

Abstract
Many epithelial—mesenchymal transition (EMT)-promoting transcription factors have been implicated in tumorigenesis and metastasis as well as chemoresistance of cancer. However, the underlying mechanisms mediating these processes are unclear. Here, we report that Foxq1, a forkhead box-containing transcription factor and EMT-inducing gene, promotes stemness traits and chemoresistance in mammary epithelial cells. Using an expression profiling assay, we identified Twist1, Zeb2, and PDGFRα and β as Foxq1 downstream targets. We further show that PDGFRα and β can be directly regulated by Foxq1 or indirectly regulated through the Foxq1/Twist1 axis. Knockdown of both PDGFRα and β results in more significant effects on reversing Foxq1-promoted oncogenesis in vitro and in vivo than knockdown of either PDGFRα or β alone. In addition, PDGFRβ is a more potent mediator of Foxq1-promoted stemness traits than PDGFRα. Finally, pharmacologic inhibition or gene silencing of PDGFRs sensitizes mammary epithelial cells to chemotherapeutic agents in vitro and in vivo. These findings collectively implicate PDGFRs as critical mediators of breast cancer oncogenesis and chemoresistance driven by Foxq1, with potential implications for developing novel therapeutic combinations to treat breast cancer.
Introduction
Cancer recurrence, metastasis, and chemoresistance correlate with each other and contribute greatly to the mortality of patients with breast cancer (1–3). For example, metastatic breast tumors tend to be more chemoresistant than primary tumors, as demonstrated by the marked decrease in the chemotherapeutic response rate in the metastatic breast cancer setting versus the neoadjuvant setting (1). In addition, chemoresistant tumors are prone to metastasize and respond poorly to neoadjuvant chemotherapy. This often correlates with earlier metastatic recurrence and shorter disease-free and overall survival (1). However, it remains undetermined whether there is a common mechanistic element linking these processes.
Recently, epithelial–mesenchymal transition (EMT) has been recognized as a mechanism for breast cancer cells to acquire metastatic properties (2, 4–8). Many transcription factors including Snail (9, 10), Twist1 (11), Foxc2 (12), Zeb1, and Zeb2 (13, 14) are capable of triggering EMT, promoting tumorigenesis and metastasis, and enhancing chemoresistance. However, how these transcription factors interact remains elusive, as do the crucial mediators of these EMT-promoting genes. Discovering these mediators could provide insight into the mechanisms of cancer recurrence, metastasis, and chemoresistance, and eventually facilitate the development of a targeted cancer therapy.
Foxq1, a human Forkhead-Box gene family member (15–17), has been shown to repress smooth muscle–specific genes (18) and is involved in hair follicle development (19, 20). Foxq1 is over-expressed in different cancer types (21–23) and known to be induced by TGFb signaling (24, 25). Consistent with these findings, our laboratory previously reported overexpression of Foxq1 in highly metastatic breast cancer cell lines and a direct correlation between Foxq1 expression and poor outcomes in patients with breast cancer. More importantly, we have also shown that ectopic expression of Foxq1 triggers EMT and contributes to breast cancer metastasis (26), a finding that was confirmed in two follow-up reports (25, 27). In our current study, we demonstrate that Foxq1 contributes to stemness traits and chemoresistance in mammary epithelial cells and that these functions are dependent on the Foxq1/Twist1/PDGFRs transcriptional axis. Using a loss-of-function assay and pharmacologic inhibition, we show that targeting PDGFRs is potentially an effective therapeutic approach to reverse oncogenesis, stemness traits, and chemoresistance driven by Foxq1 in breast cancer.
Materials and Methods
Cell culture
HMLE cells (human mammary epithelial cells immortalized with SV40 large T antigen and catalytic subunit of telomerase) and HMLER cells (HMLE cells transfected with activated Ras gene) were maintained in the culture as previously described (28). Mouse breast cancer cell line 4T1 was cultured in high-glucose DMEM and supplemented by 10% FBS, NEAA, and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin). NMuMG cells were cultured with DMEM medium with 4.5 g/L glucose, 10 mg/mL insulin and 10% FBS. All cell lines were authenticated upon receipt by comparing them to the original morphologic and growth characteristics.
Generation of gene overexpressing and knockdown stable cells
Full-length plasmids for Foxq1, Twist1, Zeb2, and Snail1 were purchased from Open Biosystems. These genes were then subcloned into a pENTR vector and recombinated into the pLenti-6/V5-DEST Vector. The lentiviruses for the full-length genes were generated using the pLentivirus-expression system (Invitrogen). The generated virus was used to infect targeted model cells. Stable cells were generated after being selected with blasticidin (10 μg/mL, Invivogen; refs. 29, 30).
A set of five shRNA clones for PDGFRα and β were purchased from Open Biosystems. The shRNA sequences for targeting PDGFRα, β and Foxq1 genes were shown in Supplementary Table S1. The lentiviruses for the shRNAs of PDGFRs were generated using the Trans-Lentiviral packaging system (Open Biosystems) for shRNA expression. The generated virus was used to infect targeted model cells. Stable cells were generated after being selected with puromycin (2.5 μg/mL, Invivogen).
Microarray analysis and RT-PCR
RNA was extracted from cells of interest using TRIzol (Invitrogen) and purified using an RNA Purification Kit (Qiagen) according to manufacturer's instructions. RNA (25 ng) was labeled with dye and applied to the microarray. Changes in gene expression were analyzed using a Sentrix human Ref-8 Expression BeadChip (Illumina, 8 array “stripes”). Data was normalized using the “average” method that simply adjusts the intensities of the two populations of gene expression values such that the means of the populations become equal. Fold enrichment values were used to obtain the list of candidate genes with greater than two-fold change. RT-PCR based on RNA from three independent cell cultures was performed as previously described (31) to validate the microarray result. Primer sequences are shown in Supplementary Table S2.
Cell proliferation, Transwell migration assay, Boyden chamber invasion assay, and clonogenic assay
Detailed methods of these assays are given in the Supplementary Materials and Methods section.
