.1431, found: 349.1436.
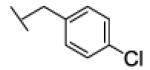
[1-[(4-chlorophenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (24c)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-4-chloro-benzene (0.14 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 50% EtOAc in Cy, to afford compound 24c as a white powder (0.13 g; 47%): 1H NMR (400 MHz, [D6]DMSO): δ=8.15 (s, 1H), 7.44 (m, 2H), 7.33 (d, J=8.5 Hz, 2H), 7.12 (d, J=7.8 Hz, 1H), 5.60 (s, 2H), 5.00 (s, 2H), 3.23 (m, 1H), 1.67 (m, 4H), 1.52 (m, 1H), 1.11 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=154.9, 143.1, 135.0, 132.8, 129.8 (2-C), 128.7 (2-C), 124.6, 56.6, 51.9, 49.4, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.55, ionization: m/z 349 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C17H21ClN4O2: 349.1431, found: 349.1427.
[1-(o-tolylmethyl)triazol-4-yl]methyl N-cyclohexylcarbamate (25a)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.37 g, 2.04 mmol) and 1-(azidomethyl)-2-methyl-benzene (0.3 g, 2.04 mmol), sodium ascorbate (0.040, 0.2 mmol), and copper (II) sulfate pentahydrate (0.005 g, 0.02 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 25a as a white powder (0.13 g; 47%): 1H NMR (400 MHz, [D6]DMSO): δ=8.03 (s, 1H), 7.29 – 7.12 (m, 4H), 7.07 (d, J=7.5 Hz, 1H), 5.60 (s, 2H), 5.00 (s, 2H), 3.22 (dt, J=10.7, 5.8 Hz, 1H), 2.30 (s, 3H), 1.82 – 1.58 (m, 4H), 1.52 (d, J=12.5 Hz, 1H), 1.31 – 0.95 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=155.4, 143.3, 136.7, 134.5, 130.8, 129.1, 128.8, 126.7, 125.1, 57.2, 51.3, 49.9, 33.0 (2-C), 25.5, 25.0 (2-C), 19.0 ppm; UPLC-MS: Method A, Rt 2.5, ionization: m/z 329 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C18H24N4O2: 329.1978, found: 329.1977.
[1-(m-tolylmethyl)triazol-4-yl]methyl N-cyclohexylcarbamate (25b)
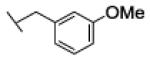
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.37 g, 2.04 mmol) and 1-(azidomethyl)-3-methyl-benzene (0.3 g, 2.04 mmol), sodium ascorbate (0.040, 0.2 mmol), and copper (II) sulfate pentahydrate (0.005 g, 0.02 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 25b as a white powder (0.17 g; 65%): 1H NMR (400 MHz, [D6]DMSO): δ=8.12 (s, 1H), 7.25 (t, J=7.8 Hz, 1H), 7.13 (m, 4H), 5.54 (s, 2H), 5.00 (s, 2H), 3.24 (m, 1H), 2.28 (s, 3H), 1.68 (dd, J=25.9, 12.5 Hz, 4H), 1.52 (d, J=12.5 Hz, 1H), 1.12 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=154.9, 142.9, 137.9, 135.8, 128.7, 128.6, 128.5, 125.0, 124.5, 56.7, 52.7, 49.4, 32.5 (2-C), 25.1, 24.5 (2-C), 20.8 ppm; UPLC-MS: Method A, Rt 2.52, ionization: m/z 329 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C18H24N4O2: 329.1978, found: 329.1981.
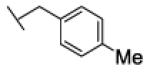
[1-(p-tolylmethyl)triazol-4-yl]methyl N-cyclohexylcarbamate (25c)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.37 g, 2.04 mmol) and 1-(azidomethyl)-4-methyl-benzene (0.3 g, 2.04 mmol), sodium ascorbate (0.040, 0.2 mmol), and copper (II) sulfate pentahydrate (0.005 g, 0.02 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 25c as a white powder (0.18 g; 67%): 1H NMR (400 MHz, [D6]DMSO): δ=8.10 (s, 1H), 7.28 – 7.08 (m, 5H), 5.53 (s, 2H), 4.99 (s, 2H), 3.30 – 3.15 (m, 1H), 2.27 (s, 3H), 1.67 (dd, J=24.7, 12.5 Hz, 4H), 1.52 (d, J=12.2 Hz, 1H), 1.15 ppm (dt, J=37.6, 12.2 Hz, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=155.4, 143.4, 137.9, 133.4, 129.7 (2-C), 128.4 (2-C), 124.9, 57.2, 53.0, 49.9, 33.0 (2-C), 25.5, 25.0 (2-C), 21.1 ppm; UPLC-MS: Method A, Rt 2.52, ionization: m/z 329 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C18H24N4O2: 329.1978, found: 329.1978.
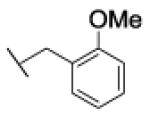
[1-[(2-methoxyphenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (26a)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-2-methoxy-benzene (0.13 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 26a as a white powder (0.18 g; 64%): 1H NMR (400 MHz, [D6]DMSO): δ=7.98 (s, 1H), 7.35 (m, 1H), 7.09 (m, 3H), 6.93 (m, 1H), 5.52 (s, 2H), 4.99 (s, 2H), 3.82 (s, 3H), 3.23 (m, 1H), 1.67 (m, 4H), 1.52 (d, J=12.6 Hz, 1H), 1.12 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=156.8, 155.0, 142.7, 130.0, 129.6, 124.6, 123.5, 120.5, 111.2, 56.7, 55.5, 49.4, 48.2, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.43, ionization: m/z 345 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C18H24N4O3: 345.1927, found: 345.1930.
