

Abstract
Sirtuins are a class of enzymes with nicotinamide adenine dinucleotide (NAD)-dependent protein lysine deacylase function. By deacylating various substrate proteins, including histones, transcription factors, and metabolic enzymes, sirtuins regulate various biological processes, such as transcription, cell survival, DNA damage and repair, and longevity. Small molecules that can inhibit sirtuins have been developed and many of them have shown anti-cancer activity. Here we summarize the major biological findings that connect sirtuins to cancer and the different types of sirtuin inhibitors developed. Interestingly, biological data suggest that sirtuins have both tumor-suppressing and tumor-promoting roles. However, most pharmacological studies with small molecule inhibitors suggest that inhibiting sirtuin is a promising anti-cancer strategy. We discuss possible explanations for this discrepancy and suggest possible future directions to further establish sirtuin inhibitors as anticancer agents.
1. Introduction
Sirtuins are a family of enzymes with nicotinamide adenine dinucleotide (NAD)-dependent protein lysine deacylase activities. Yeast Sir2, the founding member of all sirtuins, is found to be important for calorie restriction-induced life span extension in yeast [1]. Subsequent biochemical studies demonstrate that it is an NAD-dependent deacetylase that regulate histone acetylation [2]. This interesting connection between aging and metabolism (the fact that sirtuins use a metabolic important molecule, NAD, as a co-substrate) has attracted great interest into this class of enzymes. In mammals, there are seven sirtuins (SIRT1-7) that localize in different subcellular compartments, such as cytoplasm (SIRT1 and SIRT2), nucleus (SIRT1, SIRT2, SIRT6, and SIRT7) and mitochondria (SIRT3, SIRT4 and SIRT5) [3,4]. The seven sirtuins share a conserved NAD-binding and catalytic core domain, but possess distinct N- or C-terminal extensions. By regulating the activity of various substrate proteins, sirtuins are involved in many biological pathways, including transcriptional regulation, genome stability, metabolic regulation, and cell survival [3]. Small molecules that can regulate sirtuin activities are considered as promising therapeutics to treat several human diseases, including neurodegeneration and cancer. This review will focus on the connection of sirtuins with cancers and the potential of sirtuin inhibitors as anticancer agents.
Growing evidence has shown that sirtuins are important for cancer cells. Many sirtuin inhibitors have been reported to have anticancer activities. Thus, the development of small molecules targeting sirtuins as anticancer therapeutics has been a focus of many studies. Research in the past decade, however, has also disclosed that some sirtuins possess a dual role in tumorigenesis – they could have both tumor-promoting and tumor-suppressing function. Improved understanding of the function of sirtuins and molecular mechanisms underlying their function will be beneficial to further establish the utility of sirtuins as cancer targets. In this review, we will first discuss the conflicting roles of sirtuins in cancer obtained from genetic studies (knockout, knockdown, and overexpression) and then we will highlight the progress on the development of sirtuin inhibitors with anti-cancer activity. Finally, we discuss possible explanations for the differences between genetic studies and small molecule studies and suggest future directions to further establish sirtuin inhibition as an anti-cancer strategy.
2. Sirtuins biology that is related to cancer
2.1.1 Tumor-suppressing roles of SIRT1
SIRT1 is the best studied sirtuin. Due to the large number of published studies and the reference limitation of this manuscript, we restrict our citations to a few excellent review papers [4–7]. Several genetic studies provide evidence that SIRT1 suppresses tumor formation. Studies on SIRT1 transgenic mice showed that conditional SIRT1 overexpression suppresses the incidence of intestinal tumor [8], spontaneous carcinomas and sarcoma, as well as carcinogen-induced liver cancer [9].
The tumor-suppressing role of SIRT1 may come from its ability to improve genomic stability by regulating chromatin and DNA repair. Sirt1−/− mouse embryos display altered histone modification accompanied with impaired DNA damage response and reduced DNA damage repair. In line with the decreased genome stability, Sirt1+/−p53+/− mice develop tumors in various tissues [5,10].
The tumor-suppressing role of SIRT1 may come from its ability to deacetylate and inactivate certain tumor-promoting transcription factors, such as NF-κB and HIF-1α. SIRT1 deacetylates RelA/p65 subunit of NF-κB at lysine 310 and inhibits its transcription activity, thereby augmenting TNF-α-induced apoptosis [11]. Overexpression of SIRT1 suppresses the growth and angiogenesis of fibrosarcoma HT1080 tumors in a mouse xenograft model by deacetylating and inactivating HIF-1α [12].
The tumor-suppressing role of SIRT1 could also be due to its ability to suppress the transcription tumor promoting genes by deacetylation of histones [4]. BRCA1 binds to the SIRT1 promoter and increases SIRT1 expression, which in turn inhibits Survivin by deacetylating H3K9 [13]. Therefore, ablation or mutation of BRCA1 results in increased Survivin level and promotes tumor growth by suppressing SIRT1 expression.
2.1.2. Tumor-promoting roles of SIRT1
Recent genetic studies on mice provided insight into the oncogenic activity of SIRT1 in vivo [5]. SIRT1 overexpression leads to increased thyroid and prostate tumorigenesis in Pten+/− mice [14]. Based on mRNA analysis, C-MYC levels are increased when SIRT1 is overexpressed [14]. In human papillary thyroid carcinomas, SIRT1 is overexpressed and SIRT1 level positively correlates with C-MYC level. It has been shown in another study that Sirt1 knockout suppresses BCR-ABL transformation of mouse bone marrow cells and the development of a CML-like disease in a mouse model [5,15]. A recent study indicated that enterocyte-specific inactivation of Sirt1 decreases tumor number and size in the APC+/min mouse model of intestinal tumorigenesis [16]. In addition, studies in many cancer cell lines showed that inhibiting SIRT1 or decreasing SIRT1 can inhibit cancer cell proliferation [17].
The role of SIRT1 in DNA repair and genome stability can also account for SIRT1’s tumor-promoting role. A recent study revealed SIRT1 help to acquire mutations for drug resistance in CML cells [18]. SIRT1 alters both homologous recombination (HR) and error-prone, non-homologous end joining (NHEJ) DNA repair pathways by regulating the key components in these pathways, KU70 and NBS1 (Nijmegen breakage syndrome 1). SIRT1 enhances error-prone DNA damage repair, resulting in acquisition of genetic mutation for CML drug resistance [18]. As noted in previous section, role of SIRT1 in DNA repair and genome stability has also been used to explain the tumor-suppressing role of SIRT1. It is proposed that SIRT1 might differentially regulate genome stability in normal cells versus cancer cells. In normal cells, SIRT1 promotes genomic stability by activating DNA repair with high fidelity, thereby suppressing tumor progression. In cancer cells, SIRT1 activates DNA repair with low fidelity to protect cells from deleterious impact of DNA damage. However, the error-prone DNA damage repair allows mutation acquisition in cancer cells and evolution toward high-grade malignancy [4–6].
The oncogenic role of SIRT1 is further supported by its role in inhibiting cell death mediated by tumor suppressors [6]. SIRT1 has been shown to promote cell survival by deacetylating and inhibiting the function of p53 [19]. SIRT1 deacetylates p53 at lysine 382 and negatively regulates its transactivation activity. Overexpression of SIRT1 strongly attenuates p53-dependent apoptosis upon DNA damage and oxidative stress. Forkhead box O (FOXO) transcription factors are an important family of tumor suppressors that modulate the expression of genes involved in cell cycle control, apoptosis and DNA repair. SIRT1 regulates various cellular processes by deacetylating FOXO family members. For example, SIRT1 deacetylates FOXO1 and inhibits FOXO1-induced apoptosis in prostate cancer cells [20]. Furthermore, SIRT1 mediated deacetylation of FOXO3a facilitates its ubiquitination and degradation [6,21]. Interestingly, FOXO transcription factors are downstream of PTEN. PTEN acetylation level is also increased upon Sirt1 knockout and SIRT1 can directly deacetylate PTEN [4]. However, it has been shown that deacetylation of FOXO proteins by SIRT1 can also activate their transcriptional activities. Daitoku et al. showed that SIRT1 potentiates transcription mediated by FOXO1 by deacetylation in mice [22]. In line with this, FOXO1 activation via SIRT1 is closely involved in Tamoxifen-resistance in human breast cancer MCF-7 cells [23]. SIRT1 inhibits FOXO3-induced cell death but increases FOXO3-mediated cell cycle arrest and resistance to oxidative stress [4]. These findings suggest that the regulation of FOXO by SIRT1 may be complex.
SIRT1 could also promote cell proliferation by positively regulating the oncogene protein MYC. Several reports suggest that SIRT1 and c-MYC form a positive-feedback loop. c-MYC can increase SIRT1 expression, SIRT1 in turn deacetylates and stabilizes c-MYC and enhances c-MYC transcriptional activity. Constitutive activation of this SIRT1-c-Myc positive feedback loop promotes c-MYC-induced cell proliferation by suppressing apoptosis and senescence [24]. In neuroblastoma, N-MYC up-regulates SIRT1, which in turn promotes oncogenesis through a positive feedback loop involving MKP3 and ERK[25]. Preventative treatment with the SIRT1 inhibitor Cambinol reduces tumorigenesis in TH-MYCN transgenic mice. However, a negative feedback loop between SIRT1 and c-MYC has also been reported and this suppresses cancer cell proliferation [26]. SIRT1 is also reported to promote accumulation of HIF-1α and activate the transcription of HIF-1α target gene expression in hepatocellular carcinoma [5]. Moreover, recent studies indicated that SIRT1 exhibits tumor-promoting functions by regulating Dishevelled (DVL)/Wnt signaling [27], DVL/TIAM1/Rac axis [28], and estrogen-related receptor α-mediated expression of aromatase (CYP19A1) [29] in different cellular contexts.
Additionally, SIRT1 may facilitate cancer evolution by modulating the epigenetic markers. SIRT1 forms the polycomb repressor complex 4 (PRC4) with DNMT1, DNMT3B, PcG proteins, EZH2 and EED2 [30]. Cancer risk states, such as oxidative damage, induce formation and re-localization of PRC4 complex, leading to cancer-specific aberrant DNA methylation and transcriptional silencing [30]. Additionally, SIRT1 promotes H3K9me3 establishment and silent chromatin formation by deacetylating H3K9 and by regulating the H3K9me3 methyltransferase Suv39h1 activity and stability [31]. SIRT1 and Suv39h1 establish silent chromatin in the ribosomal DNA locus, inhibit rRNA transcription, thereby protecting cells from energy deprivation-dependent apoptosis [32].
2.1.3 Altered SIRT1 expression or activity in cancer
In line with tumor suppressor role of SIRT1, it was found that SIRT1 levels are lower in breast cancer compared to their normal controls. A recent analysis of 41 breast cancer cell lines reveals that allelic loss as well as mutations in the SIRT1 gene occur prevalently during breast cancer progression, supporting that SIRT1 may act as a tumor suppressor [4].