Mammosphere formation assay
A mammosphere formation assay was performed as previously described with the following modifications (32). Briefly, ten thousand cells were plated on a 6-well ultra-low attachment plate (Corning Inc.) and were grown in serum-free mammary epithelial growth medium (MEBM Basal Medium, Lonza) supplemented with B27 (Invitrogen), 20 ng/mL EGF, 1 μg/mL hydrocortisone, 5 μg/mL insulin, and 5 μg/mL β-mercaptoethanol. One milliliter of medium was added every other day for 7 to 12 days. Images of mammospheres were recorded and the number of mammospheres was manually counted. Experiments were performed in triplicate and repeated two times.
Tumor xenograft studies
All mouse experiments were carried out in accordance with approved protocols from the Institutional Animal Care and Use Committee at Wayne State University (Detroit, MI). We performed tumor xenograft studies using either BALB/c or NCR nu/nu female mice from NIH to study the effect of Foxq1 in chemoresistance, tumorigenesis and PDGFR's effect in mediating Foxq1ʹs function in chemoresistance. The detailed methods are described in the Supplementary Material and Methods.
Statistical analysis
The quantitative results were analyzed using a two-sample t test or one-way ANOVA. If the normality assumption did not hold, these parametric tests were replaced by nonparametric tests Wilcoxon or Kruskal–Wallis tests, respectively. The dose-response curves for doxorubicin or paclitaxel within each cell line type were analyzed using multiple linear regression with interactions and a dummy variable to denote the conditions of imatinib. For analysis of synergism, a 2 by 2 factorial experiment design was used. A two-way ANOVA model with two main factors imatinib and doxorubicin at day 70, or paclitaxel at day 24, and their interaction term was used. A statistically significant synergistic effect was observed if the interaction term was significant and if its effect was in the same direction.
Results
Foxq1 induces stemness traits and chemoresistance in mammary epithelial cells and breast cancer cells
To determine whether Foxq1 induces stemness traits in mammary epithelial cells, we generated HMLE stable cell model ectopically expressing the Foxq1 gene, in parallel with three known EMT promoting genes Snail, Twist1, and Zeb2 to serve as positive controls (28, 32). As anticipated, the resulting cells (HMLE/Foxq1, HMLE/Twist1, HMLE/Zeb2, and HMLE/Snail) acquired mesenchymal appearances and showed deregulation of epithelial and mesenchymal markers (Supplementary Fig. S1A and S1B). Using flow cytometry, we analyzed cells for CD44 and CD24 expression, two cell-surface markers whose expression in the CD44high/CD24low configuration are associated with both human breast cancer stem cells (CSC) and normal mammary epithelial stem cells (33, 34). More than 90% of the mesenchymal-like cells generated by Foxq1 overexpression acquired the CD44high/CD24low expression pattern, compared with less than 1% of cells expressing control LacZ vector (Fig. 1A). This same cell pattern shift was also observed for the Snail, Twist1, and Zeb2 overexpressed cells (Supplementary Fig. S1C). We then measured the cells' ability to form mammospheres to confirm acquired stemness. All HMLE cells with these transcription factors formed 8- to 10-fold more mammospheres compared with HMLE cells infected with the corresponding control vector (Fig. 1B and Supplementary Fig. S1D and S1E).
Figure 1.
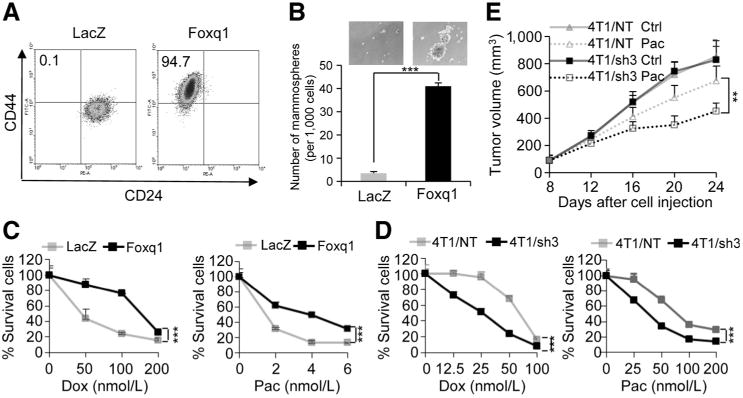
Foxq1-induced EMT, stemness traits, and chemoresistance in mammary epithelial cells and breast cancer cells. A, FACS analysis of cell-surface markers, CD44 and CD24, in HMLE cells with Foxq1 gene and LacZ control overexpression. B, in vitro quantification of mammospheres formed by cells described in A. The data are reported as the number of mammospheres formed/1,000 seeded cells ± SEM, compared with control (two experiments performed in triplicate; ***, P < 0,001). C and D, chemoresistance of HMLE cells (C; with or without Foxq1 overexpression) and 4T1 cells (D; with or without Foxq1 knockdown) was analyzed by an MTT assay after treatment with doxorubicin or paclitaxel as indicated for 24 hours. Percentages of live cells compared with the nontreatment control are shown (*, P < 0.05; **, P < 0.01). E, knockdown of Foxq1 led to significant sensitization of 4T1 cells to paclitaxel treatment in vivo (*, P < 0.05; **, P < 0.01).