[1-[(3-methoxyphenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (26b)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-3-methoxy-benzene (0.13 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 70% EtOAc in Cy, to afford compound 26b as a white powder (0.14 g; 50%): 1H NMR (400 MHz, [D6]DMSO): δ=8.14 (s, 1H), 7.28 (td, J=7.5, 1.8 Hz, 1H), 7.12 (d, J=7.8 Hz, 1H), 6.90 (d, J=6.2 Hz, 2H), 6.85 (d, J=7.8 Hz, 1H), 5.55 (s, 2H), 5.00 (s, 2H), 3.73 (s, 3H), 3.24 (d, J=7.6 Hz, 1H), 1.68 (dd, J=25.7, 12.5 Hz, 4H), 1.52 (d, J=12.4 Hz, 1H), 1.13 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=159.4, 154.9, 143.0, 137.4, 129.8, 124.6, 119.9, 113.7, 113.4, 56.7, 55.0, 52.6, 49.4, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.38, ionization: m/z 345 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C18H24N4O3: 345.1927, found: 345.1929.
[1-[(4-methoxyphenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (26c)
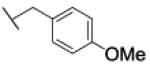
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-4-methoxy-benzene (0.13 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 60% EtOAc in Cy, to afford compound 26c as a white powder (0.15 g; 54%): 1H NMR (400 MHz, [D6]DMSO): δ=8.08 (s, 1H), 7.29 (d, J=8.6 Hz, 2H), 7.11 (d, J=7.8 Hz, 1H), 6.92 (d, J=8.6 Hz, 2H), 5.50 (s, 2H), 4.99 (s, 2H), 3.73 (s, 3H), 3.22 (m, 1H), 1.68 (dd, J=25.3, 12.6 Hz, 4H), 1.52 (d, J=12.6 Hz, 1H), 1.13 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=159.1, 154.9, 142.9, 129.5 (2-C), 127.9, 124.2, 114.0 (2-C), 56.7, 55.1, 52.2, 49.4, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.35, ionization: m/z 345 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C18H24N4O3: 345.1927, found: 345.1924.
[1-[(3,5-dimethoxyphenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (27)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-3,5-dimethoxy-benzene (0.16 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 40% EtOAc in Cy, to afford compound 27 as a white powder (0.17 g; 55%): 1H NMR (400 MHz, [D6]DMSO): δ=8.14 (s, 1H), 7.12 (d, J=7.8 Hz, 1H), 6.46 (s, 3H), 5.50 (s, 2H), 5.01 (s, 2H), 3.72 (s, 6H), 3.24 (m, 1H), 1.67 (m, 4H), 1.52 (d, J=12.4 Hz, 1H), 1.12 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=160.6, 154.9, 143.0, 138.0, 124.6, 106.0, 99.5, 56.7, 55.2, 52.7, 49.4, 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.42, ionization: m/z 375 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C19H26N4O4: 375.2032, found: 375.2047.
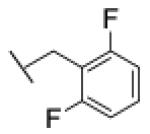
[1-[(2,6-difluorophenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (28)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 2-(azidomethyl)-1,3-difluoro-benzene (0.14 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 40% EtOAc in Cy, to afford compound 28 as a white powder (0.16 g; 56%): 1H NMR (400 MHz, [D6]DMSO): δ=8.10 (s, 1H), 7.51 (m, 1H), 7.18 (t, J=8.1 Hz, 2H), 7.13 (d, J=7.8 Hz, 1H), 5.66 (s, 2H), 4.99 (s, 2H), 3.23 (m, 1H), 1.68 (dd, J=26.0, 12.6 Hz, 4H), 1.52 (d, J=12.6 Hz, 1H), 1.12 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=161.2 (d, J=249.2 Hz), 155.4, 143.3, 132.1 (t, J=10.4 Hz), 125.1, 112.3 (d, J=24.4 Hz), 111.7 (t, J=19.3 Hz), 57.1, 49.9, 41.2 (t, J=3.7 Hz), 33.0 (2-C), 25.6, 25.0 ppm (2-C); UPLC-MS: Method A, Rt 2.38, ionization: m/z 351 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C17H20F2N4O2: 351.1633, found: 351.1631.
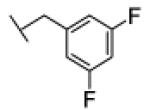
[1-[(3,5-difluorophenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (29)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-3,5-difluoro-benzene (0.14 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 29 as a white powder (0.22 g; 77%): 1H NMR (400 MHz, [D6]DMSO): δ=8.21 (s, 1H), 7.23 (tt, J=9.4, 2.3 Hz, 1H), 7.13 (d, J=7.8 Hz, 1H), 7.04 (t, J= 6.4 Hz, 2H), 5.64 (s, 2H), 5.02 (s, 2H), 3.23 (m, 1H), 1.68 (dd, J=26.4, 12.4 Hz, 4H), 1.52 (d, J=12.6 Hz, 1H), 1.13 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=162.8 (dd, J=247.1, 13.2 Hz), 155.4, 143.7, 140.6 (t, J=9.4 Hz), 125.4, 111.7 (m), 104.1 (t, J=25.7 Hz), 57.1, 52.1, 49.9, 33.0 (2-C), 25.5, 25.0 ppm (2-C); UPLC-MS: Method A, Rt 2.46, ionization: m/z 351 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C17H20F2N4O2: 351.1633, found: 351.1634.
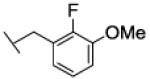
[1-[(2-fluoro-3-methoxy-phenyl)methyl]triazol-4-yl]methyl N-cyclohexylcarbamate (30)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-cyclohexylcarbamate (0.15 g, 0.82 mmol) and 1-(azidomethyl)-2-fluoro-3-methoxy-benzene (0.15 g, 0.82 mmol), sodium ascorbate (0.016, 0.08 mmol), and copper (II) sulfate pentahydrate (0.002 g, 0.008 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 30 as a white powder (0.23 g; 77%): 1H NMR (400 MHz, [D6]DMSO): δ=8.09 (s, 1H), 7.14 (m, 3H), 6.84 (m, 1H), 5.64 (s, 2H), 5.00 (s, 2H), 3.83 (s, 3H), 3.24 (m, 1H), 1.68 (dd, J=25.9, 12.5 Hz, 4H), 1.52 (d, J=12.5 Hz, 1H), 1.12 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=154.9, 149.5 (d, J=246.8 Hz), 147.3 (d, J=10.0 Hz), 142.9, 124.7, 124.6 (d, J=4.6 Hz), 123.5 (d, J=11.9 Hz), 121.1 (d, J=2.0 Hz), 114.0 (d, J=1.3 Hz), 56.7, 56.1, 49.4, 46.7 (d, J=4.6 Hz), 32.6 (2-C), 25.1, 24.5 ppm (2-C); UPLC-MS: Method A, Rt 2.37, ionization: m/z 363 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C18H23FN4O3: 363.1832, found: 363.1834.