Consistent with the tumor promoting role of SIRT1, elevated SIRT1 expression in human cancer samples has been correlated with worse prognosis and poor survival in various cancer types, including pancreas, prostate and liver cancers. Multitude mechanisms were reported to be involved in the altered SIRT1 expression in cancer. Some tumor suppressors repress SIRT1 expression. The tumor suppressor, hypermethylated in cancer 1 (HIC1), inhibits SIRT1 expression [33]. Loss of HIC1 function in mice promotes tumorigenesis by upregulating SIRT1, which deacetylates and inactivates p53, allowing cells to bypass the apoptosis induced by DNA damage. P53 binds to the SIRT1 promoter region and represses SIRT1 expression [34]. In contrast, SIRT1 expression is activated by oncogenic factors under certain circumstances. For instance, the oncogene MYC binds to the SIRT1 promoter and activates SIRT1 transcription [26]. BCR-ABL activates SIRT1 transcription partially through STAT5 and in CML cells [15]. In addition to transcription, the mRNA stability, protein stability, and enzyme activity of SIRT1 are also regulated during tumorigenesis. Various microRNAs (miRNAs) and RNA-binding proteins have been shown to bind to the 3′ untranslated regions of SIRT1 mRNA and negatively regulate SIRT1 mRNA stability. Among the miRNAs, miR-34a is the most studied one. Low levels of miR-34a were found in various human cancers [35], while overexpression of miR-34a in cancer cells decreases SIRT1 expression and triggers apoptosis [36]. The tumor suppressor, deleted in breast cancer 1 (DBC1), binds to the catalytic domain of SIRT1 and suppresses deacetylase activity of SIRT1. Knockout of Dbc1 increases SIRT1 activity and promotes tumorigenesis [6,7].
2.2. The role of SIRT2 in cancer
SIRT2 is predominantly localized in the cytoplasm but can transiently translocate to the nucleus during G2/M transition and deacetylate histone H4K16, thereby modulating chromatin condensation during metaphase[4]. Various other substrates have been recently reported, suggesting that SIRT2 is connected with multiple cellular processes [4].
SIRT2 knockout mouse studies have pointed to a tumor suppressor role of SIRT2. Sirt2 knockout mice look grossly normal, but starting from 10 months of age, they tend to have more tumors than the wild type. This effect is attributed to the role of SIRT2 in regulating the cell cycle by deacetylating APC/C [37]. The hypothesis is that SIRT2 is important for the cell cycle and thus without SIRT2, abnormal cell division occurs and thus tumor arises. However, the increased spontaneous tumor formation in Sirt2−/− mice may be strain-dependent and is not seen in another study [38]. Sirt2−/− cells have increased DNA damage and aberrant cell cycle progression compared with wild type cells. Although no increased spontaneous tumorigenesis was observed in the knockout mice up to one year of age, increase tumorigenesis was observed in an induced skin tumor model [38]. A recent report also suggested that SIRT2 can deacetylate and promote the degradation of ATP-citrate lyase, which is important for lipid biosynthesis and thus tumor growth [39]. Inhibition of SIRT2 promotes ATP-citrate lyase stability and thus may promote tumor growth.
In contrast to the tumor suppressor role of SIRT2, in many cancer cell lines, it has been demonstrated that SIRT2 knockdown or pharmacological inhibition can inhibit cancer cell proliferation and growth [40–48]. Several potential mechanisms have been suggested on the anti-proliferative effect of SIRT2 inhibition or knockdown in cancer cells. One mechanism is that SIRT2 helps to stabilize or activate oncoproteins, such as K-RAS, MYC, and FOXO. Thus, inhibiting SIRT2 will destabilize or inactivate these oncogenes and inhibit cancer. It is reported that SIRT2 can deacetylate K-RAS, for which activating mutations have been found in many cancers, and promotes it activity and cancer cell growth [49]. SIRT2 inhibition and knockdown have recently been shown to down regulate the C-MYC and N-MYC oncoproteins in neuroblastoma and pancreatic cancer cells [43]. This is achieved by releasing the inhibition of SIRT2 (via its histone deacetylase activity) on the transcription of the ubiquitin ligase NEDD4. SIRT2 has also been reported to deacetylate and decrease the level/activity of FOXO1 [50] and FOXO1 can increase cell death by activating autophagy [51]. Thus SIRT2 inhibition can promote cell death by increasing FOXO1 activity. Furthermore, SIRT2 has recently been shown to be an AKT binding partner and critical for its activation by insulin, suggesting the SIRT2 inhibition may be useful in the treatment of cancer [52].
SIRT2 inhibition has been shown to increase the levels of tumor suppressor genes, such as p53 and p21 [4,47]. Increased p53 level is achieved through deacetylation of p53, but the mechanism for increased p21 level is not clear. SIRT2 inhibition or knockdown can interfere with cancer cell metabolism, e.g. the Warburg effect. SIRT2 can deacetylate and activate lactate dehydrogenase A (LDH-A) [53]. LDH-A is over-expressed in many cancer cells and is responsible for the increased production of lactate in cancer cells. Thus inhibiting SIRT2 can potentially inhibit lactate production in cancer cells and disrupt cancer cell metabolism [53]. Additionally, SIRT2 may exert tumor-promoting function by epigenetically silencing tumor suppressors, such arrestin domain-containing 3 (ARRDC3) in basal-like breast cancer cells, by controlling histone acetylation [54].
2.3 The role of SIRT3 in cancer
SIRT3 is one of the major mitochondrial deacetylase and has been shown to regulate the activity of many mitochondrial proteins[4]. It seems that the major function of SIRT3 is to promote mitochondrial metabolism and suppress the production of reactive oxygen species (ROS), as Sirt3−/− mice or cells have decreased ATP production and increased ROS levels[4].
Strong evidence exists to support a tumor-suppressor role of SIRT3. Increased ROS levels in Sirt3−/− MEF cells are associated with increased mitochondrial DNA damage [55]. Sirt3 knockout itself does not transform MEF cells, but readily transform MEF cells when another oncogene, Ras or Myc, is overexpressed. In contrast, Sirt3+/+ MEF cells cannot be transformed by the overexpression of Ras or Myc [55]. Increased ROS level seems to be important for the tumor-permissive phenotype. Consistent with the tumor-permissive phenotype, Sirt3−/− mice develope mammary tumors over 24 months while Sirt3+/+ mice do not. Sirt3−/− MEF cells have increased ROS levels and glycolysis, similar to the Warburg effect in cancer cells [56]. Correspondingly, Sirt3−/− MEF cells proliferate faster than Sirt3+/+ cells. This effect is caused by increased ROS, decreased proline hydroxylase activity (the enzyme that hydroxylates and destabilizes HIF-1α), and thus increased HIF-1α level in the absence of SIRT3 [56]. These results are further confirmed by another study [57]. SIRT3 is heterozygously or homozygously deleted in about 20% of all human cancers and about 40% of breast and ovarian cancers. In human cancer cell lines, overexpression of SIRT3 reverses the Warburg effect and decreases cell proliferation [56]. It has also been reported that SIRT3 promotes oxidative phosphorylation (the opposite of Warburg effect) at least partially through deacetylation of cyclophilin D and the accompanied dissociation of hexokinase II from mitochondria [58,59]. Another possible molecular mechanism for the tumor suppression role of SIRT3 is suggested by studies in cardiac hypertrophy [60]. SIRT3 is shown to suppress cardiac hypertrophy by activating FOXO3a [60–62], which increases MnSOD and decreases ROS [60]. ROS can activate RAS, which further activate MAPK and AKT pathways to promote cell growth and proliferation [60]. SIRT3 can deacetylate and destabilize F-box protein S-phase kinase associated protein 2 (Skp2), a protein that supports tumorigenesis by promoting the ubiquitination and degradation of a number of tumor suppressors [63].
SIRT3 has also been reported to be present in the nucleus. Nuclear SIRT3 represses the expression of nuclear-encoded mitochondrial and some stress-related genes, including Zfat and Wapal. Both ZFAT and WAPAL have anti-apoptotic and oncogenic functions. By suppressing their expression, SIRT3 may suppress tumor formation [64]. In contrast, SIRT3 also physically binds to and deacetylates KU70, protecting cells from stress-mediated cell death in cardiomyocytes [65]. These studies indicate the previously unappreciated and sometimes contradicting roles of SIRT3 in the nucleus.
2.4 The role of SIRT4-7 in cancer
2.4.1 SIRT4
SIRT4 is one of the least understood sirtuins. SIRT4 inhibits glutamate dehydrogenase (GDH), thereby inhibiting amino acid induced insulin secretion in pancreatic beta cells [66]. The inhibitory effect on GDH seems to contribute to the tumor suppressor role of SIRT4. SIRT4 is down regulated in many cancers and mammalian target of rapamycin complex 1 (mTORC1) upregulates glutamine metabolism and cell proliferation by suppressing SIRT4 [67]. By inhibiting glutamine metabolism, SIRT4 also contributes to DNA damage and repair [68]. Transformed Sirt4−/− MEF cells form larger tumors than transformed Sirt4+/+ MEF cells in allograft tumor formation assay [68]. Sirt4−/− mice also developed more lung tumors than Sirt4+/+ mice at 18–26 months of age [68].
2.4.2 SIRT5
SIRT5 is another mitochondrial sirtuin that is not well understood and has no clear association with cancer. Sirt5−/− mice do not exhibit severe phenotype [69,70]. SIRT5 has weak deacetylase activity and has been demonstrated to have more efficient desuccinylase and demalonylase activity [71,72]. A recent proteomic study further identified hundreds of proteins with increased succinylation level when SIRT5 is knocked out, suggesting that SIRT5 regulates many metabolic pathways [73].
2.4.3 SIRT6
SIRT6 deacetylation substrates include histone H3K9 [74] and H3K56[4]. By deacetylating H3 associated as HIF-1α and MYC [4,75], SIRT6 suppresses the transcription of the target genes of these transcription factors. This mechanism has been used to explain many of the phenotype of Sirt6 knockout mice. However, given that the in vitro deacetylase activity is weak, other activity of SIRT6 has been reported. In particular, it has been shown that the activity to remove long chain fatty acyl group is hundreds-fold higher than deacetylation and this defatty-acylation activity promotes TNFα secretion [76]. The deacetylase activity of SIRT6 can be stimulated under specific conditions. For example, nucleosome [77] and free fatty acids [78] can increase in the deacetylation activity of SIRT6 in vitro, suggesting that the defatty-acylation and deacetylation activities of SIRT6 may be regulated and both can be relevant in vivo.
Most existing evidence suggests that SIRT6 acts as a tumor suppressor. Because SIRT6 promotes DNA repair and genome stability [74,79,80], it is easy to imagine that SIRT6 may suppress tumor development. A recent report indeed demonstrated that SIRT6 is a tumor suppressor and immortalized Sirt6−/− MEF cells are more tumorigenic than immortalized Sirt6+/+ MEF cells [75]. However, it has been suggested that Sirt6−/− MEF cells are more tumorigenic mainly due to the reprogramming of metabolism via the effects on two transcription factors HIF-1α and MYC, rather than the genome instability or oncogene activation [75].
2.4.4 SIRT7
SIRT7 is a nuclear sirtuin and is enriched in nucleoli [81]. Existing evidence suggests that it regulates ribosome biogenesis by controlling rRNA, tRNA, and ribosomal protein synthesis [80,82,83]. SIRT7 has been demonstrated to be a H3K18-specfic deacetylase [84]. It can be recruited by specific transcription factors, such as ELK4[84] and MYC [85], to specific genes (such as genes encoding ribosomal proteins) and repress the expression of these genes by deacetylating H3K18. It has been demonstrated that knockdown of SIRT7 inhibits the colony formation of HT1080 (a fibrosarcoma cell line) and U2OS (an osteosarcoma cell line) on soft agar, and decreases U251 (a glioma cell line) tumor size in mouse xenograft models [84]. SIRT7 knockdown also inhibits adenoviral E1A-induced cellular transformation [84]. The effects of SIRT7 knockdown might be mediated by the increased expression of certain genes, such as NME1 and ribosomal protein genes via H3K18 acetylation [84].
