

Abstract
Rationale: Eosinophils are key effector cells in allergic diseases, including allergic rhinitis, eczema, and asthma. Their tissue presence is regulated by both recruitment and increased longevity at inflamed sites.
Objectives: To investigate the ability of the flavone wogonin to induce eosinophil apoptosis in vitro and attenuate eosinophil-dominant allergic inflammation in vivo in mice.
Methods: Human and mouse eosinophil apoptosis in response to wogonin was investigated by cellular morphology, flow cytometry, mitochondrial membrane permeability, and pharmacological caspase inhibition. Allergic lung inflammation was modeled in mice sensitized and challenged with ovalbumin. Bronchoalveolar lavage (BAL) and lung tissue were examined for inflammation, mucus production, and inflammatory mediator production. Airway hyperresponsiveness to aerosolized methacholine was measured.
Measurements and Main Results: Wogonin induced time- and concentration-dependent human and mouse eosinophil apoptosis in vitro. Wogonin-induced eosinophil apoptosis occurred with activation of caspase-3 and was inhibited by pharmacological caspase inhibition. Wogonin administration attenuated allergic airway inflammation in vivo with reductions in BAL and interstitial eosinophil numbers, increased eosinophil apoptosis, reduced airway mucus production, and attenuated airway hyperresponsiveness. This wogonin-induced reduction in allergic airway inflammation was prevented by concurrent caspase inhibition in vivo.
Conclusions: Wogonin induces eosinophil apoptosis and attenuates allergic airway inflammation, suggesting that it has therapeutic potential for the treatment of allergic inflammation in humans.
Keywords: airway resistance, antiinflammatory agents, hypersensitivity, inflammation resolution, mucus
At a Glance Commentary
Scientific Knowledge on the Subject
Eosinophils form an integral part of the acute inflammatory processes that occur in allergic inflammation. At present, there are few eosinophil-specific therapies that modify the lifespan of these cells. Augmenting and accelerating eosinophil apoptosis may have promise as an alternative therapeutic approach to treating allergic inflammation.
What This Study Adds to the Field
The flavone wogonin induces caspase-dependent eosinophil apoptosis and attenuates allergic lung inflammation in mice in vivo, suggesting that it has therapeutic potential for the treatment of human allergic diseases.
Eosinophils are terminally differentiated granulocytic cells that play a central role in the pathogenesis of a variety of allergic diseases (1). Under homeostatic conditions, eosinophils are produced by the bone marrow prior to release into the circulation, where they make up only a small percentage of circulating leukocytes (approximately 1–5%). They have a short half-life in the circulation (approximately 20 h), and their usual fate is sequestration into either the thymus or the gastrointestinal tract. In pathological situations, eosinophil production is increased, and these cells migrate into inflamed tissues, where their lifespan is believed to increase by a combination of inflammatory mediators and changes to the local microenvironment (1). Although they play a beneficial role in the host defense against parasitic infections and in the response to tissue injury (2, 3), eosinophils and eosinophil-derived products are implicated in the pathology of numerous allergic disease states, including asthma, eczema, and allergic rhinitis. The results of both mouse studies and human asthmatic patients have demonstrated that eosinophil accumulation in lung tissue directly contributes to ongoing inflammation, organ dysfunction, and tissue remodeling. Eosinophil-deficient mice are protected against airway hyperresponsiveness and airway remodeling (4, 5), and targeting eosinophils by means of anti–IL-5 treatments is proving efficacious in the treatment of human asthma (6–8).
Eosinophil numbers within tissue depend not only on recruitment but also upon eosinophil lifespan and clearance. Eosinophil clearance in the lung can occur by transepithelial migration and mucociliary clearance (9) or by apoptosis (programmed cell death) and subsequent removal of apoptotic cells by surrounding cells, including macrophages, dendritic cells, and epithelial cells (10–12). The relative contribution of these two elimination pathways, as well as the role that eosinophil apoptosis may play in resolution of allergic inflammation in humans, remains controversial (13). Evidence that the control of eosinophil apoptosis and subsequent phagocytosis is dysregulated in allergic inflammation exists, with low levels of eosinophil apoptosis correlating with increased asthma severity in humans (14). Although glucocorticoid treatment (the mainstay of therapy in asthma, eczema, and allergic rhinitis) enhances eosinophil apoptosis and subsequent clearance in vitro (15, 16), it is not clear whether these effects contribute to the antiinflammatory actions of glucocorticoids in human allergic diseases. High-dose glucocorticoid treatment (frequently used in poorly controlled asthma) also causes multiple side effects, and a significant proportion of asthmatic individuals are steroid-resistant (17). Therefore, the development of novel therapies that target eosinophil apoptosis is an attractive goal for the treatment of allergic diseases such as asthma.
Flavones are polyphenolic compounds named for their bright yellow color and form a subset of flavonoids. The flavone wogonin (5,7-dihydroxy-8-methoxyflavone) has been proposed to have anticancer and antiinflammatory properties (18, 19). Recently, we showed that flavones, including wogonin, were able to drive neutrophil apoptosis by downregulating the prosurvival Bcl-2 family member Mcl-1. Furthermore, wogonin was able to enhance resolution of neutrophilic cellular inflammation in vivo in a zebrafish model of sterile tissue injury (19) and in a mouse model of LPS-induced lung injury (20). However, the effects of wogonin on eosinophil apoptosis and allergic inflammation have not previously been studied. This is important to delineate, as pharmacological induction of granulocyte apoptosis is often cell type–specific, with glucocorticoids inducing eosinophil apoptosis while delaying neutrophil apoptosis in vitro (15).
In the present study, we investigated the effects of wogonin on eosinophil lifespan and apoptosis in vitro and consequently on allergic airway inflammation in vivo. Wogonin induced primary human and mouse eosinophil apoptosis in a time-, concentration-, and caspase-dependent fashion. Furthermore, wogonin attenuated established ovalbumin (OVA)-induced allergic airway inflammation in mice. Some of the results of these studies were previously reported in the form of an abstract (21).