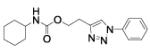
2-(1-phenyltriazol-4-yl)ethyl N-cyclohexylcarbamate (32)
2-(1-phenyltriazol-4-yl)ethanol (31a, 0.24 g, 1.26 mmol) was dissolved in dry CH3CN (5 mL) under stirring. Then, DMAP (0.15 g, 1.26 mmol) and cyclohexyl isocyanate (0.17 g, 1.38 mmol) were added and the reaction mixture was stirred overnight at 80 °C. The mixture was then diluted with EtOAc and washed once with 2N HCl, and once with brine. The organic layer was dried over sodium sulfate and concentrated in vacuo. The crude product was purified by by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM. Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 31a as a white powder (0.25 g; 63%): 1H NMR (400 MHz, [D6]DMSO): δ=8.62 (s, 1H), 7.87 (m, 2H), 7.60 (m, 2H), 7.49 (m, 1H), 7.06 (d, J=8.0 Hz, 1H), 4.25 (t, J=6.7 Hz, 2H), 3.24 (m, 1H), 3.02 (t, J=6.7 Hz, 2H), 1.69 (m, 4H), 1.53 (d, J=13.3 Hz, 1H), 1.15 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=155.7, 145.1, 137.2, 130.3 (2-C), 128.9, 121.4, 120.3 (2-C), 62.7, 49.8, 33.1 (2-C), 26.0, 25.6, 25.0 ppm (2-C); UPLC-MS: Method A, Rt 2.39, ionization: m/z 315 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C17H22N4O2: 315.1821, found: 315.1829.
2-(1-benzyltriazol-4-yl)ethyl N-cyclohexylcarbamate (33)
It was synthesized according to the procedure employed for 32, starting from 2-(1-benzyltriazol-4-yl)ethanol (31b, 0.23 g, 1.12 mmol), cyclohexyl isocyanate (0.15 g, 1.23 mmol), and DMAP (0.14 g, 1.12 mmol) in dry CH3CN (5 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 33 as a white powder (0.2 g; 54%): 1H NMR (400 MHz, [D6]DMSO): δ=7.95 (s, 1H), 7.33 (m, 5H), 7.01 (d, J=7.96 Hz, 1H), 5.55 (s, 2H), 4.16 (t, J=6.83 Hz, 2H), 3.21 (m, 1H), 2.90 (t, J=6.82 Hz, 2H), 1.67 (m, 4H), 1.52 (m, 1H), 1.12 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=155.0, 144.1, 136.6, 129.1 (2-C), 128.5, 128.3 (2-C), 123.1, 99.9, 62.8, 53.1, 49.8, 33.1 (2-C), 26.0, 25.6, 25.0 ppm (2-C); UPLC-MS: Method A, Rt 2.33, ionization: m/z 329 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C18H24N4O2: 329.1978, found: 329.1983.
[1-[(2-methoxyphenyl)methyl]triazol-4-yl]methyl N-[[1-[(2-methoxyphenyl)methyl]triazol-4-yl]methyl]carbamate (34a)
The reaction was carried out following general procedure (2), using prop-2-ynyl N-prop-2-ynylcarbamate (0.11 g, 0.767 mmol) and 1-(azidomethyl)-2-methoxy-benzene (0.25 g, 1.53 mmol), sodium ascorbate (0.030, 0.15 mmol), and copper (II) sulfate pentahydrate (0.004 g, 0.002 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 4% MeOH in DCM, to afford compound 34a as a white amorphous solid (0.2 g; 58%): 1H NMR (400 MHz, [D6]DMSO): δ=8.00 (s, 1H), 7.80 (s, 1H), 7.68 (t, J=5.7 Hz, 1H), 7.34 (m, 2H), 7.08 (m, 4H), 6.93 (m, 2H), 5.51 (s, 2H), 5.48 (s, 2H), 5.03 (s, 2H), 4.21 (d, J=5.8 Hz, 2H), 3.81 (s, 3H), 3.80 ppm (s, 3H); 13C NMR (100 MHz, [D6]DMSO): δ=156.83, 156.8, 155.9, 145.0, 142.5, 130.0, 129.9, 129.6, 129.5, 124.7, 123.6, 123.4, 122.8, 120.5 (2C), 111.2, 111.1, 57.1, 55.5 (2C), 48.2, 48.1, 35.9 ppm; UPLC-MS: Method A, Rt 2.18, ionization: m/z 464 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C23H25N7O4: 464.2046, found: 464.2056.