3. Sirtuin inhibitors
Since increasing evidence shows that sirtuins play a role in many biological processes, there has been sustained interest in developing small molecules that can regulate sirtuins. Both activators and inhibitors of sirtuins have been developed. Sirtuin activators are mainly developed for SIRT1, such as resveratrol, SRT1460, SRT1720 and SRT2183. Studies showed most of the compounds can delay age-related diseases including cancer, diabetes, inflammation, cardiovascular disease, and others, but the biochemical mechanism is still under debate [86]. Compared to sirtuin activators, more studies have been carried out towards sirtuin inhibitors, especially in the anticancer area. Below we provide a summary of different classes of sirtuin inhibitors based on their mechanism of action and structural features. Two class of sirtuin inhibitors, nicotinamide and thioacyllysine-containing compounds, can be considered as mechanism-based inhibitors [87–89]. Other sirtuin inhibitors presumably work by non-covalent binding to the sirtuin active site and block substrate binding.
3.1 Nicotinamide and its analogues
Nicotinamide (Table 1, entry 1) was one of the earliest sirtuin inhibitors discovered. Nicotinamide inhibits SIRT1, SIRT2, SIRT3, SIRT5 and SIRT6 with IC50 values varying from 50 to 184 μM [15,90,91]. It has been shown that nicotinamide can block proliferation and promote apoptosis in leukemic cells as well as inhibiting the growth and viability of human prostate cancer cells [92,93].
Table 1.
Sirtuin inhibitors:Nicotinamide and structurally similar analogs
| Entry # | Structure | IC50 | Biology Activity |
|---|---|---|---|
| 1 | nicotinamide
|
SIRT1: 120 μM[15] SIRT2: 100 μM[91] SIRT3: 50 μM[15] SIRT5: 150 μM[15] SIRT6: 184 μM[90] |
Blocked proliferation and promoted apoptosis selectively in leukemic and oral squamous cell carcinoma (OSCC) cells[92,98]; inhibited the growth and viability of human prostate cancer cells through inhibiting SIRT1[93] |
| 2 |
|
SIRT1: >300 μM SIRT2: 1 μM SIRT3: >300 μM[94] |
|
| 3 |
|
SIRT1: >300 μM SIRT2: 0.57 μM SIRT3: >300 μM[94] |
|
| 4 | AK-7
|
Sir1: ND SIRT2: 15.5 μM SIRT3: ND[95] |
Brain-permeability but limited metabolic stability[95] |
| 5 | 1,4-Dihydropyridine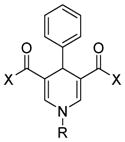
|
R=phenyl, X=OH or NH2 ~90% inhibition of SIRT1 at 50μM; R=phenyl or phenethyl, X=OEt ~60–70% inhibition of SIRT1 at 50μM and ~50% inhibition of SIRT2[96] |
|
| 6 |
|
SIRT1: 10.0 μM[97] | Inhibited MDA-MB-231 breast cancer cell lines with an IC50 of 0.25μM[97] |
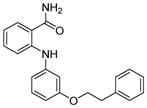
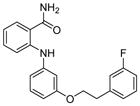
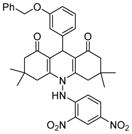
Analogues of nicotinamide and benzamide (a nicotinamide mimic) have been sought as sirtuin inhibitors. These analogs, however, most likely are not mechanism-based inhibitors. Two 3′-phenethyloxy-2-anilino benzamide analogues (Table 1, entries 2 and 3) are discovered as potent and selective SIRT2 inhibitors with IC50 value of 1 μM and 0.57 μM respectively. Selective SIRT2 inhibition by these inhibitors leads to increase of α-tubulin acetylation in human colon cancer HCT116 cells [94]. AK7 (Table 1, entry 4), another benzamide-containing compound also shows selective SIRT2 inhibition [95]. 1, 4-Dihydropyridine compounds can also inhibit sirtuins. Cyclopropyl, phenyl, or phenylethyl substituents at the N1 position of the dihydropyridine ring lead to compounds that can inhibit SIRT1 and SIRT2, but not SIRT3 (Table 1, entries 5) [96]. Another potent SIRT1 inhibitor (Table 1, entry 6) in a series of acridinedione derivatives shows anticancer activity with dose dependent increase in acetylation of SIRT1 target p53 K382[97].
3.2 Thioacyllysine-containing compounds as mechanism-based inhibitors of sirtuins
Compounds containing Nε-thioacetyllysine (Table 2, entries 1–7) can form covalent ADP-ribose-adduct (1′-S-alkylimidate intermediate) during the first step of the sirtuin-catalyzed deacetylation reaction [89,99]. This intermediate is relatively stable and does not readily undergo the normal downstream reactions. Thus, the intermediate occupies the sirtuin active site and inhibit the enzymatic activity of the sirtuin. Nε-thioacetyllysine is first incorporated into a peptide derived from the C-terminal region of the human p53 protein (amino acid residue 372–389) The resulting compound has a 2 μM IC50 value towards SIRT1 inhibition[100]. Later Nε-thioacetyllysine is also incorporated into tri-, tetra-, and pentapeptides based on the sequence of α-tubulin and p53. The p53 sequence gives better inhibition than the α-tubulin sequence. Among them, the most potent SIRT1 inhibitors have the IC50 values of 180–330 nM and the most potent SIRT2 inhibitor has an IC50 value of 1.8 μM. All the inhibitors show some selectivity for SIRT1 over SIRT2 and one of them, the tripeptide KK(N-thioacetyl)L gives the best selectivity (265-fold) while maintaining potent SIRT1 inhibition activity [101].
Table 2.
Sirtuin inhibitors: N-thiocarbamoyl lysine and N-Tfa lysine
| Entry # | Structure | IC50 | Biology Activity |
|---|---|---|---|
| 1 | H2N-KKGQSTSRHKK (N-thioacetyl)LMFKTEG-OH | SIRT1: 2 μM[100] | |
| 2 | KK(N-thioacetyl)L | SIRT1: 0.57 μM SIRT2: 151 μM[101] |
|
| 3 |
|
SIRT1: 2.7 μM SIRT2: 23 μM SIRT3: >100 μM[107] |
|
| 4 |
|
SIRT2: 0.24 μM SIRT2: 1.8 μM SIRT3: 3.9 μM[109] |
|
| 5 |

|
SIRT1: 0.89 μM SIRT2: 2.5 μM SIRT3: 8.4 μM[109] |
Antiproliferative effects on A549 lung carcinoma and MCF-7 breast carcinoma cells at μM concentrations causing cell cycle arrest at the G1 phase [109] |
| 6 |
|
SIRT1: 5.98 μM SIRT2: 25.8 μM SIRT3: 29.4 μM[109] |
|
| 7 |
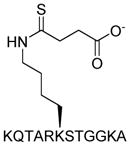
|
SIRT1: >100 μM SIRT2: >100 μM SIRT3: >100 μM SIRT5: 5 μM[15] |
|
| 8 |
|
SIRT1: 47 nM SIRT2: 3.2 nM SIRT3: 480 nM[110] |
|
| 9 |
|
SIRT1: 32 nM SIRT2: 3.7 nM SIRT3: 240 nM[110] |
|
| 10 | lin-S2iL8 AcLYSNFRIKTfaRYSNSSCR—NH2 |
SIRT1: n.d. SIRT2: 6.1 nM SIRT3: n.d.[110] |
|
| 11 | lin-S2iD7 AcDYHDYRIKTfaRYHTYPCR—NH2 |
SIRT1: n.d. SIRT2: 5.5 nM SIRT3: n.d.[110] |
|
| 12 | RIKTfaRY AcRIKTfaRY—NH2 |
SIRT1: 280 nM SIRT2: 31 nM SIRT3: 1000 nM[110] |
A lot of efforts have been invested to further investigate and improve this type of mechanism-based inhibitors, including using various N-acyl group [102–105], chemically changing the lysine side chain [106] as well as the C-terminal of the peptide [103].
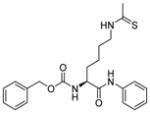
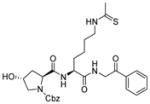
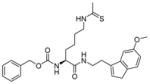
Peptide-based inhibitors are generally not very appealing as they may be unstable and not cell permeable for cellular or in vivo studies. Thus, a lot of efforts have been invested to develop non-peptide N-thioacetyllysine analogs. One N-thioacetyllysine-containing small molecule (Table 2, entry 3) showed selective SIRT1-inhibitory activity and caused a dose-dependent increase in p53 acetylation in human colon cancer HCT116 cells [107]. Structure-based computational design approach was performed in the design of N-thioacetyllysine containing pseudopeptidic inhibitors [108,109]. Three inhibitors (Table 2, entries 4–6) were picked out from a library of 30 thioacetyl pseudopeptides for further cellular studies. All three compounds showed an increase in acetylation of Lys382 of p53 after DNA damage. Two of them (Table 2, entries 5 and 6) showed anti-proliferative effect in A549 lung carcinoma and MCF-7 breast carcinoma cell lines [109].
Recently, a new twist in this type of mechanism-based inhibitors is to develop inhibitors specific for a particular sirtuin. This is prompted by the finding that human SIRT5, a mitochondrial sirtuin with weak deacetylase activity, is an efficient demalonylase and desuccinylase. Other human sirtuins do not have efficient demalonylase and desuccinylase activity. Taking the advantage of SIRT5’s unique acyl group preference, a thiosuccinyl H3K9 peptide is synthesized and shown to be a SIRT5-specific inhibitor with an IC50 value of 5 μM. The thiosuccinyl peptide does not inhibit SIRT1-3 even at 100 μM. In contrast, the thioacetyl H3K9 peptide inhibits SIRT1-3 potently, but does not inhibit SIRT5 [15]. This proof-of-principle study suggests that the mechanism-based inhibitors can be utilized to develop inhibitors specific for a particular sirtuin.
Trifluoroacetyl lysine-containing peptides have also been developed as mechanism-based sirtuin inhibitors [110]. Several cyclic and linear peptides containing trifluoroacetyl lysine (Table 2, entries 8–12) can inhibit SIRT2 selectively with low nM affinity. However, whether these compounds can inhibit cancer cell proliferation and growth has not been reported.
3. 3 β-naphthol-containing inhibitors
3.3.1 Sirtinol and its analogues
Sirtinol (Table 3, entry 1) is identified from a high throughput cell-based screen of more than 1000 compounds [111]. It inhibits yeast Sir2 and human SIRT1 with IC50 value of 70 μM and 40 μM in vitro, respectively, but it does not increase global acetylation levels of histones and tubulin in mammalian cells. SAR study shows that the hydroxyl-napthaldehyde moiety is important for the inhibition [111].
Table 3.