We next investigated the effect of Foxq1 expression on chemoresistance. Using an MTT and clonogenic assay, we observed significantly more surviving HMLE/Foxq1 cells compared with control HMLE/LacZ cells when treated with two conventional chemotherapeutic agents, doxorubicin (MTT IC50 values: 137.4 vs. 36.78 nmol/L for Foxq1 and LacZ, respectively) and paclitaxel (MTT IC50 values: 3.62 vs. 0.61 nmol/L for Foxq1 and LacZ, respectively; Fig. 1C and Supplementary Fig. S2A). Moreover, we found that 4T1/sh3 cells, a stable cell line with marked Foxq1 inhibition (4T1/Foxq1-shRNA3; ref. 26), displayed a significant decrease in mammosphere formation capability (Supplementary Fig. S2B). This line also showed lower tumor initiation capability than nontargeted control cells (4T1/NT) in an in vivo cell dilution assay (Supplementary Fig. S2C). Consistent with this, 4T1/Sh3 cells, when compared with 4T1/NT cells, were considerably more sensitive to doxorubicin (IC50: 24.05 vs. 62.58 nmol/L, respectively) and paclitaxel (IC50: 36.30 vs. 96.04 nmol/L, respectively; Fig. 1D). To evaluate chemoresistance in vivo, we injected the 4T1/sh3 and 4T1/NT cells into BALB/c mice. Both cell types grew at similar rates without drug treatment in vivo. However, with paclitaxel treatment, tumor growth was significantly inhibited by day 16 in the mice bearing 4T1/sh3 tumors compared with the 4T1/NT tumors (Fig. 1E). Immunohistochemical analysis of various apoptotic markers demonstrate that knockdown of Foxq1 facilitates apoptosis induced by paclitaxel treatment (Supplementary Fig. S2D). Together, these results are consistent with previous reports that Foxq1 expression is antiapoptotic in colorectal cancer cell lines (23), suggesting that Foxq1 expression contributes to the stemness traits of mammary epithelial cells and promotes chemoresistance of breast cancer cells.
The transcriptional axis between Foxq1, Twist1, and PDGFRs and their clinical correlation
To gain more insight into the mechanism of Foxq1, we performed expression profiling to identify downstream targets of Foxq1. A total of 293 genes (99 upregulated genes and 194 downregulated genes) were significantly deregulated (3-fold cut-off level) in HMLE/Foxq1 cells compared with HMLE/LacZ control cells. A detailed analysis of these genes reveals they are involved in different cellular biologic functions including cell-to-cell adhesion, cell motility, EMT, and drug resistance (Fig. 2A and Supplementary Table S3).
Figure 2.
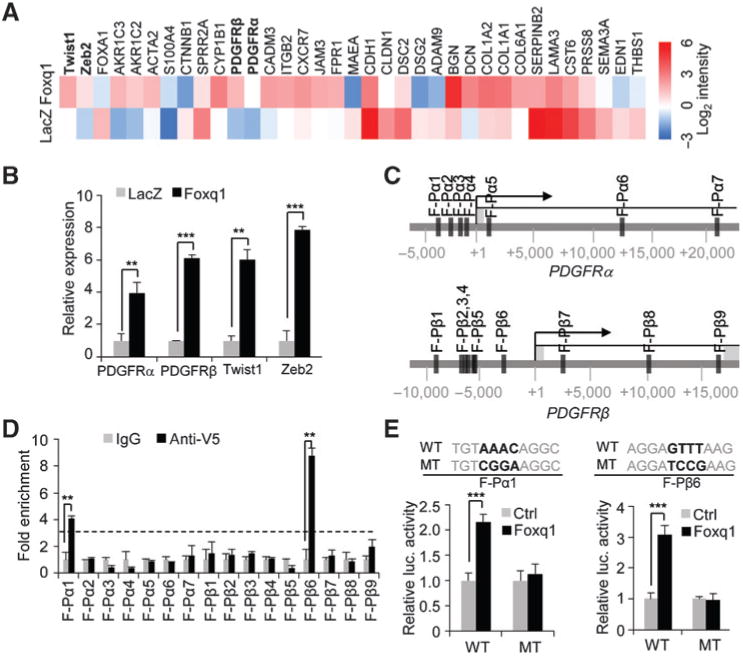
Foxq1 directly regulates PDGFRs. A, heatmap showing expression levels of the selected genes in HMLE/LacZ and HMLE/Foxq1 cells. Data are normalized to GAPDH expression. Log2 intensity scale is shown on right. B, the relative expression levels of the Twist1, Zeb2, and PDGFRα and β genes in HMLE cells with Foxq1 or control LacZ overexpression was measured by real-time RT-PCR assay (**, P < 0.01; ***, P < 0.001). C, potential binding sites of Foxq1 in the promoter regions of PDGFRα and β genes were identified by an in silico analysis. D, ChIP-qPCR analysis showed enrichment of the binding DNA in binding sites for PDGFRα (F-Pα1) and PDGFRβ (F-Pβ6; **, P < 0.01). Dotted line represents 3-fold enrichment. E, the binding of Foxq1 to the PDGFRα and β promoter region was confirmed by luciferase assay. The Foxq1 conservative binding sequence (WT) and mutant sequence (MT) in PDGFRα and β promoters was shown (***, P < 0.001).
Among the downstream targets that are upregulated by Foxq1, PDGFRα and β are two well-known receptors involved in cancer progression (Fig. 2B; refs. 35–38). We therefore explored whether Foxq1 could directly regulate PDGFRα and β promoters. Transcription factor (TF)-binding prediction assays using rVista, CONTRA v2, and the EpiTect ChIP tool revealed seven potential Foxq1 binding sites within the 25-kb PDGFRα promoter region and nine potential Foxq1-binding sites within the 25-kb PDGFRβ promoter region (Fig. 2C). By chromatin immunoprecipitation (ChIP)-PCR assay, we demonstrated significant enrichment of DNA for the first Foxq1-binding site (F-Pα1) in the PDGFRα promoter region and the sixth binding site (F-Pβ6) in the PDGFRβ promoter region, respectively (Fig. 2D). These data were further confirmed by a luciferase assay, where mutations in the core sequence of each of these two binding sites abolished the activation of these two promoters by Foxq1 (Fig. 2E). These results suggest that Foxq1 directly regulates expression of PDGFRα and β.
In parallel, Twist1 and Zeb2 genes were also identified as downstream targets of Foxq1 (Fig. 2A and B), suggesting their role as potential mediating transcription factors for Foxq1. We further identified three potential Foxq1-binding sites in the Twist1 promoter region and two Foxq1-binding sites in the Zeb2 promoter (Fig. 3A). For the Twist1 promoter, a ChIP-qPCR assay demonstrated enrichment of DNA in the first binding site (F-T1) close to the transcription start site (Fig. 3B). For the Zeb2 promoter, ChIP-qPCR data also showed that the first binding site (F-Z1) was significantly enriched in the precipitated protein–DNA complex (Fig. 3B). The results of ChIP-qPCR were further confirmed in luciferase assays in a dose- and sequence-dependent manner (Fig. 3C and D).