[1-[(3-methoxyphenyl)methyl]triazol-4-yl]methyl N-[[1-[(3-methoxyphenyl)methyl]triazol-4-yl]methyl]carbamate (34b)
The reaction was carried out following general procedure (2), using prop-2-ynyl N-prop-2-ynylcarbamate (0.11 g, 0.767 mmol) and 1-(azidomethyl)-3-methoxy-benzene (0.25 g, 1.53 mmol), sodium ascorbate (0.030, 0.15 mmol), and copper (II) sulfate pentahydrate (0.004 g, 0.002 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 5% MeOH in DCM, to afford compound 34b as a white amorphous solid (0.29 g; 81%): 1H NMR (400 MHz, [D6]DMSO): δ=8.15 (s, 1H), 7.95 (s, 1H), 7.71 (t, J=5.7 Hz, 1H), 7.28 (td, J=8.2, 7.5, 2.1 Hz, 2H), 6.89 (dd, J=5.8, 2.9 Hz, 4H), 6.85 (d, J=7.3 Hz, 2H), 5.55 (s, 2H), 5.51 (s, 2H), 5.04 (s, 2H), 4.22 (d, J=5.8 Hz, 2H), 3.73 ppm (s, 6H); 13C NMR (100 MHz, [D6]DMSO): δ=159.4 (2C), 155.9, 145.3, 142.8, 137.5, 137.4, 129.9, 129.8, 124.6, 122.8, 120.0 (2C), 113.7, 113.7, 113.4, 113.4, 57.1, 55.0 (2C), 52.7, 52.6, 36.0 ppm; UPLC-MS: Method A, Rt 2.12, ionization: m/z 464 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C23H25N7O4: 464.2046, found: 464.2052.
[1-[(2-methoxyphenyl)methyl]triazol-4-yl]methyl N-[[1-[(3-methoxyphenyl)methyl]triazol-4-yl]methyl]carbamate (37)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-[[1-[(3-methoxyphenyl)methyl]triazol-4-yl]methyl]carbamate (36b, 0.15 g, 0.5 mmol) and 1-(azidomethyl)-2-methoxy-benzene (0.08 g, 0.5 mmol), sodium ascorbate (0.010, 0.05 mmol), and copper (II) sulfate pentahydrate (0.001 g, 0.005 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 37 as a white amorphous solid (0.13 g; 58%): 1H NMR (400 MHz, [D6]DMSO): δ=8.00 (s, 1H), 7.95 (s, 1H), 7.70 (t, J=5.7 Hz, 1H), 7.34 (m, 1H), 7.27 (m, 1H), 7.11 (dd, J=7.5, 1.4 Hz, 1H), 7.05 (d, J=8.2 Hz, 1H), 6.92 (m, 3H), 6.85 (d, J=7.6 Hz, 1H), 5.51 (s, 4H), 5.03 (s, 2H), 4.21 (d, J=5.8 Hz, 2H), 3.80 (s, 3H), 3.73 ppm (s, 3H); 13C NMR (100 MHz, [D6]DMSO): δ=159.8, 157.3, 156.4, 145.8, 142.9, 137.9, 130.4, 130.3, 130.1, 125.2, 123.9, 123.3, 120.9, 120.4, 114.2, 113.8, 111.6, 57.6, 56.0, 55.5, 53.1, 48.7, 36.4 ppm; UPLC-MS: Method A, Rt 2.15, ionization: m/z 464 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C23H25N7O4: 464.2046, found: 464.2062.
[1-[(2-methoxyphenyl)methyl]triazol-4-yl]methyl N-[[1-[(4-methoxyphenyl)methyl]triazol-4-yl]methyl]carbamate (38)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-[[1-[(4-methoxyphenyl)methyl]triazol-4-yl]methyl]carbamate (36c, 0.15 g, 0.5 mmol) with 1-(azidomethyl)-2-methoxy-benzene (0.08 g, 0.5 mmol), sodium ascorbate (0.010, 0.05 mmol), and copper (II) sulfate pentahydrate (0.001 g, 0.005 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 38 as a colourless amorphous solid (0.15 g; 63%): 1H NMR (400 MHz, [D6]DMSO): δ=8.00 (s, 1H), 7.89 (s, 1H), 7.68 (t, J=5.7 Hz, 1H), 7.34 (m, 1H), 7.28 (d, J=8.5 Hz, 2H), 7.11 (dd, J=7.4, 1.3 Hz, 1H), 7.05 (d, J=8.2 Hz, 1H), 6.92 (m, 3H), 5.51 (s, 2H), 5.46 (s, 2H), 5.03 (s, 2H), 4.20 (d, J=5.8 Hz, 2H), 3.80 (s, 3H), 3.73 ppm (s, 3H); 13C NMR (100 MHz, [D6]DMSO): δ=159.5, 157.3, 156.3, 145.8, 142.9, 130.4, 130.1, 130.0 (2C), 128.4, 125.2, 123.9, 122.9, 120.9, 114.5 (2C), 111.7, 57.6, 56.0, 55.5, 52.7, 48.7, 36.4 ppm; UPLC-MS: Method A, Rt 2.13, ionization: m/z 464 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C23H25N7O4: 464.2046, found: 464.2052.
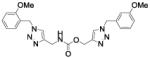
[1-[(3-methoxyphenyl)methyl]triazol-4-yl]methyl N-[[1-[(2-methoxyphenyl)methyl]triazol-4-yl]methyl]carbamate (39)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-[[1-[(2-methoxyphenyl)methyl]triazol-4-yl]methyl]carbamate (36a, 0.15 g, 0.5 mmol) and 1-(azidomethyl)-3-methoxy-benzene (0.08 g, 0.5 mmol), sodium ascorbate (0.010, 0.05 mmol), and copper (II) sulfate pentahydrate (0.001 g, 0.005 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 39 as a colourless amorphous solid (0.15 g; 66%): 1H NMR (400 MHz, [D6]DMSO): δ=8.15 (s, 1H), 7.80 (s, 1H), 7.69 (t, J=5.7 Hz, 1H), 7.34 (t, J=7.8 Hz, 1H), 7.28 (m, 1H), 7.06 (dd, J=14.4, 7.8 Hz, 2H), 6.89 (m, 4H), 5.54 (s, 2H), 5.48 (s, 2H), 5.04 (s, 2H), 4.21 (d, J=5.8 Hz, 2H), 3.80 (s, 3H), 3.73 ppm (s, 3H); 13C NMR (100 MHz, [D6]DMSO): δ=159.4, 156.8, 155.9, 145.0, 142.8, 137.4, 129.9 (2-C), 129.5, 124.6, 123.6, 122.8, 120.5, 120.0, 113.7, 113.4, 111.1, 57.1, 55.5, 55.0, 52.7, 48.1, 35.9 ppm; UPLC-MS: Method A, Rt 2.15, ionization: m/z 464 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C23H25N7O4: 464.2046, found: 464.2044.