Sirtuin inhibitors: Sirtinol, salermide and their analogues
| Entry # | Structure | IC50 | Biology Activity |
|---|---|---|---|
| 1 | Sirtinol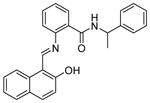
|
SIRT2: 40 μM[111] | Inhibited viability of breast, lung, prostate, and oral cancer cells[93,98,112,114] |
| 2 | JGB1741
|
SIRT1: ~15 μM SIRT2: >100 μM SIRT3: >100 μM[116] |
Inhibited proliferation of cancer cells MDA-MB-231 and K562[116] |
| 3 | Salermide
|
SIRT1: 80% inhibition at 100 μM SIRT2: 80% inhibition at 25 μM[117] |
No apparent toxicity in mice at concentrations of 100 μM[117]; induced apoptosis in MOLT4, KG1A, K562, SW480, Raji and NSCLC cells[43,117]; potent antiproliferative on MOLT4, MDA-MB-231 and colon RKO cancer cell lines and potent against colorectal carcinoma CSCs[118]; protected against OPMD[119] |
| 4 |
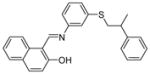
|
SIRT1: 40.3 μM SIRT2: 19.2 μM[118] |
Antiproliferative on MOLT4, MDA-MB-231 and colon RKO cancer cell lines and potent against glioblastoma multiforme CSCs [118] |
| 5 |
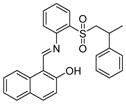
|
SIRT1: 40.3~67.3 μM SIRT2: 24.2 μM[118] |
Inhibited glioblastoma multiforme CSCs[118] |
Sirtinol is reported to have anticancer activity. It induces senescence-like growth arrest with reduced activation of RAS-MAPK pathway in human breast cancer MCF7 cells and lung cancer H1299 cells [112]. In another study, sirtinol induces cell apoptosis in MCF-7 cells in a process that requires p53 [113]. Treatment of sirtinol inhibits the growth of PC3 and Du145 cells and increases sensitivity of the cells to camptothecin and cisplatin [114]. Combined treatment of sirtinol and cisplatin also showed synergistic effect at inhibiting Hela cell proliferation [115].
JGB1741 (Table 3, entry 2) is a compound that was developed based on sirtinol [116]. It inhibits SIRT1 selectively with IC50 value of 15 μM. It inhibits the proliferation of three different cancer human cell lines, K562, HepG2 and MDA-MB-231, with IC50 values of 1, 10, and 0.5 μM, respectively. JGB1741 induces p53 level increase, cytochrome C release, and apoptosis in MDA-MB-231 cells.
3.3.2 Salermide
Salermide (Table 3, entry 3), a reversed amide based on the structure of sirtinol has stronger in vitro inhibitory effect on SIRT1 and SIRT2 than sirtinol [117]. It induces apoptosis in a wide range of human cancer cell lines but not in normal cells through inhibition of SIRT1 in a p53-independent manner. It shows much more significant inhibition of leukemia cell lines (MOLT4 and KG1A), colon cancer (SW480) and lymphoma (Raji) cells than the breast cancer cell line MDA-MB-231[117]. Salermide also induces apoptosis in non-small cell lung cancer (NSCLC) cells through up-regulation of death receptor 5 (DR5) [43]. Similar to sirtinol, the cytotoxic effect of salermide is dependent on the presence of functional p53 in the breast cancer cell line MCF-7 [113]. Several salermide analogs have also been developed and shown to have anti-proliferative effects in cancer cells (Table 3, entries 4 and 5) [118].
3.3.3 Cambinol
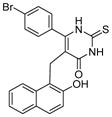
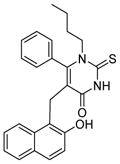
Cambinol (Table 4, entry 1) is another β-naphthol compound. It inhibits human SIRT1 and SIRT2 with IC50 values of 56 μM and 59 μM in vitro, respectively [40]. It has weak inhibition against SIRT5 and no inhibition against SIRT3. Kinetic studies reveal that cambinol is competitive with histone H4 peptide and noncompetitive with NAD. SAR studies show that the β-naphthol structure is important for the inhibitory activity of cambinol since the activity is lost when β-naphthol is replaced by phenol [40]. Changes on the phenyl ring of cambinol or functionalization of N1-position lead to improved activity and/or selectivity [120]. For example, the p-bromo-analogue (Table 4, entry 2) shows increased selectivity for inhibiting SIRT1 (IC50 =12.7 μM) against SIRT2 (IC50>90 μM). N-Alkylation with butyl group (Table 4, entry 3) significantly enhances the selectivity for SIRT2 with an IC50 value of 1μM [120]. Through virtual screening and subsequent experimental test, a series of molecules similar to cambinol are identified. These thiobarbiturate-based inhibitors (Table 4, entries 4 and 5) show inhibition of SIRT1 and SIRT2 at micromolar concentrations. Alternation of substituted groups can change the selectivity moderately [121].
Table 4.
Sirtuin inhibitors: Cambinol and its analogues
| Entry # | Structure | IC50 | Biology Activity |
|---|---|---|---|
| 1 | Cambinol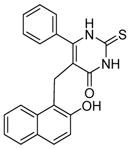
|
SIRT1: 56 μM SIRT2: 59 μM SIRT5: 42% inhibition at 300 μM[40] |
Induces apoptosis in BCL6-expressing Burkitt lymphoma cells[40]; reduces tumor growth of Burkitt lymphoma in a mouse xenograft model[40]; reduces neuroblastoma formation in N-Myc transgenic mice[25]; |
| 2 |
|
SIRT1: 12.7 μM SIRT2: >90 μM[120] |
|
| 3 |
|
SIRT1: 16.9% at 60 μM SIRT2: 1 μM[120] |
|
| 4 |
|
SIRT1: 5.9 μM SIRT2: 20.3 μM[121] |
|
| 5 |
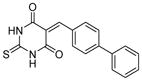
|
SIRT1: 50.5 μM SIRT2: 8.7 μM[121] |
In cellular studies, cambinol leads to hyperacetylation of tubulin, p53, KU70 and FOXO3a and promotes cell cycle arrest, presumably by inhibiting SIRT1. Treatment of BCL6-expressing Burkitt lymphoma cells with cambinol induces apoptosis and reduces tumor growth in a mouse xenograft model [40]. Preventative treatment with cambinol reduces neuroblastoma formation in N-Myc transgenic mice [25]. Cambinol markedly decreases aromatase (CYP19A1) levels in human breast cancer cells by inhibiting SIRT1-mediated deacetylation and transcription activity of estrogen-related receptor α [29].
3.3.4 Splitomicin and its derivatives
Splitomicin (Table 5, entry 1) is identified from a cell-based screening for inhibitors of Sir2 and Hst1 from yeast [122]. HR73 (Table 5, entry 2), a derivative of splitomicin which has a substitution of phenyl on 2-position and bromo on 8-position effectively inhibits SIRT1 with an IC50 of <5 μM [123]. Treatment with this molecule significantly inhibits HIV transcription through affecting acetylation of Tat [123].
Table 5.
Sirtuin inhibitors: Splitomicin and its analogues
| Entry # | Structure | IC50 | Biology Activity |
|---|---|---|---|
| 1 | splitomicin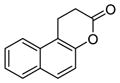
|
60 μM for Sir2[122]. No inhibition of mammalian sirtuins [123] | |
| 2 | HR73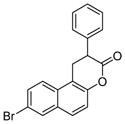
|
SIRT1: < 5μM[123] | Decreased HIV transcription through Tat acetylation[123] |
| 3 |

|
SIRT2: 5.2 μM[124] | |
| 4 |
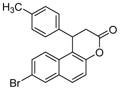
|
SIRT2: 1.5 μM[124] | Weak anti-proliferative properties in MCF7 breast cancer cells; increased tubulin acetylation in MCF7 breast cancer cells[124] |
| 5 |

|
SIRT2: 1.5 μM (racemic) 1.0 μM (R); 35.1% at 100 μM (S)[124] | Weak antiproliferative properties in MCF7 breast cancer cells; increased tubulin acetylation in MCF7 breast cancer cells[124] |
| 6 | benzodeazaoxaflavins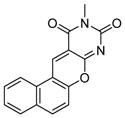
|
SIRT1: 7.0 μM SIRT2: 11.2μM[125] |
Anti-proliferative properties in cancer stem cells of colorectal carcinoma and glioblastoma multiforme[125] |
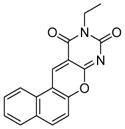
| 7 |
|
SIRT1: 20.2 μM SIRT2: 30 μM[125] |
Anti-proliferative properties in cancer stem cells of colorectal carcinoma and glioblastoma multiforme [125] |
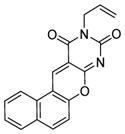
| 8 |
|
SIRT1: 6.6 μM SIRT2: 11 μM[125] |
Anti-proliferative properties in cancer stem cells of colorectal carcinoma and glioblastoma multiforme [125] |
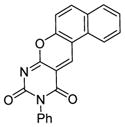
| 9 |
|
SIRT1: 8.4 μM SIRT2: 191.2 μM[119] |
Anti-proliferative effects in Raji, DLD1, and Hela cells[126] |
| 10 |
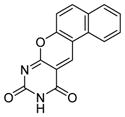
|
SIRT1: 4.2 μM Sir2: no inhibition[126] |
|
| 11 |
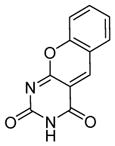
|
SIRT1: 5.3 μM SIRT2: 243.6 μM[126] |
A serials of compounds based on β-aryl splitomicins are synthesized and several SIRT2 inhibitors are identified with low IC50 values in the micromolar range [124]. β-(4-Methyl)phenyl-8-bromo-splitomicin (Table 5, entry 4) and the R-enantiomer of β-(4-methyl) phenyl-8-methyl-splitomicin (Table 5, entry 5) inhibit SIRT2 with IC50 values of 1.5 μM and 1.0 μM, respectively. These compounds inhibit the proliferation of MCF-7 breast cancer cells only modestly, which is thought to be due to their high lipophilicity.[124].
3.3.5 Splitomicin and cambinol-like inhibitors
A novel class of sirtuin inhibitors is identified with structure of tri- and tetracyclic pyrimidinediones (Table 5, entries 6–11). These inhibitors bear structural similarity to both splitomicin and cambinol. Three molecules (Table 5, entries 9–11) show selective inhibition of SIRT1 over SIRT2 with low micromolar IC50 values and one of them (Table 5, entry 9) inhibits the proliferation of several cancer cells, including Raji, DLD1 and Hela cells [126].
3.4 Indole derivatives
A series of indole compounds are discovered from a high-throughput screening of 280,000 compounds as potent SIRT1 inhibitors [127]. They are selective over SIRT2, SIRT3, and the NAD glycohydrolase CD38. One of inhibitors, EX-527 (Table 6, entry 1) has an IC50 value in the range of 60 nM to 100 nM in vitro and is cell permeable. Study of structure-activity relationships shows the importance of the primary carboxamide and its position. Small nonpolar groups at the 6-position are well tolerated and expansion of 6-membered ring to 7-membered ring gives the same activity [127]. Inhibition of SIRT1 with EX-527 leads to increased p53 acetylation after DNA damage in primary human mammary epithelial cells and several cancer cell lines, but fails to inhibit cell growth and viability [128,129].
Table 6.