Figure 3.
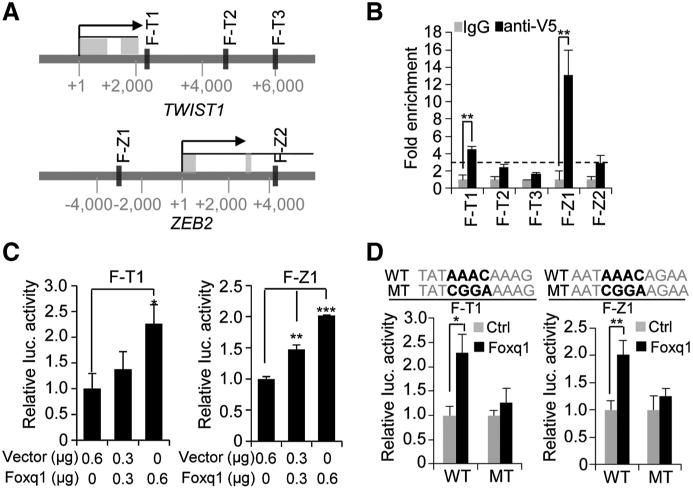
Foxq1 directly regulates Twist1 and Zeb2. A, potential binding sites of Foxq1 in the promoter regions of Twist1 and Zeb2 genes were identified by an in silico analysis. B, ChIP-qPCR analysis showed enrichment of the binding DNA in binding sites for Twist1 (F-T1) and Zeb2 (F-Z1; **, P < 0.01). Dotted line represents 3-fold enrichment. C, luciferase assay showed Foxq1 activated the Twist1 (left) and Zeb2 (right) promoter in a dose-dependent manner (*, P< 0.05; **, P< 0.01; and ***, P< 0.001). D, the binding of Foxq1 to the Twist1 and Zeb2 promoter regions was confirmed by luciferase assay. The Foxq1 conservative binding sequence (WT) and mutant sequence (MT) are shown (***, P < 0.001).
As Twist1 and Zeb2 downstream targets, the expression of PDGFRs was validated by RT-PCR in Twist1- and Zeb2-overexpressing model cell lines (Supplementary Fig. S3A and S3B). In addition, we found five Twist1-binding sites within the PDGFRα promoter region, and ten in the β promoter region, respectively (Supplementary Fig. S3C). For the PDGFRα gene, ChIP-qPCR indicated the enrichment of DNA in only the second Twist1-binding site (T-Pα2). For the PDGFRβ gene, Twist1 binding was observed in the first binding site (T-Pβ1) (Supplementary Fig. S3D). These results were further validated by luciferase assays (Supplementary Fig. S3E and S3F). Although six Zeb2-binding sites were predicted to exist in the PDGFRβ promoter region, none of these sites showed significant Zeb2 enrichment by ChIP-PCR (Supplementary Fig. S3C and S3D). Taken together, these data strongly suggest an upstream role for Foxq1 on Twist1 and Zeb2 genes, and that Foxq1 controls PDGFRα and β expression through both direct and indirect mechanisms.
We next analyzed the expression correlation of these genes in The Cancer Genome Atlas (TCGA) database. The expression of Foxq1 and Twist1 are closely correlated with PDGFRα and β (Supplementary Fig. S4A) in the level 3 gene expression (RNA-seV2) dataset for breast cancer. More importantly, the correlation of Foxq1, Twist1, and PDGFRs significantly correlated with a poor prognosis (Supplementary Fig. S4B). A similar correlation was also observed in uterine corpus endometrial carcinoma and several different cancer types (Supplementary Fig. S4C and data not shown). This discovery substantiates the clinical importance of the transcriptional cascade of Foxq1/Twist1/PDGFRs in cancer progression.
Knockdown of PDGFRs blocks Foxq1-promoted oncogenesis in vitro and in vivo
To investigate the role of PDGFRα and β in Foxq1-induced oncogenic properties, we knocked down PDGFRα and β separately, inHMLE/Foxq1 cells using shRNA technique (Supplementary Fig. S5A and S5B). Silencing of either PDGFRα or PDGFRβ significantly inhibited cell proliferation (data not shown), cell migration, and invasion (Supplementary Fig. S5C and S5D) when compared with nontarget control counterparts, suggesting that both PDGFRα and β contribute to Foxq1-induced cell proliferation and motility.
To explore potential synergistic effects of PDGFRα and β, we generated anew construct expressing two shRNAs with significant inhibitory effects on both PDGFRα and β. This construct, when packaged into a lentivirus and infected in HMLE/Foxq1 and HMLER/Foxq1 (HMLE with activated Ras gene and Foxq1) cells, produced significant inhibition of both PDGFRα and β, which is similar to what was observed for each of the individual shRNAs (Fig. 4A). The basal expression of PDGFRs in HMLE/Foxq1 and HMLER/Foxq1 are comparable (data not shown). As expected, knockdown of both PDGFRα and β displayed greater inhibitory effects on cell proliferation, cell migration, and invasion, when compared with knockdown of either PDGFRα or β alone (Fig. 4B and C)
Figure 4.