[1-[(3-methoxyphenyl)methyl]triazol-4-yl]methyl N-[[1-[(4-methoxyphenyl)methyl]triazol-4-yl]methyl]carbamate (40)
The reaction was carried out following general procedure (1), using prop-2-ynyl N-[[1-[(4-methoxyphenyl)methyl]triazol-4-yl]methyl]carbamate (36c, 0.15 g, 0.5 mmol) with 1-(azidomethyl)-3-methoxy-benzene (0.08 g, 0.5 mmol), sodium ascorbate (0.010, 0.05 mmol), and copper (II) sulfate pentahydrate (0.001 g, 0.005 mmol) in water / t-BuOH 1:1 (3 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 2% MeOH in DCM, to afford compound 40 as a colourless amorphous solid (0.16 g; 68%): 1H NMR (400 MHz, [D6]DMSO): δ=8.15 (s, 1H), 7.89 (s, 1H), 7.69 (t, J=5.7 Hz, 1H), 7.28 (m, 3H), 6.90 (m, 4H), 6.85 (d, J=7.7 Hz, 1H), 5.55 (s, 2H), 5.46 (s, 2H), 5.04 (s, 2H), 4.20 (d, J=5.8 Hz, 2H), 3.73 ppm (s, 6H); 13C NMR (100 MHz, [D6]DMSO): δ=159.4, 159.1, 155.9, 145.3, 142.8, 137.4, 129.9, 129.6 (2C), 127.9, 124.6, 122.4, 120.0, 114.0 (2C), 113.7, 113.4, 57.1, 55.1, 55.0, 52.6, 52.2, 35.9 ppm; UPLC-MS: Method A, Rt 2.09, ionization: m/z 464 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C23H25N7O4: 464.2046, found: 464.2052.
(1-(1-phenyl-1,2,4-triazol-3-yl)methyl N-cyclohexylcarbamate (43)
It was synthesized according to the procedure employed for 32, starting from (1-phenyl-1,2,4-triazol-3-yl)methanol (42a, 0.05 g, 0.28 mmol), cyclohexyl isocyanate (0.04 g, 0.30 mmol), and DMAP (0.03 g, 0.28 mmol) in dry CH3CN (5 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 100% EtOAc in Cy, to afford compound 43 as a white powder (0.05 g; 58%): 1H NMR (400 MHz, [D6]DMSO): δ=9.26 (s, 1H), 7.89 – 7.77 (m, 2H), 7.62 – 7.49 (m, 2H), 7.48 – 7.37 (m, 1H), 7.26 (d, J=7.8 Hz, 1H), 5.08 (s, 2H), 3.26 (ddd, J=10.5, 7.2, 3.2 Hz, 1H), 1.86 – 1.60 (m, 4H), 1.53 (d, J=12.8 Hz, 1H), 1.30 – 1.01 ppm (m, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=161.0, 155.2, 143.5, 137.0, 130.2, 128.3, 119.7, 58.8, 50.0, 33.1 (2C), 25.6, 25.0 ppm (2C); UPLC-MS: Method A, Rt 2.34, ionization: m/z 301 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C16H20N4O2: 301.1665, found: 301.1674.
Benzyl-1,2,4-triazol-3-yl)methyl N-cyclohexylcarbamate (46)
It was synthesized according to the procedure employed for 32, starting from (1-benzyl-1,2,4-triazol-3-yl)methanol (42b, 0.06 g, 0.32 mmol), cyclohexyl isocyanate (0.044 g, 0.35 mmol), and DMAP (0.04 g, 0.32 mmol) in dry CH3CN (5 mL). Purification was performed by flash chromatography (SiO2) eluting with a gradient from 0 to 100% EtOAc in Cy, to afford compound 46 as a white powder (0.07 g; 68%): 1H NMR (400 MHz, [D6]DMSO): δ=8.62 (s, 1H), 7.42 – 7.25 (m, 5H), 7.17 (d, J=7.7 Hz, 1H), 5.37 (s, 2H), 4.93 (s, 2H), 3.27 – 3.15 (m, 1H), 1.68 (dd, J=28.7, 12.5 Hz, 4H), 1.52 (d, J=12.6 Hz, 1H), 1.13 ppm (tdd, J=32.8, 24.1, 12.1 Hz, 5H); 13C NMR (100 MHz, [D6]DMSO): δ=159.7, 154.7, 145.0, 136.1, 128.6, 127.9, 127.9, 58.4, 52.1, 49.4, 32.5 (2C), 25.1, 24.5 ppm (2C); UPLC-MS: Method A, Rt 2.27, ionization: m/z 315 [M+H]+; HRMS–ESI: m/z [M+H]+ calcd for C17H22N4O2: 315.1821, found: 315.1823.
Pharmacology
In vitro rat FAAH radiometric assay
Rat FAAH was prepared from male Sprague Dawley rat brains, homogenized in a potter in 20 mM of Tris HCl pH 7.4, 0.32 M sucrose. The radiometric assay used to measure FAAH activity was performed in Eppendorf tubes: 50 µg of total rat brain homogenate were pre-incubated in 445.5 µL of assay buffer (50 mM Tris-HCl pH 7.4, 0.05% Fatty acid-free -bovine serum albumin (BSA)) with 4.5 µL of inhibitor (at appropriate concentration in DMSO) or DMSO alone (to measure FAAH total activity) for 10 min at 37 °C. The blank (no activity control) was prepared using 445.5 µL of assay buffer and 4.5 µL of DMSO without the 50 µg of total rat brain homogenate. After 10 min of pre-incubation with test compounds, the reaction was started by adding 50 µL of substrate and incubating for 30 min at 37 °C. The substrate was prepared in assay buffer in order to achieve the final concentration of 1 µM arachidonoyl ethanolamide (Cayman Chemical N. 90050) and 0.6nM anandamide [ethanolamine-1-3H] (American Radiolabeled Chemicals Inc., ART. 0626, conc. 1 mCi/mL, S.A. 60 Ci/mmol). The reaction was stopped by adding cold 1:1 CHCl3/methanol. After 10 min of centrifugation (845xg at 4 °C) 600 µL of aqueous phase were transferred into scintillation vials previously filled with 3mL of scintillation fluid (Ultima GoldTM, Perkin Elmer Inc., Cat. 6013329). Radioactivity was measured by liquid scintillation counting (MicroBeta2 LumiJET Perkin Elmer Inc.).