Methods
Human Eosinophil Isolation
Granulocytes were isolated from healthy donor blood (Lothian Research Ethics Committee, 08/S1103/38) by dextran sedimentation and Percoll gradient (22). Eosinophils were subsequently separated by negative selection (23), with purity routinely greater than 95%. Cells were resuspended in Iscove’s modified Dulbecco’s medium (IMDM) (PAA Laboratories, Pasching, Austria) with 10% autologous serum at a final concentration of 2 × 106/ml (37°C, 5% CO2) and incubated with wogonin (Sigma, Dorset, UK), z-VAD-fmk (z-VAD; Bachem, Bubendorf, Switzerland), Q-VD-OPh (Q-VD; R&D Systems, Abingdon, UK), or vehicle control as described in the figures.
Assessment of Cell Viability, Apoptosis, and Caspase Activation
Light microscopy, flow cytometry (fluorescein isothiocyanate-annexin-V/propidium iodide [PI] or anti–Ly6G-Pacific Blue/anti–Siglec-F-phycoerythrin/annexin-V/4′,6-diamidino-2-phenylindole [DAPI]; Invitrogen), and mitochondrial membrane permeability (MitoCapture; BioVision, Milpitas, CA) were assessed as previously described (20) or according to the manufacturer’s instructions. Eosinophils were lysed, and Western blotting for caspase activation was performed as described previously (19).
In Vivo Model of Allergic Inflammation
The animal work was carried out in accordance with the UK Home Office under the Animals (Scientific Procedures) Act 1986 and following review by the local ethics committee. OVA-induced allergic inflammation was modeled using a modification of a previously described method (24). Female BALB/c mice 8–14 weeks of age were sensitized by intraperitoneal (i.p.) alum-precipitated (Alum Imject; Pierce Biotechnology, Rockford, IL) OVA (Sigma) (20 μg of OVA and 50 μl of alum per mouse) on Days 1 and 10. Challenge was initiated by intratracheal OVA (50 μg) on Days 22, 23, and 24. Mice received wogonin (1 mg/mouse i.p.) or vehicle on Days 25–28, with acquisition of bronchoalveolar lavage (BAL) fluid, lung tissue, blood, and bone marrow on Day 29 and analyzed as described previously (20). Cells were incubated with combinations of CD45/Siglec-F/Ly6G/CD11b/CD117, with flow cytometric analysis performed in the presence of Flow-Check fluorospheres to allow absolute quantification of cell numbers (Beckman Coulter, Brea, CA). Following left lung homogenization (Precellys 24 homogenizer; Bertin Technologies, Montigny-le-Bretonneux, France), cytokines were measured by ELISA (R&D Systems). Lungs from separate animals were taken for histology and fixed with 10% formalin (Sigma) prior to hematoxylin and eosin (H&E) or periodic acid–Schiff (PAS) staining. A histological scoring system for quantifying mucus production was performed by two independent blinded observers using an adaptation of a previously described method (25). After PAS staining, at least 15 consecutive airways (×200 magnification) were scored as follows: 1 = nil (<5% PAS+ve circumferential goblet cell staining), 2 = mild (<30% PAS+ve goblet cells), 3 = moderate (30–60% PAS+ve goblet cells), and 4 = severe (>60% PAS+ve goblet cells). The total score was then divided by the number of airways examined to give an average severity score per airway, the mucus-goblet index. Airway responsiveness to aerosolized methacholine was assessed in anesthetized and mechanically ventilated mice, with lung resistance determined and expressed relative to baseline values after phosphate-buffered saline exposure (Buxco Research Systems, Wilmington, NC). For experiments with z-VAD, 5 mg/kg z-VAD (26) was administered intraperitoneally every 4 hours from 32 to 44 hours after the last intratracheal administration of OVA, with lungs retrieved at 48 hours for histological analysis. Features of interstitial and alveolar inflammatory cell infiltration were assessed semiquantitatively on H&E-stained lung sections by two independent observers blinded to the experimental conditions. Five consecutive views (×100 magnification) were scored using the following scale, with the mean value taken: 0 = normal; 1 = mild, infrequent inflammatory cell accumulation; 2 = moderate, frequent inflammatory cell accumulation; and 3 = severe, widespread inflammatory cell accumulation with consequent marked loss of alveolar definition. For ex vivo mouse eosinophil experiments, BAL cells, interstitial lung cells following collagenase D (Roche, Mannheim, Germany) digestion, and bone marrow cells were incubated at 2 × 106/ml in IMDM with 10% fetal calf serum.
Statistical Analysis
Flow cytometric analysis was performed using FlowJo software (TreeStar, Ashland, OR). All data are expressed as mean ± SEM, analyzed using GraphPad Prism software (La Jolla, CA) by unpaired t test or analysis of variance as appropriate.
Results
Wogonin Induces Time-, Concentration-, and Caspase-Dependent Apoptosis of Primary Human Eosinophils
Isolated human eosinophils were incubated for 0–20 hours with increasing concentrations of wogonin (0–125 μM) prior to viability and apoptosis assessment by flow cytometry and morphological examination. Eosinophils underwent constitutive apoptosis in 4.0 ± 2.5% of cells at 0 hours and 11.1 ± 2.3% after 20 hours incubation (P < 0.05). Wogonin accelerated eosinophil death in a time- and concentration-dependent fashion (Figure 1A; for clarity, not all concentrations used are shown). For example, survival after 20-hour incubation was 75.2 ± 3.3% in control versus 34.3 ± 8.0% with 75 μM wogonin (P < 0.001). The accelerated eosinophil death observed with wogonin was secondary to the induction of apoptosis (Figures 1B and 1C). At later time points, apoptotic eosinophils underwent secondary necrosis due to absence of phagocytes in the culture system (data not shown). Representative flow cytometry plots and morphological appearances are shown at 6 hours for control and wogonin-treated eosinophils, demonstrating increased numbers of eosinophils with changes typical of apoptosis (cellular shrinkage and nuclear condensation) in the wogonin-treated group (Figures 1D and 1E). The induction of eosinophil apoptosis by wogonin appeared to be a cell type–specific phenomenon, and not just general cytotoxicity, as wogonin did not induce apoptosis in mouse thymocytes (see Figure E1 in the online supplement). Wogonin-induced eosinophil apoptosis was caspase-dependent, as caspase inhibition by the broad-spectrum pharmacological caspase inhibitors z-VAD and Q-VD prevented wogonin-induced eosinophil apoptosis (Figures 2A–2C), whereas wogonin treatment led to increased caspase-3 activation (cleavage) in a time-dependent manner (Figure 2D). In addition, wogonin led to loss of mitochondrial outer membrane potential (observed at 4 h) prior to the onset of apoptosis (observed from 6 h onward) (Figures 1A and 1B), suggesting that apoptosis was occurring via the intrinsic pathway of apoptosis (Figure 2E).