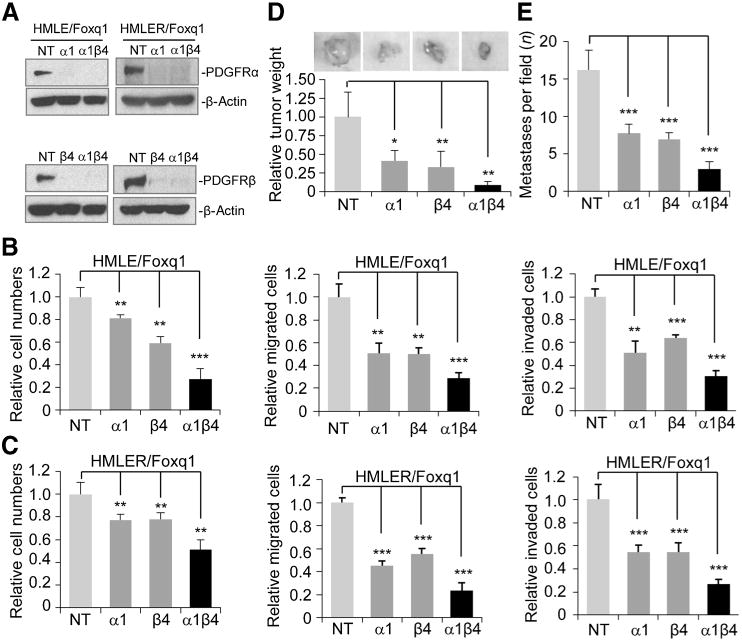
Knockdown of PDGFRs reverses Foxq1-promoted oncogenesis in vitro and in vivo. A, Western blot analysis showed individual or double knockdown of PDGFRα and β in HMLE/Foxq1 (left) and HMLER/Foxq1 (right) cells. β-Actin was used as a protein loading control. B and C, the effect of PDGFR silencing on cell proliferation (left), migration (middle), and invasion (right) in Foxq1-overexpressed HMLE (B) and HMLER (C) cells. Each bar represents the mean ± SEM (two experiments performed in triplicate; **, P < 0.01; ***, P < 0.001). D and E, the effect of PDGFRs on Foxq1-promoted breast cancer tumorigenesis (D) and metastasis (E; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
To examine the effects of PDGFRα and β on Foxq1-induced tumorigenesis and metastasis in vivo, HMLER/Foxq1 cells expressing nontarget control (NT), α, β, or both α and β PDGFR-silencing shRNA were injected into the fat pads of NCR nu/nu mice. In the absence of PDGFRα and β, we observed a significant decrease in tumor burdens compared with NT-expressing HMLER/Foxq1 cells, with the double knockdown of α and β showing the greatest effect (Fig. 4D). Moreover, HMLER/Foxq1 cells with a NT control showed an average of 16.3 metastatic loci in the lung section. In contrast, cells with either PDGFRα, PGGFRβ, or both PDGFRα and β silencing, showed an average of 7.8, 7, and 3 metastatic loci in the lung section, respectively, as evidenced by positive staining with anti-V5 antibody (Fig. 4E).
The effects PDGFRα and β on Foxq1-promoted stemness traits
Next, we assessed the effects of PDGFRβ on Foxq1-induced stemness in HMLE cells. We found that knockdown of both PDGFRs, or PDGFRβ alone, resulted in an approximate 25% decrease of CD44high/CD24low cells compared with the nontarget vector control, while knockdown of PDGFRα produced a much smaller effect (<5%; Fig. 5A and Supplementary Fig. S5E). Consistent with these results, knockdown of both PDGFRβ and β, or PDGFRβ alone, but not PDGFRα alone, significantly inhibited Foxq1-induced mammosphere formation (Fig. 5B and C and Supplementary Fig. S5F and S5G), suggesting a possible difference of PDGFRβ and PDGFRα in stemness characteristics. This result requires further validation of an in vivo cell dilution assay. Interestingly, knockdown of PDGFRα or β, alone or together, could not reverse Foxq1-induced EMT at the molecular or morphologic level (Fig. 5D and Supplementary Fig. S5H), suggesting Foxq1-induced EMT does not totally depend on PDGFRs.
Figure 5.
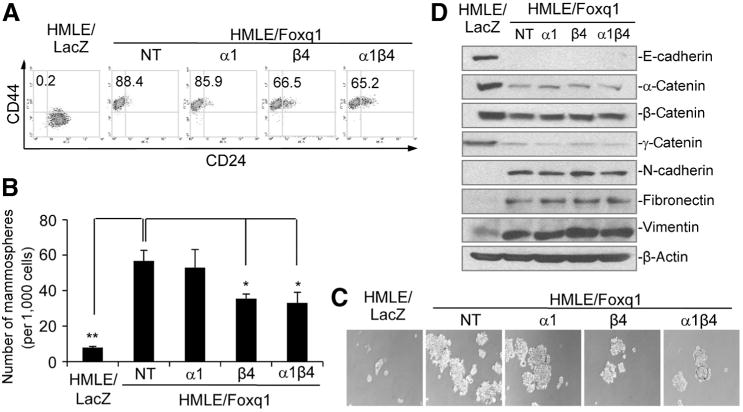
Different effects of PDGFRα and β on Foxq1-promoted stemness traits. A, FACS analysis of cell-surface markers, CD44 and CD24, in the HMLE/Foxq1 cells with individual or double PDGFRα and β knockdown. B, mammosphere formation promoted by Foxq1 was significantly reversed by PDGFRβ knockdown and PDGFRα and β double knockdown (*, P < 0.05; **, P < 0.01), but not by PDGFRα knockdown alone. C, representative figures for mammosphere formation by HMLE/Foxq1 cells with individual or double PDGFRα and β knockdown. D, Western blot analysis demonstrated no expression change for epithelial and mesenchymal markers in HMLE/Foxq1 cells with individual or double knockdown of PDGFRα and β.
To explore the clinical implications of the aforementioned discoveries, we investigated the therapeutic effect of the tyrosine kinase inhibitor imatinib, which targets PDGFRs, c-ABL, and Kit (39–41), on Foxq1-driven properties. We found that, in contrast to PDGFRs, there was no significant difference in c-ABL and Kit expression between HMLE/Foxq1 and HMLE/LacZ cells (Supplementary Fig. S6A). Similarly, the phosphorylation level of PDGFRs, but not ABL and Kit, was markedly higher in HMLE/Foxq1 compared with HMLE/LacZ cells (Supplementary Fig. S6B). Furthermore, imatinib treatment led to a significant decrease in the phosphorylation of PDGFα and β (Supplementary Fig. S6C), suggesting that PDGFRs are major targets for imatinib in HMLE/Foxq1 cells. We further showed that cell proliferation of HMLE/Foxq1 cells was significantly inhibited with a high-dose imatinib treatment after day 2 (Fig. 6A), whereas cell migration and invasion were markedly inhibited within one day even with low-dose imatinib treatment (Fig. 6B). Moreover, the CD44high/CD24low cell population and mammosphere formation in HMLE/Foxq1 cells were significantly decreased. There is a positive correlation between imatinib concentration and the magnitude of these effects (Fig. 6C and 6D and Supplementary Fig. S6D). Similar to PDGFR knockdown, imatinib treatment was unable to reverse EMT at morphologic or molecular levels (Supplementary Fig. S6E and S6F). Similar results were a achieved when we investigated the effects of imatinib treatment on 4T1 cells, which are known to express high level of Foxq1 (data not shown; ref. 26).