In vitro human FAAH fluorescent assay
Human recombinant FAAH was obtained from a HEK-293 cell line stably overexpressing human FAAH-1 enzyme. Cells were grown in Dulbecco's Modified Eagle Medium (DMEM) containing 10% FBS, 1% pen/strep, 1% glutamine and 500 µg /mL G418. To obtain membrane preparation cells were scraped off with cold PBS and collected by centrifugation (500xg, 10 minutes, 4°C); the cell pellet was re-suspended in 20mM Tris-HCl pH 7.4, 0.32M sucrose, disrupted by sonication (10 pulses, 5 times) and centrifuged (800xg, 15 minutes, 4°C); the collected supernatant was centrifuged at 105,000xg for 1h at 4°C and the pellet was re-suspended in PBS. The fluorescent assay to measure FAAH activity was performed in 96 wells black plates: 2.5 µg of human FAAH-1 membrane preparation were pre-incubated for 50 min at 37 °C, in 180 µL of assay buffer (50mM Tris-HCl pH 7.4, 0.05% Fatty acid-free BSA) with 10 µL of inhibitor or 10 µL DMSO to measure FAAH total activity. The background (no activity) samples were prepared using 180 µL of assay buffer without human FAAH-1 and 10 µL of DMSO. The reaction was then started by addition of 10 µL of the substrate (7-amino-4-methyl coumarin-arachidonamideN. 10005098, Cayman Chemical) dissolved in ethanol and used at a final concentration of 2 µM. The reaction was carried out for 30 min at 37 °C and fluorescence was measured with a Tecan Infinite M200 nanoquant plate reader (excitation wavelength 350 nm / emission wavelength 460nm). IC50 values (concentrations causing half-maximal inhibition) were determined by non-linear regression analysis of the Log [concentration]/response curves generated with mean replicate values using a four parameter Hill equation curve fitting with GraphPad Prism 5 (GraphPad Software Inc., CA – USA).
Ex vivo FAAH inhibition assay
CD1 male mice from Charles River Italia were treated intraperitoneally (i.p.) with the test compound (3 mg/kg) or vehicle (1:1:8, PEG400, Tween® 80 and Saline 0.9%). One hour after treatment, the animals were killed by decapitation and the brain and liver were collected. Samples were homogenized in 1.5 mL of 20 mM Tris-HCl buffer pH 7.4, containing 0.32 M sucrose and the homogenates were centrifuged at 1000xg for 10 min (4°C). The supernatants were collected and the protein concentration was measured by Bradford method (Bio Rad Protein Assay Kit). FAAH activity was measured using 50 µg of total brain or liver homogenate in 450 µL of assay buffer (50 mM Tris-HCl pH 7.4, 0.05% Fatty acid-free BSA); the blank (no activity sample) was prepared with 450 µL of assay buffer. The reaction was started by adding 50 µL of substrate for 30 min at 37 °C. The substrate was prepared in assay buffer in order to obtain a final concentration of 1 µM arachidonoyl-ethanolamide (N.90050, Cayman Chemical) and 0.6 nM anandamide [ethanolamine-1-3H] (American Radiolabeled Chemicals Inc., ART.0626, 1 mCi/mL, specific activity 60 Ci/mmoL). The reaction was stopped by adding cold 1:1 chloroform/methanol. After 10 min centrifugation (845xg at 4°C) 600 µL of the aqueous phase was transferred into scintillation vials previously filled with 3 mL of scintillation fluid (ULTIMA GOLD, Cat.6013329, Perkin Elmer). Radioactivity was measured by liquid scintillation counting (Microbeta2 Lumijet, Perkin Elmer Inc.).
MGL activity assay
10 ng of the purified rat MGL was pre-incubated with appropriate drug for 10 min at 37 °C in 50 mM Tris-HCl, pH 8.0 containing 0.5 mg/mL fatty acid-free bovine serum albumin (BSA, Sigma-Aldrich). The final concentration of vehicle (1% DMSO) had no effect on MGL activity. Then, 2-oleoylglycerol (2-OG, 10 μM final) was added to the mixture and incubated for additional 30 min at 37 °C. Reactions were stopped by adding chloroform:methanol (2:1, vol:vol), containing heptadecanoic acid (5 nmol/sample) as an internal standard. After centrifugation at 2,000 x g at 4 °C for 10 min, the organic layers were collected and dried under a stream of N2. The lipid extracts were then suspended in chloroform:methanol (1:3, vol:vol) and analyzed by a liquid chromatography/mass spectrometry (LC/MS) method.
In vitro Rat Plasma stability assay
Compounds were diluted in rat plasma added with 10 % DMSO to help solubilization. Plasma was already pre-heated at 37° C (30 min). The final compound concentration was 1.0 μM. At time points (immediately after dilution, 30, 60, 120, 240, 360 and 420 min) a 40 µL aliquot of the incubation solution was diluted in 150 µL of cold CH3CN spiked with 200 nM warfarin as internal standard. After vortexing for 30 s, the solution was centrifuged at 3500 g for 15 min at 4 °C and the supernatant transferred for LC-MS analysis on a Waters ACQUITY UPLC/MS TQD system consisting of a TQD (Triple Quadrupole Detector) Mass Spectrometer equipped with an Electrospray Ionization interface. Briefly, 3.0 µL of the supernatant were injected on a reversed phase column (BEH C18 1.7 µm 2.1X50 mm) and separated with a linear acetonitrile gradient. Compounds were quantified on the basis of their MRM (Multiple Reaction Monitoring) peak areas. The response factors, calculated on the basis of the internal standard peak area, were then plotted over time. For each compound, analyses were conducted in triplicate: compound remaining (%) with corresponding standard deviation at 420 minutes is reported.