Figure 1.

Wogonin induces time- and concentration-dependent apoptosis of human eosinophils. (A–C) Eosinophils were cultured for (A) 4–20 hours, (B) 6 hours, or (C) 20 hours with increasing concentrations of the flavone wogonin prior to eosinophil viability and apoptosis determination by flow cytometry (annexin V/propidium iodide [AnnV/PI] binding) (n ≥ 4). (D, E) Representative flow cytometry plots (x axis: AnnV; y axis: PI) and representative cytocentrifuge preparations (×1,000 original magnification) at 6 hours for (D) control and (E) 75 μM wogonin-treated eosinophils. Black arrow points to an eosinophil with apoptotic morphology. Data are expressed as mean ± SEM as analyzed by two-way analysis of variance (ANOVA) with a Bonferroni test (A) or one-way ANOVA with a Dunnett test (B, C). *P < 0.05, **P < 0.01, and ***P < 0.001 versus control.
Figure 2.

Wogonin-induced eosinophil apoptosis is caspase dependent. Eosinophils were cultured for either (A) 6 hours or (B) 20 hours with the flavone wogonin (75 μM) with and without the broad-spectrum caspase inhibitors Q-VD (20 μM) or z-VAD (100 μM) prior to apoptosis determination by flow cytometry (annexin V/propidium iodide [AnnV/PI] binding) (n ≥ 5). (C) Representative overlay histogram of AnnV binding of eosinophils after 6 hours of treatment with wogonin (75 μM; blue line), wogonin plus z-VAD (100 μM; red line), and wogonin plus Q-VD (20 μM; green line). (D) Eosinophils were cultured for 4 hours or 6 hours in either control medium or wogonin (75 μM) before lysing and Western blotting for cleaved caspase-3 (17/19 kD) and β-actin (42 kD). (E) Eosinophils were cultured for 4 hours in either control medium or wogonin (100 μM) prior to analysis of mitochondrial membrane potential (Δψm), with loss of Δψm indicated by an increase in fluorescence channel (FL-1) fluorescence. A representative overlay histogram of FL-1 fluorescence is shown in (E, i), and cumulative data expressed relative to control are represented in (E, ii) (n = 7). Data are expressed as mean ± SEM as analyzed by analysis of variance with a Newman-Keuls multiple-comparisons test (A, B) or by t test (E). **P < 0.01 and ***P < 0.001 versus control; ##P < 0.01 and ###P < 0.001 versus wogonin-treated sample.
Wogonin Induces Apoptosis of Mouse Eosinophils Recruited to Sites of Allergic Inflammation
To investigate whether wogonin was able to induce apoptosis in eosinophils recruited into sites of allergic inflammation, and as a precursor to in vivo experiments, allergic inflammation was induced in mouse lungs by OVA sensitization and challenges (Figure 3A). BAL cells were recovered and then incubated ex vivo. Eosinophils were easily distinguished on flow cytometry by Siglec-F and Ly6G staining (Siglec-F+ve/Ly6G−ve cells) (Figure 3B) and by cellular morphology on cytocentrifuge preparations (Figure 3E). Wogonin induced concentration-dependent apoptosis of mouse eosinophils (Figure 3C), and, similar to the process in human eosinophils, this was caspase-dependent, as it was inhibited by the presence of the caspase inhibitor Q-VD (Figure 3C) and was associated with caspase-3 cleavage (Figure 3D). Representative cytocentrifuge preparations are shown in Figure 3E, with increased numbers of apoptotic eosinophils observed in the wogonin-treated group.
Figure 3.

Mouse eosinophils recruited to sites of allergic inflammation undergo wogonin-induced apoptosis ex vivo. (A) Schema of the experimental protocol. (B) Representative flow cytometry plot (x axis: Siglec-F; y axis: Ly6G) demonstrating delineation of the eosinophil population (Ly6G−ve/Siglec-F+ve) as well as a smaller neutrophil population (Ly6G+ve/Siglec-F−ve). (C) Eosinophil apoptosis (AnnV+ve/DAPI−ve/Ly6G−ve/Siglec-F+ve) at 6 hours with increasing concentrations of wogonin (10–100 μM) with or without the caspase inhibitor Q-VD (20 μM) are shown (n = 3 or 4 separate experiments). (D) Bronchoalveolar lavage (BAL) cells incubated in control media or wogonin (100 μM) for 4 hours prior to lysing and Western blotting for cleaved caspase-3 (17/19 kD) and β-actin (42 kD). (E) Representative cytocentrifuge preparations (×1,000 original magnification) at 6 hours for (i) control cells and cells treated with (ii) wogonin (100 μM) and (iii) combined wogonin (100 μM) and Q-VD (20 μM). Black arrows indicate apoptotic eosinophils. Data are expressed as mean ± SEM as analyzed by analysis of variance with a Newman-Keuls multiple-comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001 versus control, ###P < 0.001 versus 100 μM wogonin-treated sample. AnnV = annexin V; DAPI = 4′,6-diamidino-2-phenylindole; i.p. = intraperitoneal; i.t. = intratracheal; OVA = ovalbumin.