Figure 6.
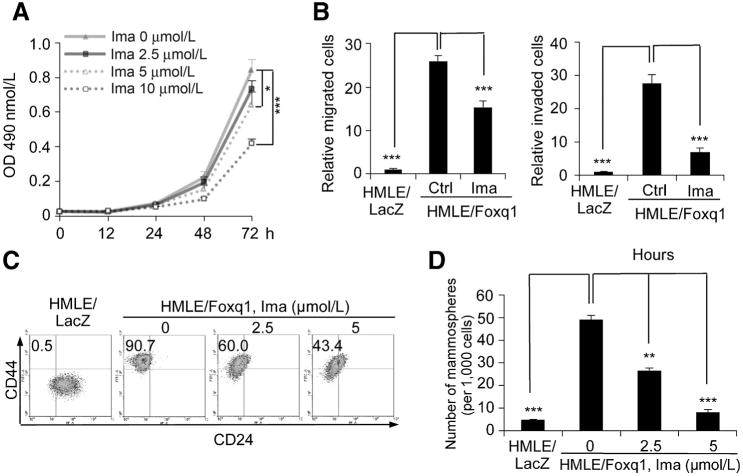
Pharmacologic inhibition of PDGFRs reverses Foxq1-promoted oncogenic properties and stemness traits. A, the effect of imatinib treatment on cel proliferation measured with a MTT assay in three days (*, P < 0.05; ***, P < 0.001). B, the effects of low-dose imatinib treatment (2.5 mmol/L) on HMLE/Foxq1 cell migration (left) and invasion (right; ***, P< 0.001). C, FACS analysis of cell-surface markers, CD44 and CD24, in the cells treated with different doses of imatinib. D, mammosphere formation promoted by Foxq1 was significantly inhibited by the treatment of imatinib in a dose-dependent manner (**, P < 0.01; ***, P < 0.001).
Targeting PDGFRs sensitizes Foxq1-overexpressing human mammary epithelial cells and breast cancer cells to chemotherapeutic agents
We next examined the effects of PDGFR on Foxq1-driven chemoresistance. Similar to the effects on stemness traits, knockdown of PDGFRβ showed more significant effects than PDGFRα on sensitizing Foxq1-overexpressing cells to both doxorubicin and paclitaxel, with double knockdown of PDGFRα and β showing the most potent effect (Supplementary Fig. S7A and S7B). In addition, low or intermediate doses of imatinib generated minimal effects on cell proliferation, but significantly sensitized Foxq1-overexpressing cells, including 4T1, HMLER/Foxq1, and HMLE/Foxq1 cells, to doxorubicin and paclitaxel. This is demonstrated by the statistically significant differences in the percentage of surviving cells in all cell models with and without imatinib treatment (Fig. 7A and B and Supplementary Fig. S7C). As expected, imatinib treatment had no significant effect on sensitizing NMuMG, a cell with low levels of PDGFRs, to either doxorubicin or paclitaxel (Supplementary Fig. S7D).
Figure 7.
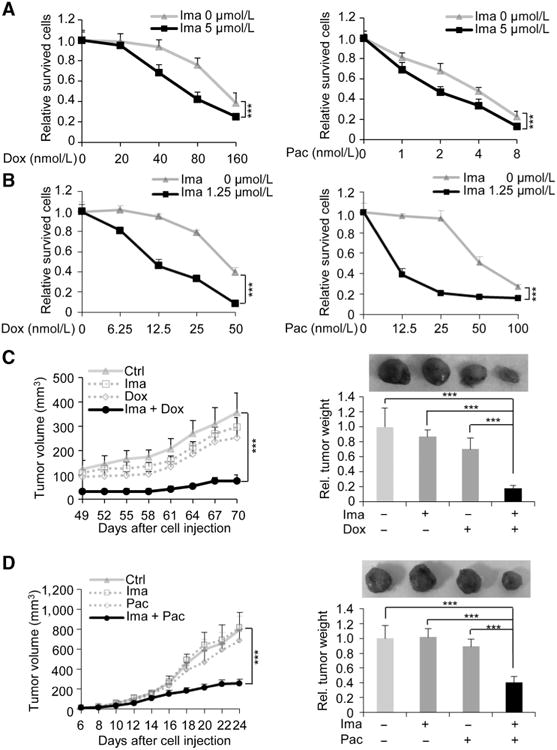
Targeting PDGFRs sensitizes breast cancer cells with Foxq1 overexpression to chemotherapeutic agents. A and B, the effect of imatinib on HMLER/Foxq1 (A) or 4T1 (B) cells with treatment of various doses of doxorubicin (left) and paclitaxel (right; ***, P < 0.001). C and D, synergistic inhibitory effects of imatinib with doxorubicin (C) or with paclitaxel (D) on HMLER/Foxq1 cells (C) or 4T1 cells (D) induced tumor growth (left) and tumor weight (right) were shown (***, P < 0.001).