In vitro Mouse Liver Microsomes (MLM) stability assay
Compounds were preincubated with microsomes in 100mM TRIS-HCl pH 7.4 for 15 minutes. At time zero, cofactors were added. The final incubation conditions for each sample were: 1.25mg/mL liver microsomes, 5μM compound (final DMSO 0.1%), NADP 1mM, G6P 20mM, MgCl2 2mM, G6P dehydrogenase 2 Units. The mixture was kept at 37°C under shaking. Aliquots (30μL) were taken at various time points (typically 0, 5, 15, 30, and 60 minutes) and crashed with 200μL of acetonitrile spiked with 200nM warfarin (internal standard). A reference incubation, with microsomes but without cofactors, was kept at 37°C and sampled at the end of the time course. After vortexing and centrifugation, 3μL of superrnatant were analyzed by LC-MS/MS by multiple reaction monitoring (MRM).
Aqueous Kinetic Solubility assay
The kinetic solubility in Phosphate Buffered Saline (PBS) at pH 7.4 was determined starting from a 10 mM DMSO solution of the test compounds. The study was performed by incubation of an aliquot of 10 mM DMSO solution in PBS (pH 7.4) at 25°C for 24h, under shaking, followed by centrifugation and quantification of dissolved compound in the supernatant by UPLC/MS. The compound target concentration in the solutionwas 250 µM, resulting in a final DMSO concentration of 2.5%. The supernatant was analyzed by UPLC/MS and the quantification of the dissolved compound was determined by monitoring the UV trace at 215nm. The kinetic solubility (µM) was calculated by dividing the peak area of the test compound in the supernatant by the peak area of a reference solution (250 μM) of the test compound in 1:1 CH3CN-H2O, and further multiplied by the concentration of the test compound reference and the dilution factor. The UPLC/MS analyses were performed on a Waters ACQUITY UPLC/MS system consisting of a SQD (Single Quadrupole Detector) Mass Spectrometer equipped with an Electrospray Ionization interface and a Photodiode Array Detector. The PDA range was 210-400 nm. The analyses were run on an ACQUITY UPLC BEH C18 column (50x2.1 mmID, particle size 1.7 µm) with a VanGuard BEH C18 pre-column (5x2.1 mmID, particle size 1.7 µm). The mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in CH3CN-H2O (95:5) at pH 5 (B). Electrospray ionization in positive and negative mode was applied in the mass scan range 100-500Da.
Supplementary Material
Acknowledgements
The authors thank Sine Mandrup Bertozzi for determination of compound’s solubility in buffer and for HRMS analyses, Dr. Andrea Armirotti for discussions on the rat plasma stability data, Luca Goldoni for NMR technical support, and Silvia Venzano for compounds handling.
References
- 1.a) Desarnaud F, Cadas H, Piomelli D. J Biol Chem. 1995;270:6030–6035. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]; b) Hillard CJ, Wilkison DM, Edgemond WS, Campbell WB. Biochim Biophys Acta. 1995;1257:249–256. doi: 10.1016/0005-2760(95)00087-s. [DOI] [PubMed] [Google Scholar]; c) Ueda N, Kurahashi Y, Yamamoto S, Tokunaga T. J Biol Chem. 1995;270:23823–23827. doi: 10.1074/jbc.270.40.23823. [DOI] [PubMed] [Google Scholar]; d) Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 2.a) Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]; b) Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 3.Bisogno T, Maurelli S, Melck D, DePetrocellis L, DiMarzo V. J Biol Chem. 1997;272:3315–3323. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- 4.a) Rodriguez de Fonseca F, Navarro M, Gomez R, Escuredo L, Nava F, Fu J, Murillo-Rodriguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. Nature. 2001;414:209–212. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]; b) Piomelli D. Trends Endocrinol Metab. 2013;24:332–341. doi: 10.1016/j.tem.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodriguez De Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]; b) Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. Mol Pharmacol. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- 6.Di Marzo V. Nat Rev Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 7.a) Lambert DM, Muccioli GG. Curr Opin Clin Nutr Metab Care. 2007;10:735–744. doi: 10.1097/MCO.0b013e3282f00061. [DOI] [PubMed] [Google Scholar]; b) Ahn K, Johnson DS, Cravatt BF. Expert Opin Drug Dis. 2009;4:763–784. doi: 10.1517/17460440903018857. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hansen HS. Exp Neurol. 2010;224:48–55. doi: 10.1016/j.expneurol.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 8.a) Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. J Med Chem. 2003;46:2352–2360. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]; b) Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. J Med Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]; c) Clapper JR, Vacondio F, King AR, Duranti A, Tontini A, Silva C, Sanchini S, Tarzia G, Mor M, Piomelli D. ChemMedChem. 2009;4:1505–1513. doi: 10.1002/cmdc.200900210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]; b) Johnson DS, Ahn K, Kesten S, Lazerwith SE, Song Y, Morris M, Fay L, Gregory T, Stiff C, Dunbar JB, Jr., Liimatta M, Beidler D, Smith S, Nomanbhoy TK, Cravatt BF. Bioorg Med Chem Lett. 2009;19:2865–2869. doi: 10.1016/j.bmcl.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, Beidler D, Liimatta MB, Smith SE, Dudley DT, Sadagopan N, Bhattachar SN, Kesten SJ, Nomanbhoy TK, Cravatt BF, Ahn K. ACS Med Chem Lett. 2011;2:91–96. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexander JP, Cravatt BF. Chem Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Boger DL, Sato H, Lerner AE, Hedrick MP, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF. Proc Natl Acad Sci U S A. 2000;97:5044–5049. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Otrubova K, Ezzili C, Boger DL. Bioorg Med Chem Lett. 2011;21:4674–4685. doi: 10.1016/j.bmcl.2011.06.096. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Otrubova K, Boger DL. ACS Chem Neurosci. 2012;3:340–348. doi: 10.1021/cn2001206. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Otrubova K, Brown M, McCormick MS, Han GW, O'Neal ST, Cravatt BF, Stevens RC, Lichtman AH, Boger DL. J Am Chem Soc. 2013;135:6289–6299. doi: 10.1021/ja4014997. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Otrubova K, Cravatt BF, Boger DL. J Med Chem. 2014;57:1079–1089. doi: 10.1021/jm401820q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feledziak M, Lambert DM, Marchand-Brynaert J, Muccioli GG. Recent Pat CNS Drug Discov. 2012;7:49–70. doi: 10.2174/157488912798842223. [DOI] [PubMed] [Google Scholar]
- 13.Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 14.Tarzia G, Duranti A, Gatti G, Piersanti G, Tontini A, Rivara S, Lodola A, Plazzi PV, Mor M, Kathuria S, Piomelli D. ChemMedChem. 2006;1:130–139. doi: 10.1002/cmdc.200500017. [DOI] [PubMed] [Google Scholar]
- 15.a) Mor M, Lodola A, Rivara S, Vacondio F, Duranti A, Tontini A, Sanchini S, Piersanti G, Clapper JR, King AR, Tarzia G, Piomelli D. J Med Chem. 2008;51:3487–3498. doi: 10.1021/jm701631z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vacondio F, Silva C, Lodola A, Fioni A, Rivara S, Duranti A, Tontini A, Sanchini S, Clapper JR, Piomelli D, Mor M, Tarzia G. ChemMedChem. 2009;4:1495–1504. doi: 10.1002/cmdc.200900120. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, Hohmann AG, Calignano A, Mor M, Tarzia G, Piomelli D. Nat Neurosci. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Vacondio F, Silva C, Lodola A, Carmi C, Rivara S, Duranti A, Tontini A, Sanchini S, Clapper JR, Piomelli D, Tarzia G, Mor M. Eur J Med Chem. 2011;46:4466–4473. doi: 10.1016/j.ejmech.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Proc Natl Acad Sci U S A. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, Dasse O, Monaghan EP, Parrott JA, Putman D. Cns Drug Rev. 2006;12:21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Russo R, Loverme J, La Rana G, Compton TR, Parrott J, Duranti A, Tontini A, Mor M, Tarzia G, Calignano A, Piomelli D. J Pharmacol Exp Ther. 2007;322:236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- 17.a) McKinney MK, Cravatt BF. Annu Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]; b) Ahn K, McKinney MK, Cravatt BF. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Lodola A, Mor M, Hermann JC, Tarzia G, Piomelli D, Mulholland AJ. Chem Commun. 2005:4399–4401. doi: 10.1039/b503887a. [DOI] [PubMed] [Google Scholar]; b) Lodola A, Mor M, Zurek J, Tarzia G, Piomelli D, Harvey JN, Mulholland AJ. Biophys J. 2007;92:L20–L22. doi: 10.1529/biophysj.106.098434. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chudyk EI, Dyguda-Kazimierowicz E, Langner KM, Sokalski WA, Lodola A, Mor M, Sirirak J, Mulholland AJ. J Phys Chem B. 2013;117:6656–6666. doi: 10.1021/jp401834v. [DOI] [PubMed] [Google Scholar]; d) Lodola A, Capoferri L, Rivara S, Tarzia G, Piomelli D, Mulholland A, Mor M. J Med Chem. 2013;56:2500–2512. doi: 10.1021/jm301867x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mileni M, Kamtekar S, Wood DC, Benson TE, Cravatt BF, Stevens RC. J Mol Biol. 2010;400:743–754. doi: 10.1016/j.jmb.2010.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Edit. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; b) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Edit. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; c) Tornoe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]; d) Kolb HC, Sharpless KB. Drug Discov Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 21.Colombano G, Travelli C, Galli U, Caldarelli A, Chini MG, Canonico PL, Sorba G, Bifulco G, Tron GC, Genazzani AA. J Med Chem. 2010;53:616–623. doi: 10.1021/jm9010669. [DOI] [PubMed] [Google Scholar]
- 22.Fischer C, Zultanski SL, Zhou H, Methot JL, Brown WC, Mampreian DM, Schell AJ, Shah S, Nuthall H, Hughes BL, Smotrov N, Kenific CM, Cruz JC, Walker D, Bouthillette M, Nikov GN, Savage DF, Jeliazkova-Mecheva VV, Diaz D, Szewczak AA, Bays N, Middleton RE, Munoz B, Shearman MS. Bioorg Med Chem Lett. 2011;21:4083–4087. doi: 10.1016/j.bmcl.2011.04.089. [DOI] [PubMed] [Google Scholar]
- 23.Ramesh R, Bhat RG, Chandrasekaran S. J Org Chem. 2005;70:837–840. doi: 10.1021/jo048777o. [DOI] [PubMed] [Google Scholar]
- 24.Sarmiento-Sanchez JI, Ochoa-Teran A, Rivero IA. Arkivoc. 2011:177–188. [Google Scholar]
- 25.Curran TP, Yoon RSH, Volk BR. J Organomet Chem. 2004;689:4837–4847. [Google Scholar]
- 26.The half-life of URB524 is comparable to the value reported in ref. 15b for the same compound in 80% rat plasma.
- 27.Dinh TP, Freund TF, Piomelli D. Chem Phys Lipids. 2002;121:149–158. doi: 10.1016/s0009-3084(02)00150-0. [DOI] [PubMed] [Google Scholar]
- 28.Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.