Wogonin Attenuates Allergic Airway Inflammation In Vivo
To assess the ability of wogonin to attenuate allergic airway inflammation, mice were sensitized and challenged with OVA, and then wogonin was administered on 4 consecutive days prior to acquisition of tissue (Figure 4A). Wogonin administration led to reductions in total BAL cells (Figure 4B), BAL eosinophils (Figure 4C), and interstitial eosinophils (Figures 4D and 4E). Wogonin did not significantly reduce numbers of circulating or bone marrow eosinophils (Figure 4F) or alter interstitial mast cell numbers (CD45+ve/CD117+ve cells) (Figure 4G). To examine whether eosinophil activation status was altered in vivo following wogonin administration, CD11b (a cell surface β2-integrin) was measured on BAL and interstitial eosinophils. CD11b expression was unaltered following wogonin administration (Figure 4H).
Figure 4.

Wogonin attenuates allergic airway inflammation in vivo. (A) Following ovalbumin (OVA) sensitization and challenges, mice were treated with wogonin (1 mg) or vehicle control on Days 25–28, with acquisition and analysis of tissue taking place on Day 29. (B) Total bronchoalveolar lavage (BAL) cells, (C) BAL eosinophils (BAL eos), and (D) interstitial eosinophils are shown, with (E) representative overlay histogram of Siglec-F binding of lung interstitial inflammatory cells from mice treated with OVA (red line) or OVA + wogonin (blue line). (F) Blood and bone marrow eosinophils, (G) interstitial mast cells, and (H) interstitial and BAL eosinophil CD11b expression are shown. n = 7 or 8 mice. Data are expressed as mean ± SEM as analyzed by unpaired t test. *P < 0.05 versus vehicle. BALF = bronchoalveolar fluid; i.p. = intraperitoneal; i.t. = intratracheal; MFI = mean fluorescence intensity; n.s. = not significant.
To investigate the functional consequences of the changes in cellular inflammation seen following wogonin administration, lungs were fixed and stained with PAS to determine mucus production, and separate mice underwent airway hyperresponsiveness testing. Following OVA administration, widespread bronchial cell mucus production was observed, which was greatly attenuated by administration of wogonin (Figures 5A and 5B). Airway hyperresponsiveness to aerosolized methacholine was assessed in anesthetized, mechanically ventilated mice. Mice that received wogonin had attenuated methacholine-induced increases in lung resistance (Figure 5C). A small but significant reduction in the cytokine IL-17 was observed following wogonin administration (Figure 5D), whereas levels of IL-5 and CCL11 (eotaxin) were unchanged (data not shown). The reductions in mucus production or airway hyperresponsiveness did not appear to be secondary to any toxicity of wogonin, as BAL protein, a marker of alveolar–capillary barrier integrity, was unaltered by wogonin administration (Figure 5F), and mouse weights were not significantly different between groups (data not shown).
Figure 5.
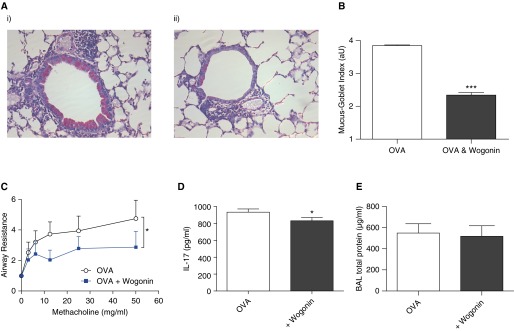
Wogonin improves allergic airway responses. Following ovalbumin (OVA) sensitization and challenges, mice were treated with wogonin (1 mg) or vehicle control on Days 25–28, with acquisition and analysis of tissue and lung function performed on Day 29. (A) Representative lung tissue sections stained with periodic acid–Schiff stain in (i) OVA- and (ii) OVA- and wogonin-treated animals (×200 original magnification). (B) Quantification of mucus production was assessed by the mucus-goblet index, an average severity score per airway, expressed in arbitrary units (aU) (n = 4 or 5 mice per group). (C) Airway responsiveness was assessed in anesthetized and mechanically ventilated mice in response to aerosolized methacholine, with lung resistance determined and expressed relative to baseline values after phosphate-buffered saline exposure. (D) Lung IL-17 concentration and (E) bronchoalveolar lavage (BAL) protein (n ≥ 6). Data are expressed as mean ± SEM as analyzed by two-way analysis of variance (C) or unpaired t test (B, D, and E). *P < 0.05 and ***P < 0.001 versus control.
To investigate whether eosinophil apoptosis was occurring in vivo, mice were treated with wogonin on a single occasion (Day 25), and BAL and lung tissue were acquired the following day (16 h after wogonin treatment). At this time point, wogonin administration led to a small reduction in total BAL cells (Figure 6A), with an increase in apoptotic eosinophils observed (Figure 6B). BAL monocyte and macrophage cell numbers were unchanged following wogonin administration (Figure 6C), but there was evidence of increased clearance of apoptotic cells (apoptotic bodies observed within macrophages) (Figures 6D and 6E). An increased number of DAPI−ve/annexin-V+ve eosinophils within the interstitial compartment were also observed after a single treatment with wogonin (Figure E2). Given that wogonin induced eosinophil apoptosis in vivo within the inflamed lung, it was surprising that we observed little effect on the numbers of blood and bone marrow eosinophils (Figure 4F). We therefore cultured BAL fluid, interstitial cells, and bone marrow cells ex vivo with increasing concentrations of wogonin prior to eosinophil apoptosis assessment. This revealed that BAL eosinophils were most sensitive to wogonin-induced apoptosis, followed by lung and then bone marrow eosinophils (Figure E3).
Figure 6.

Wogonin accelerates eosinophil apoptosis in vivo. Following ovalbumin (OVA) sensitization and challenges, mice were treated with wogonin (1 mg) or vehicle control on Day 25, with acquisition and analysis of tissue performed on Day 26 (16 h after wogonin treatment). (A) Total bronchoalveolar lavage (BAL) inflammatory cells, (B) apoptotic eosinophils, (C) total macrophage/monocytes, and (D) macrophages containing apoptotic bodies (n ≥ 6). (E) BAL cells (×1,000 original magnification) demonstrate (i) normal eosinophil morphology in the control group, (ii) a macrophage containing apoptotic bodies in the wogonin-treated group (black arrow), and (iii) an apoptotic body (white arrow) and apoptotic eosinophils (black arrows) in the wogonin-treated group. Data are expressed as mean ± SEM, analyzed by unpaired t test. *P < 0.05 versus control. BALF = bronchoalveolar fluid.