We then implanted HMLER/Foxq1 cells into the fat pads of NCR nu/nu mice and examined the effects of imatinib, alone or together with doxorubicin, on tumor growth. In the presence of imatinib, doxorubicin inhibited tumor growth by 80%, but neither treatment alone inhibited xenograft growth significantly. Thus, a synergistic effect was observed for treatment with imatinib and doxorubicin in vivo (P < 0.001; Fig. 7C). Consistent with these results, treatment of 4T1 tumor-bearing BALB/c mice with imatinib and paclitaxel also generated significant synergistic effects on tumor growth in 24 days (P < 0.001; Fig. 7D). Furthermore, combinational treatment produced marked tumor apoptosis compared with single drug treatment as shown using immuno-histochemistry against apoptotic markers (Supplementary Fig. S8A and S8B). Overall, these results suggest that mammary cancer cells can become sensitized to various chemotherapeutic agents by targeting PDGFRs.
Discussion
In our current study, we have demonstrated that Foxq1, a downstream target of TGFb, triggers EMT and induces stemness traits and chemoresistance in mammary epithelial cells. This function is at least partially due to the transcriptional regulation of Foxq1 on PDGFRs directly or indirectly (Supplementary Fig. S8C). Knockdown or pharmacologic inhibition of PDGFR results in not only reversion of stemness traits, but also increased sensitivity of cells to chemotherapeutic agents. This discovery suggests an important signaling transition between TGFβ and PDGF, which is mediated by a novel transcriptional axis, as a major underlying mechanism for the acquisition of stemness traits and chemoresistant phenotype in mammary epithelial cells and breast cancer cells.
Several published articles suggest a role of PDGF/PDGFR signaling in various developmental processes and cellular functions, including EMT. For example, autocrine PDGF/PDGFR signaling contributes to maintenance of EMT, possibly through activation of STAT1 and other distinct pathways (42). Inhibition of PDGFR signaling interfered with EMT and caused apoptosis in murine and human mammary carcinoma cell lines. In addition, targeting PDGFRs, either by overexpression of a dominant-negative PDGFR or application of various drugs, can interfere with tumor growth and metastasis in mice and human patients (37, 43, 44). To our surprise, we demonstrated that genetic manipulation or pharmacologic inhibition of PDGFRs could reverse Foxq1-promoted oncogenesis and chemoresistance, but could not reverse Foxq1-triggered EMT at either the cellular or molecular level, suggesting that, under certain conditions, oncogenic properties of cancer cells are not necessarily bound to EMT status. This serendipitous finding is in contrast to the currently popular notion that EMT is a prerequisite for acquiring oncogenic properties during cancer progression, because most EMT promoting genes also promote stemness, oncogenesis, and chemoresistance. However, the results of our study provide intriguing evidence of the disassociation of EMT and oncogenic characteristics in cancer cells, suggesting a more complicated regulatory mechanism in EMT and cancer progression.
We show for the first time that PDGFRα and β have similar functions in oncogenic properties, but have different functions in stemness traits induced by Foxq1 in mammary epithelial cells and breast cancer cells. This result resembles a discovery 30 years ago indicating that PDGFRα and β demonstrate functional specificity during embryogenesis (45, 46). In line with these observations, a recent study showed that PDGFRα, but not PDGFRβ, plays a critical role in invadopodia formation (47). In another study, PDGFRβ, but not PDGFRα, informs intertumoral and intratumoral heterogeneity in glioblastoma (36). Taken together, these studies highlight the convergent and divergent functions of PDGFRα and β in cancer development. The underlying mechanism could be distinct ligand affinities and significant low structure homology in the kinase insert and carboxy-terminal region of these two isoforms, which lead to different downstream signaling activation (48).
More importantly, anti–PDGFR-based approaches sometimes result in a mixed clinical response, suggesting the importance of identifying specific patient subgroups (36, 49). The results of our current study demonstrate that PDGFRs are not only important for the oncogenic properties of cancer cells, but also critical for chemoresistance in human breast cancers over-expressing EMT-promoting genes like Foxq1. These findings provide a rational basis for targeting PDGFRs in conjunction with chemotherapeutic agents for suppressing tumor growth, blocking metastasis or enhancing the efficacy of chemotherapeutic treatments. It is worth to mention that the high doses of imatinib used in our study may not be appropriate for direct transition to the clinic. More specific and less toxic PDGFR inhibitors are needed in this scenario and could be a future direction of our study. In summary, the innovative drug combination we identified provides proof of principle for a novel combinational therapy for patients with breast cancer with aberrant expression of EMT promoting genes such as Foxq1 and Twist1.
Supplementary Material
Acknowledgments
The authors thank Dr. Robert A. Weinberg at the Massachusetts Institute of Technology (Boston, MA) for providing the HMLE and HMLER cell lines. The authors also thank Ms. Elizabeth A. Katz of the Karmanos Cancer Institute's Marketing and Communications Department for editing the manuscript.
Grant Support: This work was supported by ACS IRG grant from the Karmanos Cancer Institute, Wayne State University (G. Wu), and NIH/NCI grant RO1CA163772 (B. Zhang). The Biostatistics Core and the Biorepository Core of the Karmanos Cancer Institute are supported by grant number P30-CA022453-29.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
The datasets generated in the article have been deposited in GEO under the accession number GSE46834.