Sirtuin inhibitors: Indole derivatives
| Entry # | Structure | IC50 | Biology Activity |
|---|---|---|---|
| 1 | EX527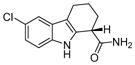
|
SIRT1: 60–100 nM[127] | Induced apoptosis in leukemia cell when combined with HDAC inhibitors[135]; protected against OPMD [119] |
| 2 | AC-93253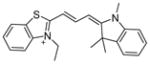
|
SIRT1: 45.3 μM SIRT2: 6 μM SIRT3: 24.6 μM[48] |
Cytotoxic effects in prostate DU145, pancreas MiaPaCa, lung A549 and NCI-H460 cancer cell lines[48] |
| 3 | Inauhzin (INZ)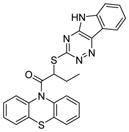
|
SIRT1: 0.7–2 μM[130] | Inhibited cell proliferation, induced senescence and apoptosis of human cancer cell without genotoxicity, and repressed the growth of xenograft tumors derived from human lung cancer H460 and colon cancer HCT116 cells harbouring p53[130]. |
| 4 | Ro31-8220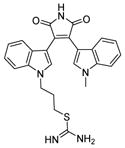
|
SIRT1: 3.5 μM SIRT2: 0.8 μM[132] |
|
| 5 | GW5074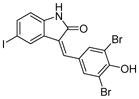
|
SIRT1: 41.6 μM [133] SIRT2: 15.6 μM [133] SIRT3: 25.1 μM [133] SIRT5: 19.5 μM [134] |
AC-93253 (Table 6, entry 3), a compound containing a modified indole ring, preferentially inhibits SIRT2 and has cytotoxic effect in several cancer cell lines [48]. Another indole derivative, inauhzin (Table 6, entry 4) is identified via virtual screening for compounds that can inhibit p53 and MDM2/MDMX interaction. Inauhzin has potent anti-cancer activity and represses the growth of xenograft tumors derived from H460 and HCT116 cells [130].
Sirtuins use NAD as a co-substrate and kinases use ATP as a co-substrate. Given that both NAD and ATP contain an adenosine moiety, kinase inhibitors may inhibit sirtuins. Several kinase inhibitors are identified as sirtuin inhibitors. Ro31-8220 (Table 6, entry 5), a nM protein kinase C inhibitor [131], is one of the most active inhibitors based on bis(indolyl)maleimides with IC50 values of 0.8 μM against SIRT2 and 3.5 μM against SIRT1. Ro31-8220 induces hyperacetylation of tubulin in A549 human lung adenocarcinoma cells [132]. GW5074 (Table 6, entry 6), a nanomolar c-RAF1 kinase inhibitor, shows inhibition of SIRT2 with an IC50 value of 15.6 μM [133] and SIRT5 with an IC50 value of 19.5 μM [134]. However, given that these compounds are much better kinase inhibitors, the utility of them as sirtuin inhibitors may be limited.
3.5 Suramin
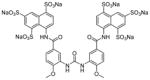
Suramin is developed for the treatment of sleeping sickness and onchocerciasis. It inhibits the deacetylase activity of SIRT5 with an IC50 value of 22 μM [136]. Structural study showed suramin binds between NAD binding site and the substrate binding site to inhibit sirtuin activity [136]. A set of suramin analogues were synthesized to study the potency and selectivity towards various sirtuins [137]. Most of the inhibitors, including suramin, showed preference for SIRT1. One of them (Table 7, entry 2) is the aminoanthranilic acid derivative that exhibits IC50 values of 93 nM against SIRT1 and 2.3 μM for SIRT2. The most selective but still potent inhibitor is a urea compound with a 170-fold selectivity between SIRT1 and SIRT2 (Table 7, entry 3). However, suramin targets many other proteins, including cell surface receptors. Suramin and its analogues also likely suffer from cell impermeability due to the highly polar sulfonic acid structure. These features may limit their utility as sirtuin inhibitors [137].
Table 7.
Sirtuin inhibitors: Suramin and its analogues

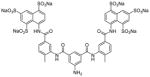
3.6 Tenovin and its analogues
A cell-based screen designed to detect small molecules that activated the tumor suppressor p53 leads to the discovery of tenovin-1 (Table 8, entry 1) and its water-soluble analogue tenovin-6 (Table 8, entry 2) [138,139]. Biochemical assays suggest that the targets of the tenovins are SIRT1 and SIRT2. At low micromolar concentrations, tenovin-1 shows cytotoxic effects to the BL2 Burkitt’s lymphoma cells and ARN8 melanoma cells expressing wild type p53, and reduced tumor growth derived from those cells [138]. Treatment of tenovin-1 with mutant p53 breast cancer cell lines (MDA-MB-231 and MDA-MB-468) and p53 null cells suggests that functional p53 likely contributes to the cytotoxicity of tenovin-1 but is not essential for the long-term killing effect [138]. Tenovin-6 is more active than tenovin-1 to ARN8 melanoma cells and it delays the growth of ARN8-derived xenograft tumors [138]. Treatment with the tenovin-6 in mice deters the disease progression of chronic myelogenous leukemia [15]. A series of analogues of tenovin-1 and tenovin-6 are synthesized to obtain more water-soluble molecules. The tenovins adopt a preferred conformation in solution containing an intramolecular hydrogen bond based on 1H NMR and X-ray crystallographic analysis. The ability to form this intramolecular hydrogen bond correlated to the inhibition of SIRT1 [139].
Table 8.
Sirtuin inhibitors: Tenovin and its analogues
| Entry # | Structure | IC50 | Biology Activity |
|---|---|---|---|
| 1 | tenovin-1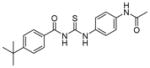
|
Not determined due to lack of water solubility[139] | Cytotoxic to the BL2 Burkitt’s lymphoma cells and ARN8 melanoma cells; reduced tumor growth in the BL2 and ARN8 mouse xenograft model [138] |
| 2 | tenovin-6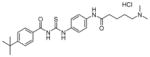
|
SIRT1: 37.5 μM SIRT2: 10.4 μM[139] |
Cytotoxic to the ARN8 melanoma cells; delayed the growth of xenograft tumors derived from ARN8 cells [138]; detered the disease progression of chronic myelogenous leukemia in mice model [15] |
3.7 Other sirtuin inhibitors
Many other types of compounds have been reported as sirtuin inhibitors (Table 9, entries 1–10). Several of these are worth commenting. AGK2 (Table 9, entry 1) is discovered from a focused compound library as a selective SIRT2 inhibitor which rescues α-synuclein toxicity and protects against Parkinson’s disease in a cellular model [140]. This study suggests that SIRT2 may be a good target for Parkinson’s disease. Phloroglucinol derivatives, such as aristoforin (Table 9, entry 2), is shown to inhibit SIRT1 and SIRT2 [141]. Aristoforin has anti-proliferative activity on human umbilical vein endothelia cells (HUVEC), but reports on its anti-cancer activity are lacking. Aurone derivatives, such as entry 3 in Table 9, have also been shown to inhibit SIRT1 and the proliferation of breast cancer cells [142].
Table 9.
Other sirtuin inhibitors
| Entry # | Structure | IC50 | Biology Activity |
|---|---|---|---|
| 1 | AGK2
|
SIRT1: >50 μM SIRT2: 3.5 μM SIRT3: >50 μM[140] |
Protective against Parkinson’s disease[140,143]; potent against glioblastoma multiforme CSCs[118] |
| 2 | Aristoforin
|
SIRT1: 7 μM SIRT2: 21 μM[141] |
Anti-proliferative activity on HUVEC cells[141] |
| 3 |

|
SIRT1: 1 μM[142] | Anti-proliferative in MAD-MB-231 and MCF7 cancer cell lines[142]; |
| 4 | Tanikolide Dimer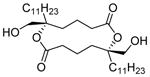
|
SIRT1: 36.4 μM SIRT2: 3.3 μM[144] |
|
| 5 |

|
SIRT2: 1.5 μM[145] | |
| 6 |
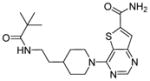
|
SIRT1: 15 nM SIRT2: 10 nM SIRT3: 33 nM [146] |
|
| 7 |
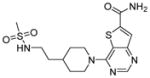
|
SIRT1: 4.3 nM SIRT2: 1.1 nM SIRT3: 7.2 nM [146] |
The most potent sirtuin inhibitors reported to date are thieno[3,2-d]pyrimidine-6-carboxamides (Table 9, entries 6 and 7) that are developed using encoded library screen technology. These compounds inhibit SIRT1-3 with low nM IC50 values [146]. Extensive structural-activity relationship studies are carried out and many active sirtuin inhibitors are obtained. These studies reveal that the carboxamide moiety is important for the sirtuin inhibition activity. Crystal structures of SIRT3 in complex with several of these inhibitors showed that the carboxamide moiety occupies the nicotinamide pocket and forms several hydrogen bonding with Sirt3, providing a nice explanation for the importance of the carboxamide moiety [146]. However, the biological activities of these compounds have not been reported. It would be very interesting to see whether these compounds exhibit anti-cancer activity.
4. Future Perspective
It seems that the biological data described in Section 2 and the sirtuin inhibitor studies described in Section 3 above do not completely agree with each other. One of the most striking differences between the biological data and pharmacological data is that while the former points to conflicting (both tumor-suppressing and tumor-promoting) roles of different sirtuins, the majority of sirtuin inhibitors studies suggest that inhibiting sirtuin is a promising anti-cancer strategy. We think there are several possible reasons for this, as explained below.
First, inhibiting an enzyme with a small molecule is different from knocking down or knocking out the corresponding gene. The enzyme concentration generally remains unchanged in cells with a small molecule inhibitor, while the concentration is lower when the gene is knocked out or knocked down. In one simple scenario, if the enzyme also has other roles (e.g. mediating protein-protein interactions) that are not dependent on its enzymatic activity, using small molecule inhibitors likely will not affect these other roles. Many examples in the kinase inhibitor literature have documented that inhibitors may produce different effects from genetic knockout or knockdown [147]. In this sense, we would like to emphasize those efforts to develop sirtuin inhibitors as potential anti-cancer agents should not be discouraged by genetic evidence showing that sirtuins may be tumor suppressors. Ultimately, pharmacological data would be the most important for therapeutics development.
Second, it is possible that the anti-cancer effects of sirtuin inhibitors are due to the inhibition of multiple sirtuins. This is very likely especially because most of the sirtuin inhibitors that show anticancer effects are not very selective for a particular sirtuin. Peck et al. have proposed that inhibiting both SIRT1 and SIRT2 is important for the anticancer effect of salermide and sirtinol [113]. Most of the inhibitors shown in Tables (1–9) only have modest selectivity (≤ 10-fold) toward different sirtuins. Small molecule inhibitors of the zinc-dependent histone deacetylases that are FDA-approved for treating cancers, such as SAHA, can non-specifically inhibit several different HDACs and it was speculated that this lack of specificity may be beneficial [148]. This situation may also be true for other enzyme inhibitors that were once thought to be very specific. For example, Gleevec, was originally considered to exert its anti-cancer effect by inhibiting BCR-ABL, but later it was demonstrated that the therapeutic effects also come from the inhibition of another kinase KIT [149]. These observations have generated a new concept of “polypharmacology”, which is the treatment of diseases by affecting multiple protein targets [150]. Thus, the broader specificity of small molecule inhibitors may be a desirable property that genetic knockdown or knockout cannot easily achieve. In the future, the field should try to develop a series of sirtuin inhibitors with different specificity profiles and to test the hypothesis that inhibiting several sirtuins may be better than inhibiting only one sirtuin in cancer treatment. Of course, the key is to find the right balance in specificity, potency, and toxicity. It is possible that inhibitors that non-specifically inhibit all sirtuins are too toxic and thus not therapeutically useful.
Third, there is a possibility that the anti-cancer effects of sirtuin inhibitor are due to off-target effects. This is less likely as it is hard to imagine that off-target effects would occur to so many sirtuin inhibitors. However, it does raise a deficiency in most sirtuin inhibitors studies. In most sirtuin inhibitor studies, few experiments are carried out to carefully examine whether the anti-cancer effect of sirtuin inhibitors is due to sirtuin inhibition or not. RNA interference and overexpression could serve as a simple way to test this. For example, if the effect of a sirtuin inhibitor is due to the inhibition of SIRT1, then overexpression of SIRT1 should decrease while knockdown of SIRT1 should increase the sensitivity of cancer cells to the sirtuin inhibitor. In contrast, knockdown or overexpression of an irrelevant sirtuin should not significantly affect the sensitivity of cells to the sirtuin inhibitor.