To investigate whether apoptosis was important for the attenuated inflammation seen following wogonin, mice were treated with the caspase inhibitor z-VAD alone and in combination with wogonin (Figure 7). H&E staining of fixed lungs demonstrated that the reduction in inflammatory cell infiltrate seen with wogonin could be blocked by concurrent z-VAD administration, confirming an essential role for apoptosis in the proresolution effects of wogonin.
Figure 7.

Caspase inhibition prevents wogonin-induced attenuation of allergic airway inflammation. Following ovalbumin (OVA) sensitization and challenges, mice were treated with vehicle control, wogonin (1 mg at 32 h), z-VAD (5 mg/kg at 32, 36, 40, and 44 h), or combined wogonin and z-VAD, with acquisition and analysis of lungs for histology performed at 48 hours (Day 26). Representative lung tissue sections (hematoxylin and eosin stain) in (A) control/OVA, (B) wogonin-treated, (C) z-VAD-treated, and (D) combined wogonin- and z-VAD-treated animals (all images obtained at ×100 original magnification). (E) Quantification of interstitial and alveolar inflammatory cell infiltration, expressed in arbitrary units (aU) (n = 6 or 7 per group). Data are expressed as mean ± SEM, analyzed by analysis of variance with a Newman-Keuls multiple-comparison test. ***P < 0.001 versus control; ###P < 0.001 versus wogonin-treated mice.
Discussion
Inflammation is a highly orchestrated process that is crucial to maintaining tissue homeostasis and repairing damage following infection and injury. These events must be self-limiting; otherwise, they can result in great metabolic energy expenditure, host tissue damage, and organ failure. Resolution of inflammation is an active and controlled process regulated by changes in local autacoid production, inflammatory cell apoptosis, and subsequent clearance by surrounding phagocytes (12, 27). It is increasingly recognized that dysregulation of the resolution process contributes to human inflammatory diseases (28, 29) and that the processes involved can be targeted for potential therapeutic benefit (30, 31). Certainly, neutrophils (20), and more recently mast cells (32), have been targeted with pharmacological agents to increase their rates of apoptosis, with consequent benefits seen in preclinical models of inflammation. Eosinophils are terminally differentiated granulocytes whose main role is thought to be the defense against parasitic infection. However, they also play a central role in allergic diseases, including hay fever, eczema, and asthma (2). During an exacerbation of asthma, eosinophils are recruited into lung tissue, where they are believed to contribute directly to disease pathogenesis (1), although most of the evidence for the role of eosinophils in allergic inflammation comes from models using allergen-sensitized mice. Low levels of eosinophil apoptosis in sputum correlate with asthma severity in humans (14), and evidence from mice demonstrates that eosinophils exacerbate airway remodeling, mucus production, and airway hyperresponsiveness (4, 5). Glucocorticoids are widely used to treat allergic diseases and are known to induce eosinophil apoptosis in vitro, which may contribute to their antiallergy and antiinflammatory effects. Therefore, pharmacological agents that counteract the prolongation of eosinophil lifespan in allergic inflammation have potential as novel treatments for human allergic disease.
In the present study, we show that the flavone wogonin induces prompt time- and concentration-dependent induction of eosinophil apoptosis. Wogonin-induced eosinophil apoptosis was rapid; it was observed after 6 hours. This is in contrast to numerous other eosinophil apoptosis-inducing agents, including glucocorticoids (15), antibody-mediated Fas ligation (33), or transforming growth factor β (34), which tend to cause eosinophil apoptosis at later time points. Wogonin-induced eosinophil apoptosis was dependent upon activation of caspase enzymes, as it was inhibited by the presence of the broad-spectrum caspase inhibitors z-VAD and Q-VD, and was associated with accelerated activation of caspase-3, as determined by Western blot analysis. Wogonin treatment also augmented the loss of eosinophil mitochondrial membrane potential, suggesting activation of the intrinsic pathway of apoptosis.
Following the transmigration of granulocytes into an area of inflammation, their sensitivity toward apoptosis-inducing agents is altered by a combination of the process of transendothelial migration and exposure to a proinflammatory environment with high local concentrations of inflammatory mediators (35). Certainly, the eosinophil apoptosis-inducing potential of glucocorticoids is reduced in the presence of factors such as IL-5 (36), which may partly contribute to the steroid resistance that is seen in severe allergic disease in humans. Interestingly, ligation of Siglec-8, which can drive eosinophil apoptosis, causes necrosis in IL-5-primed human eosinophils in vitro (37), and evidence exists for primary lysis of eosinophils in human asthmatic airways (13). This suggests that activated eosinophils may be primed to undergo alternative death pathways, including primary lysis and/or necrosis, with potentially deleterious and pathogenic consequences. Similarities and differences between mouse and human eosinophil functions have been described (38), and there are likely to be differences in the fate of eosinophils between those described in vitro compared with what may occur in vivo, especially between eosinophils located in different anatomical compartments (e.g., airway lumen versus tissue dwelling eosinophils). We observed, however, that ex vivo mouse eosinophils, which had already migrated toward and been exposed to a proinflammatory environment within the inflamed lung prior to isolation, underwent wogonin-induced, caspase-dependent apoptosis. Prior to ascertaining whether this observation is similar in humans, the study of wogonin’s effect on the cell death of transmigrated human eosinophils would be worth investigating.
Eosinophil apoptosis-inducing pharmacological agents have been tested previously in preclinical models of allergic inflammation, with varying success. Cyclin-dependent kinase inhibitors (CDKis), which induce eosinophil apoptosis in vitro (23), have been tested in allergic inflammation. Although one study group found that the CDKi roscovitine induced eosinophil apoptosis in vitro, they did not find any significant effect of roscovitine when administered in a model of allergic inflammation in vivo with respect to airway or tissue eosinophilia, nor was any difference in goblet cell mucus production noted (39). This is in partial contrast to a previous study by our group in which we used a different CDKi (AT7519) where administration in a model of allergic pleurisy was able to reduce eosinophil numbers at the site of inflammation secondary to enhanced eosinophil apoptosis (40), although the functional effects of this CDKi-mediated reduction in eosinophils were not investigated. Similarly, in a recent investigation, mice deficient in Siglec-F signaling demonstrated enhanced eosinophilic airway inflammation, presumed secondary to a reduction in eosinophil apoptosis (41). Functional consequences of this increased eosinophilic inflammation were not measured, although previous studies of the role of Siglec-F have not shown a significant effect on airway hyperresponsiveness, with mixed results in relation to mucus production (42, 43).
To investigate the ability of wogonin to reduce allergic inflammation in vivo, we used an OVA model of allergic airway inflammation. Because the results of our in vitro experiments demonstrated that wogonin altered eosinophil apoptosis, a key component of the resolution of inflammation, we chose to administer our drug after the onset of inflammation. Wogonin administration in established allergic inflammation led to reductions in both BAL and tissue eosinophils, with a corresponding increase in BAL apoptotic eosinophils and increased numbers of apoptotic bodies present within macrophages. Wogonin had a greater effect on reducing alveolar eosinophils compared with interstitial eosinophils (30.3% reduction vs. 23.2%), which is similar to observations made by others that antieosinophil therapies, including glucocorticoids and anti–IL-5 treatment, have a greater effect on limiting BAL eosinophilia compared with tissue eosinophilia in mice (44, 45). We found that alveolar eosinophils were more sensitive than lung interstitial or circulating eosinophils to wogonin-induced apoptosis, although the underlying molecular mechanisms of this observation remain to be elucidated.
In confirmation of our in vitro and ex vivo observations, we found that the ability of wogonin to attenuate inflammation in vivo was blocked by coadministration of the caspase inhibitor z-VAD, suggesting that apoptosis was the major mechanism behind the antiinflammatory effect of wogonin in our model. In addition, administration of wogonin not only reduced inflammatory cell numbers but also had functional effects on lung tissue, with a significant reduction in airway mucus production and attenuated airway hyperresponsiveness, two key pathological and functional hallmarks of human asthma. Determining whether such functional tissue changes are a direct consequence of enhanced eosinophil apoptosis or represent additional antiinflammatory properties of wogonin requires further experimentation. Although we did not examine the downstream cellular consequences of enhanced eosinophil apoptosis, including alterations in macrophage phenotype and phagocytic capacity, these represent important areas for future experiments. Certainly, it is known that the clearance of apoptotic cells by lung epithelial cells leads to an antiinflammatory environment in allergic inflammation, whereas engulfment of apoptotic cells by surrounding macrophages leads to a phenotypic switch with reduced proinflammatory and increased antiinflammatory mediator production (11). Levels of the cytokine IL-17, known to enhance eosinophilic airway inflammation (46), were reduced by wogonin, whereas the eosinophil-recruiting cytokines IL-5 and CCL11 (eotaxin) were unchanged. This further suggests that the reductions in eosinophils seen in vivo were due to enhanced apoptosis rather than being secondary to alterations in eosinophil recruitment to lung tissue. Although the pathogenic role of eosinophils in allergic inflammation relates not only to absolute numbers but also to other factors, including activation status, we found no difference in CD11b surface expression on eosinophils in vivo following wogonin administration.
In summary, we show that wogonin induces caspase-dependent eosinophil apoptosis via the intrinsic pathway of apoptosis and, in doing so, reduces inflammatory cell numbers at sites of allergic inflammation. Although wogonin had a relatively low potency, we observed that eosinophils recruited to sites of allergic inflammation had increased sensitivity toward wogonin-induced apoptosis. In addition, wogonin has functional effects on lung tissue, with reduced mucus production and attenuated airway hyperresponsiveness, key pathological features present in allergic airway inflammation. The ability to intervene with wogonin after the onset of inflammation, and not having to rely on pretreatment or administration close to inflammation onset, has obvious additional benefits. Therefore, wogonin and related eosinophil apoptosis-inducing compounds may have therapeutic potential for the treatment of human allergic inflammatory diseases.
Acknowledgments
Acknowledgment
The authors thank Fiona Rossi for assistance with flow cytometry, Dr. Paul Fitch for assistance with performing in vivo lung function measurements, and Calum Robb for laboratory assistance.
Footnotes
Supported by the Wellcome Trust (WT094415 [C.D.L.] and WT096497 [D.A.D.]) and the UK Medical Research Council (MR/K013386/1 [A.G.R., R.D., and C.H.]).