In the next 5–10 years, more and better sirtuin inhibitors will be developed and help to address a number of questions raised above. Most of the sirtuin inhibitors reported are still not potent enough with IC50 values in the μM range. Recently, sirtuin inhibitors with low nM IC50 values are reported [146], suggesting that highly potent sirtuin inhibitors are on the rise. More potent sirtuin inhibitors will help to reduce the concern about off-target effects and help to directly test whether inhibiting sirtuins is a good strategy for treating cancers. Currently, most of the sirtuin inhibitors are not tested for their specificity toward the seven human sirtuins. For a long time, the inhibitors are only tested for SIRT1 and SIRT2 inhibition. In the future, the inhibitors will have to be tested against all the sirtuins. This will allow the development of a set of inhibitors with distinct selectivity, which will in turn allow the community to find out inhibiting which sirtuins provide maximum benefit in combating cancer. Most of the existing sirtuin inhibitors only inhibit SIRT1-3 and there are no good inhibitors for SIRT4-7 yet. The lack of SIRT4-7 inhibitors is mainly due to the lack of efficient enzymatic activity assays for these sirtuins as they have weak and sometimes undetectable deacetylase activity. With the finding of more efficient activities for SIRT5 and SIRT6 [71] [76], better enzyme assays are now available and we expect that potent inhibitors for SIRT4-7 will soon emerge. The inhibitors developed will in turn help to establish the feasibility of targeting sirtuins as anti-cancer therapies.
Executive Summary.
-
Genetic studies have revealed both tumor-suppressing and tumor-promoting roles for SIRT1-3. Molecular pathways that can explain these roles are discussed.
Genetic studies on SIRT1 revealed both tumor-suppressing and tumor-promoting roles for SIRT1;
Genectic studies in mice support a tumor-suppressing role for SIRT2, while small molecule inhibition studies and knockdown in cancer cell lines support a tumor-promoting role for SIRT2;
Most biological studies support that SIRT3 is a tumor suppressor.
-
The role of SIRT4-7 in cancers are less well studied, but existing evidence suggests that
SIRT4 has a tumor suppressing role;
SIRT6 has a tumor suppressing role;
SIRT7 has a tumor-promoting role.
-
Reported sirtuin inhibitors are summarized into different structural classes:
Nicotinamide and its analogues;
Thioacyllysine-containing compounds;
β-naphthol-containing inhibitors, including sirtinol, cambinol, and splitomicin and derivatives;
Indole derivatives;
Suramin and its analogs;
Tenovin and its analogs;
Other sirtuin inhibitors.
The discovery, development, and biological activities of the inhibitors are discussed.
In contrast to the conflicting genetic evidence, most sirtuin inhibitor studies suggest that inhibiting sirtuins is a promising strategy to inhibit cancer cell proliferation and growth. However, inhibiting which sirtuin(s) underlies the anticancer effects or whether these sirtuins inhibitors have off-target effects are not well known in many cases.
We propose that the difference between genetic (knockout or knockdown) and small molecule inhibitor studies can be explained in several different ways. Future studies should aim to develop more potent and more specific sirtuin inhibitors, which can help to understand the discrepancy and further establish the utility of sirtuin inhibitors as anticancer agents.
Key terms
- Oncogene
genes that can cause cancer. They are typically mutated or overexpressed in cancer
- Epigenetic
changing gene function or expression without changing the DNA sequence. Typically refers to modifications on DNA or histones that affect transcription
- Knockdown
using short hairpin RNA or short double-strand interfering RNA to turn down the expression of genes that have complementary sequence to the RNA used
- Knockout
eliminate the expression of a gene by deleting part of or the entire gene
- Warburg effect
the observation that cancer cells takes up a lot of glucose and most of the glucose is metabolized to lactate via aerobic fermentation
- Off-target effect
The biological effect of a small molecule inhibitor is commonly believed to be achieved by targeting a specific protein. However, in certain cases, the biological effect could also come from targeting other unidentified proteins. The effect from targeting these unidentified proteins is termed off-target effect
- IC50 value (of a sirtuin inhibitor)
The concentration of the inhibitor required to inhibit 50% of the enzymatic activity. The IC50 value is dependent on the exact assay condition used, such as enzyme and substrate concentrations. Thus, comparison of IC50 values is only meaningful if the assays are carried out under the same conditions
Footnotes
Disclosure. Work in our lab on sirtuin inhibitors is supported by NIH grant R01 CA163255. H.L and Cornell University have patents pending on sirtuin inhibitors.
References
- 1.Lin S-J, Defossez P-A, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289(5487):2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 2.Imai S-i, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403(6771):795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 3.Haigis MC, Sinclair DA. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu Rev Pathol. 2010;5(1):253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med. 2013;56(0):133–171. doi: 10.1016/j.freeradbiomed.2012.10.525. [DOI] [PubMed] [Google Scholar]
- 5.Roth M, Chen WY. Sorting out functions of sirtuins in cancer. Oncogene. 2013 doi: 10.1038/onc.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin Z, Fang D. The Roles of SIRT1 in Cancer. Genes Cancer. 2013;4(3–4):97–104. doi: 10.1177/1947601912475079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song NY, Surh YJ. Janus-faced role of SIRT1 in tumorigenesis. Ann N Y Acad Sci. 2012;1271:10–19. doi: 10.1111/j.1749-6632.2012.06762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Firestein R, Blander G, Michan S, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS One. 2008;3(4):e2020. doi: 10.1371/journal.pone.0002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herranz D, Munoz-Martin M, Canamero M, et al. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun. 2010;1:3. doi: 10.1038/ncomms1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang RH, Sengupta K, Li C, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14(4):312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-eB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim J-H, Lee Y-M, Chun Y-S, Chen J, Kim J-E, Park J-W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol Cell. 2010;38(6):864–878. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 13.Wang R-H, Zheng Y, Kim H-S, et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-Associated Tumorigenesis. Mol Cell. 2008;32(1):11–20. doi: 10.1016/j.molcel.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herranz D, Maraver A, Canamero M, et al. SIRT1 promotes thyroid carcinogenesis driven by PTEN deficiency. Oncogene. 2013;32(34):4052–4056. doi: 10.1038/onc.2012.407. [DOI] [PubMed] [Google Scholar]
- 15.Yuan H, Wang Z, Li L, et al. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood. 2012;119(8):1904–1914. doi: 10.1182/blood-2011-06-361691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leko V, Park GJ, Lao U, Simon JA, Bedalov A. Enterocyte-specific inactivation of SIRT1 reduces tumor load in the APC(+/min) mouse model. PLoS One. 2013;8(6):e66283. doi: 10.1371/journal.pone.0066283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L. Medicinal chemistry of sirtuin inhibitors. Curr Med Chem. 2011;18(13):1936–1946. doi: 10.2174/092986711795590057. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, Yuan H, Roth M, Stark JM, Bhatia R, Chen WY. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene. 2013;32(5):589–598. doi: 10.1038/onc.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo J, Nikolaev AY, Imai S-i, et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107(2):137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Hou H, Haller EM, Nicosia SV, Bai W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005;24(5):1021–1032. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang F, Chan CH, Chen K, Guan X, Lin HK, Tong Q. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene. 2012;31(12):1546–1557. doi: 10.1038/onc.2011.347. [DOI] [PubMed] [Google Scholar]
- 22.Daitoku H, Hatta M, Matsuzaki H, et al. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004;101(27):10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi HK, Cho KB, Phuong NT, et al. SIRT1-Mediated FoxO1 Deacetylation Is Essential for Multidrug Resistance-Associated Protein 2 Expression in Tamoxifen-Resistant Breast Cancer Cells. Mol Pharm. 2013;10(7):2517–2527. doi: 10.1021/mp400287p. [DOI] [PubMed] [Google Scholar]
- 24.Menssen A, Hydbring P, Kapelle K, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci U S A. 2012;109(4):E187–196. doi: 10.1073/pnas.1105304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marshall GM, Liu PY, Gherardi S, et al. SIRT1 promotes N-Myc oncogenesis through a positive feedback loop involving the effects of MKP3 and ERK on N-Myc protein stability. PLoS Genet. 2011;7(6):e1002135. doi: 10.1371/journal.pgen.1002135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan J, Minter-Dykhouse K, Lou Z. A c-Myc SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol. 2009;185(2):203–211. doi: 10.1083/jcb.200809167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holloway KR, Calhoun TN, Saxena M, et al. SIRT1 regulates Dishevelled proteins and promotes transient and constitutive Wnt signaling. Proc Natl Acad Sci U S A. 2010;107(20):9216–9221. doi: 10.1073/pnas.0911325107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saxena M, Dykes SS, Malyarchuk S, Wang AE, Cardelli JA, Pruitt K. The sirtuins promote Dishevelled-1 scaffolding of TIAM1, Rac activation and cell migration. Oncogene. 2013 doi: 10.1038/onc.2013.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holloway KR, Barbieri A, Malyarchuk S, et al. SIRT1 positively regulates breast cancer associated human aromatase (CYP19A1) expression. Mol Endocrinol. 2013;27(3):480–490. doi: 10.1210/me.2012-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Hagan HM, Wang W, Sen S, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20(5):606–619. doi: 10.1016/j.ccr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bosch-Presegu?? L, Raurell-Vila H, Marazuela-Duque A, et al. Stabilization of Suv39H1 by SirT1 Is Part of Oxidative Stress Response and Ensures Genome Protection. Mol Cell. 2011;42(2):210–223. doi: 10.1016/j.molcel.2011.02.034. [DOI] [PubMed] [Google Scholar]
- 32.Murayama A, Ohmori K, Fujimura A, et al. Epigenetic Control of rDNA Loci in Response to Intracellular Energy Status. Cell. 2008;133(4):627–639. doi: 10.1016/j.cell.2008.03.030. [DOI] [PubMed] [Google Scholar]
- 33.Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123(3):437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 34.Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306(5704):2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 35.Gallardo E, Navarro A, Vinolas N, et al. miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis. 2009;30(11):1903–1909. doi: 10.1093/carcin/bgp219. [DOI] [PubMed] [Google Scholar]