Author Contributions: C.D.L., D.A.D., S.S., J.R., J.M.F., A.L.A., R.D., and A.G.R.: performed research; C.D.L., D.A.D., J.S., C.H., and A.G.R.: designed the research; C.D.L., D.A.D., and A.G.R.: wrote the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201408-1565OC on January 28, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Rosenberg HF, Dyer KD, Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol. 2013;13:9–22. doi: 10.1038/nri3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fulkerson PC, Rothenberg ME. Targeting eosinophils in allergy, inflammation and beyond. Nat Rev Drug Discov. 2013;12:117–129. doi: 10.1038/nrd3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goh YP, Henderson NC, Heredia JE, Red Eagle A, Odegaard JI, Lehwald N, Nguyen KD, Sheppard D, Mukundan L, Locksley RM, et al. Eosinophils secrete IL-4 to facilitate liver regeneration. Proc Natl Acad Sci USA. 2013;110:9914–9919. doi: 10.1073/pnas.1304046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, O’Neill KR, Protheroe C, Pero R, Nguyen T, Cormier SA, et al. Defining a link with asthma in mice congenitally deficient in eosinophils. Science. 2004;305:1773–1776. doi: 10.1126/science.1099472. [DOI] [PubMed] [Google Scholar]
- 5.Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, Ghiran S, Gerard NP, Yu C, Orkin SH, et al. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–1779. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 6.Castro M, Mathur S, Hargreave F, Boulet LP, Xie F, Young J, Wilkins HJ, Henkel T, Nair P. Reslizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am J Respir Crit Care Med. 2011;184:1125–1132. doi: 10.1164/rccm.201103-0396OC. [DOI] [PubMed] [Google Scholar]
- 7.Bel EH, Wenzel SE, Thompson PJ, Prazma CM, Keene ON, Yancey SW, Ortega HG, Pavord ID SIRIUS Investigators. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N Engl J Med. 2014;371:1189–1197. doi: 10.1056/NEJMoa1403291. [DOI] [PubMed] [Google Scholar]
- 8.Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, Humbert M, Katz LE, Keene ON, Yancey SW, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371:1198–1207. doi: 10.1056/NEJMoa1403290. [DOI] [PubMed] [Google Scholar]
- 9.Persson C, Uller L. Transepithelial exit of leucocytes: inflicting, reflecting or resolving airway inflammation? Thorax. 2010;65:1111–1115. doi: 10.1136/thx.2009.133363. [DOI] [PubMed] [Google Scholar]
- 10.Leitch AE, Duffin R, Haslett C, Rossi AG. Relevance of granulocyte apoptosis to resolution of inflammation at the respiratory mucosa. Mucosal Immunol. 2008;1:350–363. doi: 10.1038/mi.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L, Ravichandran KS. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. 2013;493:547–551. doi: 10.1038/nature11714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14:166–180. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Persson C, Uller L. Theirs but to die and do: Primary lysis of eosinophils and free eosinophil granules in asthma. Am J Respir Crit Care Med. 2014;189:628–633. doi: 10.1164/rccm.201311-2069OE. [DOI] [PubMed] [Google Scholar]
- 14.Duncan CJ, Lawrie A, Blaylock MG, Douglas JG, Walsh GM. Reduced eosinophil apoptosis in induced sputum correlates with asthma severity. Eur Respir J. 2003;22:484–490. doi: 10.1183/09031936.03.00109803a. [DOI] [PubMed] [Google Scholar]
- 15.Meagher LC, Cousin JM, Seckl JR, Haslett C. Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J Immunol. 1996;156:4422–4428. [PubMed] [Google Scholar]
- 16.Liu Y, Cousin JM, Hughes J, Van Damme J, Seckl JR, Haslett C, Dransfield I, Savill J, Rossi AG. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol. 1999;162:3639–3646. [PubMed] [Google Scholar]
- 17.Fahy JV. Eosinophilic and neutrophilic inflammation in asthma: insights from clinical studies. Proc Am Thorac Soc. 2009;6:256–259. doi: 10.1513/pats.200808-087RM. [DOI] [PubMed] [Google Scholar]
- 18.Polier G, Ding J, Konkimalla BV, Eick D, Ribeiro N, Köhler R, Giaisi M, Efferth T, Desaubry L, Krammer PH, et al. Wogonin and related natural flavones are inhibitors of CDK9 that induce apoptosis in cancer cells by transcriptional suppression of Mcl-1. Cell Death Dis. 2011;2:e182. doi: 10.1038/cddis.2011.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lucas CD, Allen KC, Dorward DA, Hoodless LJ, Melrose LA, Marwick JA, Tucker CS, Haslett C, Duffin R, Rossi AG. Flavones induce neutrophil apoptosis by down-regulation of Mcl-1 via a proteasomal-dependent pathway. FASEB J. 2013;27:1084–1094. doi: 10.1096/fj.12-218990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lucas CD, Dorward DA, Tait MA, Fox S, Marwick JA, Allen KC, Robb CT, Hirani N, Haslett C, Duffin R, et al. Downregulation of Mcl-1 has anti-inflammatory pro-resolution effects and enhances bacterial clearance from the lung. Mucosal Immunol. 2014;7:857–868. doi: 10.1038/mi.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma S, Dorward DA, Rossi AG, Lucas CD. Wogonin induces caspase-dependent human eosinophil apoptosis. Poster presented at the British Pharmacological Society Conference. London; 2013 [accessed 2015 Feb 4]. Available from: http://www.pa2online.org/abstracts/vol11issue3abst013p.pdf. [Google Scholar]
- 22.Dorward DA, Lucas CD, Alessandri AL, Marwick JA, Rossi F, Dransfield I, Haslett C, Dhaliwal K, Rossi AG. Technical advance: autofluorescence-based sorting: rapid and nonperturbing isolation of ultrapure neutrophils to determine cytokine production. J Leukoc Biol. 2013;94:193–202. doi: 10.1189/jlb.0113040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duffin R, Leitch AE, Sheldrake TA, Hallett JM, Meyer C, Fox S, Alessandri AL, Martin MC, Brady HJ, Teixeira MM, et al. The CDK inhibitor, R-roscovitine, promotes eosinophil apoptosis by down-regulation of Mcl-1. FEBS Lett. 2009;583:2540–2546. doi: 10.1016/j.febslet.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 24.Mackenzie KJ, Fitch PM, Leech MD, Ilchmann A, Wilson C, McFarlane AJ, Howie SE, Anderton SM, Schwarze J. Combination peptide immunotherapy based on T-cell epitope mapping reduces allergen-specific IgE and eosinophilia in allergic airway inflammation. Immunology. 2013;138:258–268. doi: 10.1111/imm.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grünig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, Caldicott A, Martinez-Losa M, Walker TR, Duffin R, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis Nat Med 2006121056–1064.[Published erratum appears in Nat Med 2006;12:1434.] [DOI] [PubMed] [Google Scholar]
- 27.El Kebir D, József L, Pan W, Wang L, Petasis NA, Serhan CN, Filep JG. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med. 2009;180:311–319. doi: 10.1164/rccm.200810-1601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levy BD, Bonnans C, Silverman ES, Palmer LJ, Marigowda G, Israel E. Diminished lipoxin biosynthesis in severe asthma. Am J Respir Crit Care Med. 2005;172:824–830. doi: 10.1164/rccm.200410-1413OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Planagumà A, Kazani S, Marigowda G, Haworth O, Mariani TJ, Israel E, Bleecker ER, Curran-Everett D, Erzurum SC, Calhoun WJ, et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. Am J Respir Crit Care Med. 2008;178:574–582. doi: 10.1164/rccm.200801-061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dorward DA, Lucas CD, Allen KC, Rossi AG.Resolution of allergic inflammation Adkinson NF, Jr, Bochner BS, Burks W, Busse WW, Holgate ST, Lemanske RF, Jr, O’Hehir RE.Middleton’s allergy: principles & practice8th ed.Vol. 1Oxford: Saunders; 2013389–396. [Google Scholar]
- 31.Felton JM, Lucas CD, Rossi AG, Dransfield I. Eosinophils in the lung – modulating apoptosis and efferocytosis in airway inflammation. Front Immunol. 2014;5:302. doi: 10.3389/fimmu.2014.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wechsler JB, Hsu CL, Bryce PJ. IgE-mediated mast cell responses are inhibited by thymol-mediated, activation-induced cell death in skin inflammation. J Allergy Clin Immunol. 2014;133:1735–1743. doi: 10.1016/j.jaci.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsumoto K, Schleimer RP, Saito H, Iikura Y, Bochner BS. Induction of apoptosis in human eosinophils by anti-Fas antibody treatment in vitro. Blood. 1995;86:1437–1443. [PubMed] [Google Scholar]
- 34.Alam R, Forsythe P, Stafford S, Fukuda Y. Transforming growth factor β abrogates the effects of hematopoietins on eosinophils and induces their apoptosis. J Exp Med. 1994;179:1041–1045. doi: 10.1084/jem.179.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watson RW, Rotstein OD, Nathens AB, Parodo J, Marshall JC. Neutrophil apoptosis is modulated by endothelial transmigration and adhesion molecule engagement. J Immunol. 1997;158:945–953. [PubMed] [Google Scholar]
- 36.Brode S, Farahi N, Cowburn AS, Juss JK, Condliffe AM, Chilvers ER. Interleukin-5 inhibits glucocorticoid-mediated apoptosis in human eosinophils. Thorax. 2010;65:1116–1117. doi: 10.1136/thx.2009.124909. [DOI] [PubMed] [Google Scholar]
- 37.Kano G, Almanan M, Bochner BS, Zimmermann N. Mechanism of Siglec-8-mediated cell death in IL-5-activated eosinophils: role for reactive oxygen species-enhanced MEK/ERK activation. J Allergy Clin Immunol. 2013;132:437–445. doi: 10.1016/j.jaci.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JJ, Jacobsen EA, Ochkur SI, McGarry MP, Condjella RM, Doyle AD, Luo H, Zellner KR, Protheroe CA, Willetts L, et al. Human versus mouse eosinophils: “that which we call an eosinophil, by any other name would stain as red.”. J Allergy Clin Immunol. 2012;130:572–584. doi: 10.1016/j.jaci.2012.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farahi N, Uller L, Juss JK, Langton AJ, Cowburn AS, Gibson A, Foster MR, Farrow SN, Marco-Casanova P, Sobolewski A, et al. Effects of the cyclin-dependent kinase inhibitor R-roscovitine on eosinophil survival and clearance. Clin Exp Allergy. 2011;41:673–687. doi: 10.1111/j.1365-2222.2010.03680.x. [DOI] [PubMed] [Google Scholar]
- 40.Alessandri AL, Duffin R, Leitch AE, Lucas CD, Sheldrake TA, Dorward DA, Hirani N, Pinho V, de Sousa LP, Teixeira MM, et al. Induction of eosinophil apoptosis by the cyclin-dependent kinase inhibitor AT7519 promotes the resolution of eosinophil-dominant allergic inflammation. PLoS ONE. 2011;6:e25683. doi: 10.1371/journal.pone.0025683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiwamoto T, Brummet ME, Wu F, Motari MG, Smith DF, Schnaar RL, Zhu Z, Bochner BS. Mice deficient in the St3gal3 gene product α2,3 sialyltransferase (ST3Gal-III) exhibit enhanced allergic eosinophilic airway inflammation. J Allergy Clin Immunol. 2014;133:240–247. doi: 10.1016/j.jaci.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang M, Angata T, Cho JY, Miller M, Broide DH, Varki A. Defining the in vivo function of Siglec-F, a CD33-related Siglec expressed on mouse eosinophils. Blood. 2007;109:4280–4287. doi: 10.1182/blood-2006-08-039255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho JY, Song DJ, Pham A, Rosenthal P, Miller M, Dayan S, Doherty TA, Varki A, Broide DH. Chronic OVA allergen challenged Siglec-F deficient mice have increased mucus, remodeling, and epithelial Siglec-F ligands which are up-regulated by IL-4 and IL-13. Respir Res. 2010;11:154. doi: 10.1186/1465-9921-11-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Birrell MA, Battram CH, Woodman P, McCluskie K, Belvisi MG. Dissociation by steroids of eosinophilic inflammation from airway hyperresponsiveness in murine airways. Respir Res. 2003;4:3. doi: 10.1186/rr197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kung TT, Stelts DM, Zurcher JA, Adams GK, III, Egan RW, Kreutner W, Watnick AS, Jones H, Chapman RW. Involvement of IL-5 in a murine model of allergic pulmonary inflammation: prophylactic and therapeutic effect of an anti-IL-5 antibody. Am J Respir Cell Mol Biol. 1995;13:360–365. doi: 10.1165/ajrcmb.13.3.7654390. [DOI] [PubMed] [Google Scholar]
- 46.Wakashin H, Hirose K, Maezawa Y, Kagami S, Suto A, Watanabe N, Saito Y, Hatano M, Tokuhisa T, Iwakura Y, et al. IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med. 2008;178:1023–1032. doi: 10.1164/rccm.200801-086OC. [DOI] [PubMed] [Google Scholar]