- 36.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105(36):13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim H-S, Vassilopoulos A, Wang R-H, et al. SIRT2 Maintains Genome Integrity and Suppresses Tumorigenesis through Regulating APC/C Activity. Cancer Cell. 2011;20(4):487–499. doi: 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serrano L, Martinez-Redondo P, Marazuela-Duque A, et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013;27(6):639–653. doi: 10.1101/gad.211342.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin R, Tao R, Gao X, et al. Acetylation Stabilizes ATP-Citrate Lyase to Promote Lipid Biosynthesis and Tumor Growth. Mol Cell. 2013;51(4):506–518. doi: 10.1016/j.molcel.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heltweg B, Gatbonton T, Schuler AD, et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006;66(8):4368–4377. doi: 10.1158/0008-5472.CAN-05-3617. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Matsumori H, Nakayama Y, et al. SIRT2 down-regulation in HeLa can induce p53 accumulation via p38 MAPK activation-dependent p300 decrease, eventually leading to apoptosis. Genes Cells. 2011;16(1):34–45. doi: 10.1111/j.1365-2443.2010.01460.x. [DOI] [PubMed] [Google Scholar]
- 42.Liu G, Su L, Hao X, et al. Salermide up-regulates death receptor 5 expression through the ATF4-ATF3-CHOP axis and leads to apoptosis in human cancer cells. J Cell Mol Med. 2012;16(7):1618–1628. doi: 10.1111/j.1582-4934.2011.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu PY, Xu N, Malyukova A, et al. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ. 2013;20(3):503–514. doi: 10.1038/cdd.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sunami Y, Araki M, Hironaka Y, et al. Inhibition of the NAD-dependent protein deacetylase SIRT2 induces granulocytic differentiation in human leukemia cells. PLoS One. 2013;8(2):e57633. doi: 10.1371/journal.pone.0057633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Chan AW, To KF, et al. SIRT2 overexpression in hepatocellular carcinoma mediates epithelial to mesenchymal transition by protein kinase B/glycogen synthase kinase-3beta/beta-catenin signaling. Hepatology. 2013;57(6):2287–2298. doi: 10.1002/hep.26278. [DOI] [PubMed] [Google Scholar]
- 46.He X, Nie H, Hong Y, Sheng C, Xia W, Ying W. SIRT2 activity is required for the survival of C6 glioma cells. Biochem Biophys Res Comm. 2012;417(1):468–472. doi: 10.1016/j.bbrc.2011.11.141. [DOI] [PubMed] [Google Scholar]
- 47.McCarthy AR, Sachweh MC, Higgins M, et al. Tenovin-D3, a novel small-molecule inhibitor of sirtuin SirT2, increases p21 (CDKN1A) expression in a p53-independent manner. Mol Cancer Ther. 2013;12(4):352–360. doi: 10.1158/1535-7163.MCT-12-0900. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Au Q, Zhang M, Barber JR, Ng SC, Zhang B. Identification of a small molecule SIRT2 inhibitor with selective tumor cytotoxicity. Biochem Biophys Res Commun. 2009;386(4):729–733. doi: 10.1016/j.bbrc.2009.06.113. [DOI] [PubMed] [Google Scholar]
- 49.Yang MH, Laurent G, Bause AS, et al. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol Cancer Res. 2013;11(9):1072–1077. doi: 10.1158/1541-7786.MCR-13-0040-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jing E, Gesta S, Kahn CR. SIRT2 Regulates Adipocyte Differentiation through FoxO1 Acetylation/Deacetylation. Cell Metab. 2007;6(2):105–114. doi: 10.1016/j.cmet.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao Y, Yang J, Liao W, et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12(7):665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 52.Ramakrishnan G, Davaakhuu G, Kaplun L, et al. Sirt2 Deacetylase Is a Novel AKT Binding Partner Critical for AKT Activation by Insulin. J Biol Chem. 2014;289(9):6054–6066. doi: 10.1074/jbc.M113.537266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao D, Zou S-W, Liu Y, et al. Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell. 2013;23(4):464–476. doi: 10.1016/j.ccr.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soung YH, Pruitt K, Chung J. Epigenetic silencing of ARRDC3 expression in basal-like breast cancer cells. Sci Rep. 2014;4:3846. doi: 10.1038/srep03846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim HS, Patel K, Muldoon-Jacobs K, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17(1):41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finley LW, Carracedo A, Lee J, et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011;19(3):416–428. doi: 10.1016/j.ccr.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bell EL, Emerling BM, Ricoult SJ, Guarente L. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011;30(26):2986–2996. doi: 10.1038/onc.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shulga N, Wilson-Smith R, Pastorino JG. Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. J Cell Sci. 2010;123(Pt 6):894–902. doi: 10.1242/jcs.061846. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Hafner AV, Dai J, Gomes AP, et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2(12):914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119(9):2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jacobs KM, Pennington JD, Bisht KS, et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci. 2008;4(5):291–299. doi: 10.7150/ijbs.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tseng AH, Shieh SS, Wang DL. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic Biol Med. 2013;63:222–234. doi: 10.1016/j.freeradbiomed.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 63.Inuzuka H, Gao D, Finley LW, et al. Acetylation-dependent regulation of Skp2 function. Cell. 2012;150(1):179–193. doi: 10.1016/j.cell.2012.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iwahara T, Bonasio R, Narendra V, Reinberg D. SIRT3 functions in the nucleus in the control of stress-related gene expression. Mol Cell Biol. 2012;32(24):5022–5034. doi: 10.1128/MCB.00822-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. 2008;28(20):6384–6401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haigis MC, Mostoslavsky R, Haigis KM, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126(5):941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 67.Csibi A, Fendt SM, Li C, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. 2013;153(4):840–854. doi: 10.1016/j.cell.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jeong SM, Xiao C, Finley LW, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell. 2013;23(4):450–463. doi: 10.1016/j.ccr.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu J, Sadhukhan S, Noriega LG, et al. Metabolic characterization of a Sirt5 deficient mouse model. Sci Rep. 2013;3:2806. doi: 10.1038/srep02806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009;137(3):560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Du J, Zhou Y, Su X, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334(6057):806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peng C, Lu Z, Xie Z, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics. 2011;10(12):M111 012658. doi: 10.1074/mcp.M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park J, Chen Y, Tishkoff DX, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell. 2013;50(6):919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Michishita E, McCord RA, Berber E, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452(7186):492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sebastian C, Zwaans BM, Silberman DM, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151(6):1185–1199. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang H, Khan S, Wang Y, et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013;496(7443):110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gil R, Barth S, Kanfi Y, Cohen HY. SIRT6 exhibits nucleosome-dependent deacetylase activity. Nucleic Acids Res. 2013;41(18):8537–8545. doi: 10.1093/nar/gkt642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feldman JL, Baeza J, Denu JM. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem. 2013;288(43):31350–31356. doi: 10.1074/jbc.C113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mostoslavsky R, Chua KF, Lombard DB, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124(2):315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 80.Chen S, Seiler J, Santiago-Reichelt M, Felbel K, Grummt I, Voit R. Repression of RNA Polymerase I upon Stress Is Caused by Inhibition of RNA-Dependent Deacetylation of PAF53 by SIRT7. Mol Cell. 2013;52(3):303–313. doi: 10.1016/j.molcel.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 81.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16(10):4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006;20(9):1075–1080. doi: 10.1101/gad.1399706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsai YC, Greco TM, Cristea IM. SIRT7 plays a role in ribosome biogenesis and protein synthesis. Mol Cell Proteomics. 2013 doi: 10.1074/mcp.M113.031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barber MF, Michishita-Kioi E, Xi Y, et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature. 2012;487(7405):114–118. doi: 10.1038/nature11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shin J, He M, Liu Y, et al. SIRT7 Represses Myc Activity to Suppress ER Stress and Prevent Fatty Liver Disease. Cell Rep. 2013;5(3):654–665. doi: 10.1016/j.celrep.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. 2014;35:146–154. doi: 10.1016/j.tips.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J Biol Chem. 2003;278(51):50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- 88.Smith BC, Denu JM. Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide. Biochemistry. 2007;46(50):14478–14486. doi: 10.1021/bi7013294. [DOI] [PubMed] [Google Scholar]
- 89.Hawse WF, Hoff KG, Fatkins DG, et al. Structural insights into intermediate steps in the Sir2 deacetylation reaction. Structure. 2008;16(9):1368–1377. doi: 10.1016/j.str.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hu J, He B, Bhargava S, Lin H. A fluorogenic assay for screening Sirt6 modulators. Org Biomol Chem. 2013;11(32):5213–5216. doi: 10.1039/c3ob41138a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tervo AJ, Kyrylenko S, Niskanen P, et al. An in silico approach to discovering novel inhibitors of human sirtuin type 2. J Med Chem. 2004;47(25):6292–6298. doi: 10.1021/jm049933m. [DOI] [PubMed] [Google Scholar]
- 92.Audrito V, Vaisitti T, Rossi D, et al. Nicotinamide blocks proliferation and induces apoptosis of chronic lymphocytic leukemia cells through activation of the p53/miR-34a/SIRT1 tumor suppressor network. Cancer Res. 2011;71(13):4473–4483. doi: 10.1158/0008-5472.CAN-10-4452. [DOI] [PubMed] [Google Scholar]
- 93.Jung-Hynes B, Nihal M, Zhong W, Ahmad N. Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition? J Biol Chem. 2009;284(6):3823–3832. doi: 10.1074/jbc.M807869200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Suzuki T, Khan MN, Sawada H, et al. Design, synthesis, and biological activity of a novel series of human sirtuin-2-selective inhibitors. J Med Chem. 2012;55(12):5760–5773. doi: 10.1021/jm3002108. [DOI] [PubMed] [Google Scholar]
- 95.Taylor DM, Balabadra U, Xiang Z, et al. A brain-permeable small molecule reduces neuronal cholesterol by inhibiting activity of sirtuin 2 deacetylase. ACS Chem Biol. 2011;6(6):540–546. doi: 10.1021/cb100376q. [DOI] [PubMed] [Google Scholar]
- 96.Mai A, Valente S, Meade S, et al. Study of 1,4-dihydropyridine structural scaffold: discovery of novel sirtuin activators and inhibitors. J Med Chem. 2009;52(17):5496–5504. doi: 10.1021/jm9008289. [DOI] [PubMed] [Google Scholar]
- 97.Alvala M, Bhatnagar S, Ravi A, et al. Novel acridinedione derivatives: design, synthesis, SIRT1 enzyme and tumor cell growth inhibition studies. Bioorg Med Chem Lett. 2012;22(9):3256–3260. doi: 10.1016/j.bmcl.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 98.Alhazzazi TY, Kamarajan P, Joo N, et al. Sirtuin-3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer. 2011;117(8):1670–1678. doi: 10.1002/cncr.25676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith BC, Denu JM. Acetyllysine analog peptides as mechanistic probes of protein deacetylases. J Biol Chem. 2007;282(51):37256–37265. doi: 10.1074/jbc.M707878200. [DOI] [PubMed] [Google Scholar]
- 100.Fatkins DG, Monnot AD, Zheng W. Nepsilon-thioacetyllysine: a multi-facet functional probe for enzymatic protein lysine Nepsilon-deacetylation. Bioorg Med Chem Lett. 2006;16(14):3651–3656. doi: 10.1016/j.bmcl.2006.04.075. [DOI] [PubMed] [Google Scholar]
- 101.Kiviranta PH, Suuronen T, Wallen EA, et al. N(epsilon)-thioacetyllysine-containing tri-, tetra-, and pentapeptides as SIRT1 and SIRT2 inhibitors. J Med Chem. 2009;52(7):2153–2156. doi: 10.1021/jm801401k. [DOI] [PubMed] [Google Scholar]
- 102.Jamonnak N, Fatkins DG, Wei L, Zheng W. N(epsilon)-methanesulfonyllysine as a non-hydrolyzable functional surrogate for N(epsilon)-acetyllysine. Org Biomol Chem. 2007;5(6):892–896. doi: 10.1039/b617185k. [DOI] [PubMed] [Google Scholar]
- 103.Huhtiniemi T, Suuronen T, Lahtela-Kakkonen M, et al. N(epsilon)-Modified lysine containing inhibitors for SIRT1 and SIRT2. Bioorg Med Chem. 2010;18(15):5616–5625. doi: 10.1016/j.bmc.2010.06.035. [DOI] [PubMed] [Google Scholar]
- 104.Chakrabarty SP, Ramapanicker R, Mishra R, Chandrasekaran S, Balaram H. Development and characterization of lysine based tripeptide analogues as inhibitors of Sir2 activity. Bioorg Med Chem. 2009;17(23):8060–8072. doi: 10.1016/j.bmc.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 105.Hirsch BM, Hao Y, Li X, Wesdemiotis C, Wang Z, Zheng W. A mechanism-based potent sirtuin inhibitor containing Nepsilon-thiocarbamoyllysine (TuAcK) Bioorg Med Chem Lett. 2011;21(16):4753–4757. doi: 10.1016/j.bmcl.2011.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jamonnak N, Hirsch BM, Pang Y, Zheng W. Substrate specificity of SIRT1-catalyzed lysine Nepsilon-deacetylation reaction probed with the side chain modified Nepsilon-acetyllysine analogs. Bioorg Chem. 2010;38(1):17–25. doi: 10.1016/j.bioorg.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 107.Suzuki T, Asaba T, Imai E, Tsumoto H, Nakagawa H, Miyata N. Identification of a cell-active non-peptide sirtuin inhibitor containing N-thioacetyl lysine. Bioorg Med Chem Lett. 2009;19(19):5670–5672. doi: 10.1016/j.bmcl.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 108.Huhtiniemi T, Salo HS, Suuronen T, et al. Structure-based design of pseudopeptidic inhibitors for SIRT1 and SIRT2. J Med Chem. 2011;54(19):6456–6468. doi: 10.1021/jm200590k. [DOI] [PubMed] [Google Scholar]
- 109.Mellini P, Kokkola T, Suuronen T, et al. Screen of pseudopeptidic inhibitors of human sirtuins 1–3: two lead compounds with antiproliferative effects in cancer cells. J Med Chem. 2013;56(17):6681–6695. doi: 10.1021/jm400438k. [DOI] [PubMed] [Google Scholar]
- 110.Morimoto J, Hayashi Y, Suga H. Discovery of macrocyclic peptides armed with a mechanism-based warhead: isoform-selective inhibition of human deacetylase SIRT2. Angew Chem Int Ed Engl. 2012;51(14):3423–3427. doi: 10.1002/anie.201108118. [DOI] [PubMed] [Google Scholar]
- 111.Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem. 2001;276(42):38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- 112.Ota H, Tokunaga E, Chang K, et al. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2006;25(2):176–185. doi: 10.1038/sj.onc.1209049. [DOI] [PubMed] [Google Scholar]
- 113.Peck B, Chen CY, Ho KK, et al. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther. 2010;9(4):844–855. doi: 10.1158/1535-7163.MCT-09-0971. [DOI] [PubMed] [Google Scholar]
- 114.Kojima K, Ohhashi R, Fujita Y, et al. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem Biophys Res Commun. 2008;373(3):423–428. doi: 10.1016/j.bbrc.2008.06.045. [DOI] [PubMed] [Google Scholar]
- 115.Jin KL, Park JY, Noh EJ, et al. The effect of combined treatment with cisplatin and histone deacetylase inhibitors on HeLa cells. J Gynecol Oncol. 2010;21(4):262–268. doi: 10.3802/jgo.2010.21.4.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kalle AM, Mallika A, Badiger J, Alinakhi, Talukdar P, Sachchidanand Inhibition of SIRT1 by a small molecule induces apoptosis in breast cancer cells. Biochem Biophys Res Commun. 2010;401(1):13–19. doi: 10.1016/j.bbrc.2010.08.118. [DOI] [PubMed] [Google Scholar]
- 117.Lara E, Mai A, Calvanese V, et al. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene. 2009;28(6):781–791. doi: 10.1038/onc.2008.436. [DOI] [PubMed] [Google Scholar]
- 118.Rotili D, Tarantino D, Nebbioso A, et al. Discovery of salermide-related sirtuin inhibitors: binding mode studies and antiproliferative effects in cancer cells including cancer stem cells. J Med Chem. 2012;55(24):10937–10947. doi: 10.1021/jm3011614. [DOI] [PubMed] [Google Scholar]
- 119.Pasco MY, Rotili D, Altucci L, et al. Characterization of sirtuin inhibitors in nematodes expressing a muscular dystrophy protein reveals muscle cell and behavioral protection by specific sirtinol analogues. J Med Chem. 2010;53(3):1407–1411. doi: 10.1021/jm9013345. [DOI] [PubMed] [Google Scholar]
- 120.Medda F, Russell RJ, Higgins M, et al. Novel cambinol analogs as sirtuin inhibitors: synthesis, biological evaluation, and rationalization of activity. J Med Chem. 2009;52(9):2673–2682. doi: 10.1021/jm8014298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Uciechowska U, Schemies J, Neugebauer RC, et al. Thiobarbiturates as sirtuin inhibitors: virtual screening, free-energy calculations, and biological testing. ChemMedChem. 2008;3(12):1965–1976. doi: 10.1002/cmdc.200800104. [DOI] [PubMed] [Google Scholar]
- 122.Bedalov A, Gatbonton T, Irvine WP, Gottschling DE, Simon JA. Identification of a small molecule inhibitor of Sir2p. Proc Natl Acad Sci U S A. 2001;98(26):15113–15118. doi: 10.1073/pnas.261574398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pagans S, Pedal A, North BJ, et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 2005;3(2):e41. doi: 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Neugebauer RC, Uchiechowska U, Meier R, et al. Structure-activity studies on splitomicin derivatives as sirtuin inhibitors and computational prediction of binding mode. J Med Chem. 2008;51(5):1203–1213. doi: 10.1021/jm700972e. [DOI] [PubMed] [Google Scholar]
- 125.Rotili D, Tarantino D, Carafa V, et al. Benzodeazaoxaflavins as sirtuin inhibitors with antiproliferative properties in cancer stem cells. J Med Chem. 2012;55(18):8193–8197. doi: 10.1021/jm301115r. [DOI] [PubMed] [Google Scholar]
- 126.Rotili D, Tarantino D, Carafa V, et al. Identification of tri- and tetracyclic pyrimidinediones as sirtuin inhibitors. ChemMedChem. 2010;5(5):674–677. doi: 10.1002/cmdc.201000030. [DOI] [PubMed] [Google Scholar]
- 127.Napper AD, Hixon J, McDonagh T, et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem. 2005;48(25):8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- 128.Solomon JM, Pasupuleti R, Xu L, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol. 2006;26(1):28–38. doi: 10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stunkel W, Peh BK, Tan YC, et al. Function of the SIRT1 protein deacetylase in cancer. Biotechnol J. 2007;2(11):1360–1368. doi: 10.1002/biot.200700087. [DOI] [PubMed] [Google Scholar]
- 130.Zhang Q, Zeng SX, Zhang Y, et al. A small molecule Inauhzin inhibits SIRT1 activity and suppresses tumour growth through activation of p53. EMBO Mol Med. 2012;4(4):298–312. doi: 10.1002/emmm.201100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Davis PD, Hill CH, Keech E, et al. Potent selective inhibitors of protein kinase C. FEBS Lett. 1989;259(1):61–63. doi: 10.1016/0014-5793(89)81494-2. [DOI] [PubMed] [Google Scholar]
- 132.Trapp J, Jochum A, Meier R, et al. Adenosine mimetics as inhibitors of NAD+-dependent histone deacetylases, from kinase to sirtuin inhibition. J Med Chem. 2006;49(25):7307–7316. doi: 10.1021/jm060118b. [DOI] [PubMed] [Google Scholar]
- 133.Huber K, Schemies J, Uciechowska U, et al. Novel 3-arylideneindolin-2-ones as inhibitors of NAD+-dependent histone deacetylases (sirtuins) J Med Chem. 2010;53(3):1383–1386. doi: 10.1021/jm901055u. [DOI] [PubMed] [Google Scholar]
- 134.Suenkel B, Fischer F, Steegborn C. Inhibition of the human deacylase Sirtuin 5 by the indole GW5074. Bioorg Med Chem Lett. 2013;23(1):143–146. doi: 10.1016/j.bmcl.2012.10.136. [DOI] [PubMed] [Google Scholar]
- 135.Cea M, Soncini D, Fruscione F, et al. Synergistic interactions between HDAC and sirtuin inhibitors in human leukemia cells. PLoS One. 2011;6(7):e22739. doi: 10.1371/journal.pone.0022739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schuetz A, Min J, Antoshenko T, et al. Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure. 2007;15(3):377–389. doi: 10.1016/j.str.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 137.Trapp J, Meier R, Hongwiset D, Kassack MU, Sippl W, Jung M. Structure-activity studies on suramin analogues as inhibitors of NAD+-dependent histone deacetylases (sirtuins) ChemMedChem. 2007;2(10):1419–1431. doi: 10.1002/cmdc.200700003. [DOI] [PubMed] [Google Scholar]
- 138.Lain S, Hollick JJ, Campbell J, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13(5):454–463. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McCarthy AR, Pirrie L, Hollick JJ, et al. Synthesis and biological characterisation of sirtuin inhibitors based on the tenovins. Bioorg Med Chem. 2012;20(5):1779–1793. doi: 10.1016/j.bmc.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 140.Outeiro TF, Kontopoulos E, Altmann SM, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317(5837):516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 141.Gey C, Kyrylenko S, Hennig L, et al. Phloroglucinol derivatives guttiferone G, aristoforin, and hyperforin: inhibitors of human sirtuins SIRT1 and SIRT2. Angew Chem Int Ed Engl. 2007;46(27):5219–5222. doi: 10.1002/anie.200605207. [DOI] [PubMed] [Google Scholar]
- 142.Manjulatha K, Srinivas S, Mulakayala N, et al. Ethylenediamine diacetate (EDDA) mediated synthesis of aurones under ultrasound: their evaluation as inhibitors of SIRT1. Bioorg Med Chem Lett. 2012;22(19):6160–6165. doi: 10.1016/j.bmcl.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 143.Garske AL, Smith BC, Denu JM. Linking SIRT2 to Parkinson’s disease. ACS Chem Biol. 2007;2(8):529–532. doi: 10.1021/cb700160d. [DOI] [PubMed] [Google Scholar]
- 144.Gutierrez M, Andrianasolo EH, Shin WK, et al. Structural and synthetic investigations of tanikolide dimer, a SIRT2 selective inhibitor, and tanikolide seco-acid from the Madagascar marine cyanobacterium Lyngbya majuscula. J Org Chem. 2009;74(15):5267–5275. doi: 10.1021/jo900578j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Friden-Saxin M, Seifert T, Landergren MR, et al. Synthesis and evaluation of substituted chroman-4-one and chromone derivatives as sirtuin 2-selective inhibitors. J Med Chem. 2012;55(16):7104–7113. doi: 10.1021/jm3005288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Disch JS, Evindar G, Chiu CH, et al. Discovery of Thieno[3,2-d]pyrimidine-6-carboxamides as Potent Inhibitors of SIRT1, SIRT2, and SIRT3. J Med Chem. 2013;56(9):3666–3679. doi: 10.1021/jm400204k. [DOI] [PubMed] [Google Scholar]
- 147.Knight ZA, Shokat KM. Chemical Genetics: Where Genetics and Pharmacology Meet. Cell. 2007;128(3):425–430. doi: 10.1016/j.cell.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 148.Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotech. 2007;25(1):84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 149.Wong S, McLaughlin J, Cheng D, Zhang C, Shokat KM, Witte ON. Sole BCR-ABL inhibition is insufficient to eliminate all myeloproliferative disorder cell populations. Proc Natl Acad Sci U S A. 2004;101(50):17456–17461. doi: 10.1073/pnas.0407061101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hopkins AL, Mason JS, Overington JP. Can we rationally design promiscuous drugs? Curr Opin Struct Biol. 2006;16(1):127–136. doi: 10.1016/j.sbi.2006.01.013. [DOI] [PubMed] [Google Scholar]