Authors' Contributions: Conception and design: F. Meng, G. Wu
Development of methodology: F. Meng, C.L. Speyer, G. Wu
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): F. Meng, F.R. Miller, G. Wu
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): C.L. Speyer, B. Zhang, Y. Zhao, W. Chen, G. Wu
Writing, review, and/or revision of the manuscript: F. Meng, C.L. Speyer, B. Zhang, D.H. Gorski, F.R. Miller, G. Wu
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): F. Meng, Y. Zhao, G. Wu
Study supervision: G. Wu
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed
References
- 1.Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22. doi: 10.1007/978-0-387-74039-3_1. [DOI] [PubMed] [Google Scholar]
- 2.Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, Nakamori S, et al. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010;101:293–9. doi: 10.1111/j.1349-7006.2009.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah AN, Gallick GE. Src, chemoresistance and epithelial to mesenchymal transition: are they related? Anticancer Drugs. 2007;18:371–5. doi: 10.1097/CAD.0b013e32801265d7. [DOI] [PubMed] [Google Scholar]
- 4.Chakrabarti R, Hwang J, Andres Blanco M, Wei Y, Lukacisin M, Romano RA, et al. Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat Cell Biol. 2012;14:1212–22. doi: 10.1038/ncb2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kallergi G, Papadaki MA, Politaki E, Mavroudis D, Georgoulias V, Agelaki S. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res. 2011;13:R59. doi: 10.1186/bcr2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meng F, Wu G. The rejuvenated scenario of epithelial-mesenchymal transition (EMT) and cancer metastasis. Cancer Metastasis Rev. 2012;31:455–67. doi: 10.1007/s10555-012-9379-3. [DOI] [PubMed] [Google Scholar]
- 7.Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15:117–34. doi: 10.1007/s10911-010-9178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parvani JG, Schiemann WP. Sox4, EMT programs, and the metastatic progression of breast cancers: mastering the masters of EMT. Breast Cancer Res. 2013;15:R72. doi: 10.1186/bcr3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoshino H, Miyoshi N, Nagai K, Tomimaru Y, Nagano H, Sekimoto M, et al. Epithelial-mesenchymal transition with expression of SNAI1-induced chemoresistance in colorectal cancer. Biochem Biophys Res Commun. 2009;390:1061–5. doi: 10.1016/j.bbrc.2009.10.117. [DOI] [PubMed] [Google Scholar]
- 10.Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doi-phode RY, et al. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells. 2009;27:2059–68. doi: 10.1002/stem.154. [DOI] [PubMed] [Google Scholar]
- 11.Pham CG, Bubici C, Zazzeroni F, Knabb JR, Papa S, Kuntzen C, et al. Upregulation of Twist-1 by NF-kappaB blocks cytotoxicity induced by chemotherapeutic drugs. Mol Cell Biol. 2007;27:3920–35. doi: 10.1128/MCB.01219-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, et al. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. 2013;73:1981–92. doi: 10.1158/0008-5472.CAN-12-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–9. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JY, Park MK, Park JH, Lee HJ, Shin DH, Kang Y, et al. Loss of the polycomb protein Mel-18 enhances the epithelial-mesenchymal transition by ZEB1 and ZEB2 expression through the downregulation of miR-205 in breast cancer. Oncogene. 2014;33:1325–35. doi: 10.1038/onc.2013.53. [DOI] [PubMed] [Google Scholar]
- 15.Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol. 2002;250:1–23. doi: 10.1006/dbio.2002.0780. [DOI] [PubMed] [Google Scholar]
- 16.Jonsson H, Peng SL. Forkhead transcription factors in immunology. Cell Mol Life Sci. 2005;62:397–409. doi: 10.1007/s00018-004-4365-8. [DOI] [PubMed] [Google Scholar]
- 17.Katoh M, Katoh M. Human FOX gene family (Review) Int J Oncol. 2004;25:1495–500. [PubMed] [Google Scholar]
- 18.Hoggatt AM, Kriegel AM, Smith AF, Herring BP. Hepatocyte nuclear factor-3 homologue 1 (HFH-1) represses transcription of smooth muscle-specific genes. J Biol Chem. 2000;275:31162–70. doi: 10.1074/jbc.M005595200. [DOI] [PubMed] [Google Scholar]
- 19.Martinez-Ceballos E, Chambon P, Gudas LJ. Differences in gene expression between wild type and Hoxa1 knockout embryonic stem cells after retinoic acid treatment or leukemia inhibitory factor (LIF) removal. J Biol Chem. 2005;280:16484–98. doi: 10.1074/jbc.M414397200. [DOI] [PubMed] [Google Scholar]
- 20.Potter CS, Peterson RL, Barth JL, Pruett ND, Jacobs DF, Kern MJ, et al. Evidence that the satin hair mutant gene Foxq1 is among multiple and functionally diverse regulatory targets for Hoxc13 during hair follicle differentiation. J Biol Chem. 2006;281:29245–55. doi: 10.1074/jbc.M603646200. [DOI] [PubMed] [Google Scholar]
- 21.Bieller A, Pasche B, Frank S, Glaser B, Kunz J, Witt K, et al. Isolation and characterization of the human forkhead gene FOXQ1. DNA Cell Biol. 2001;20:555–61. doi: 10.1089/104454901317094963. [DOI] [PubMed] [Google Scholar]
- 22.Cao D, Hustinx SR, Sui G, Bala P, Sato N, Martin S, et al. Identification of novel highly expressed genes in pancreatic ductal adenocarcinomas through a bioinformatics analysis of expressed sequence tags. Cancer Biol Ther. 2004;3:1081–9. doi: 10.4161/cbt.3.11.1175. [DOI] [PubMed] [Google Scholar]
- 23.Kaneda H, Arao T, Tanaka K, Tamura D, Aomatsu K, Kudo K, et al. FOXQ1 is overexpressed in colorectal cancer and enhances tumorigenicity and tumor growth. Cancer Res. 2010;70:2053–63. doi: 10.1158/0008-5472.CAN-09-2161. [DOI] [PubMed] [Google Scholar]
- 24.Bierie B, Chung CH, Parker JS, Stover DG, Cheng N, Chytil A, et al. Abrogation of TGF-beta signaling enhances chemokine production and correlates with prognosis in human breast cancer. J Clin Invest. 2009;119:1571–82. doi: 10.1172/JCI37480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feuerborn A, Srivastava PK, Kuffer S, Grandy WA, Sijmonsma TP, Gretz N, et al. The Forkhead factor FoxQ1 influences epithelial differentiation. J Cell Physiol. 2011;226:710–9. doi: 10.1002/jcp.22385. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Meng F, Liu G, Zhang B, Zhu J, Wu F, et al. Forkhead transcription factor foxq1 promotes epithelial-mesenchymal transition and breast cancer metastasis. Cancer Res. 2011;71:1292–301. doi: 10.1158/0008-5472.CAN-10-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiao Y, Jiang X, Lee ST, Karuturi RK, Hooi SC, Yu Q. FOXQ1 regulates epithelial-mesenchymal transition inhuman cancers. Cancer Res. 2011;71:3076–86. doi: 10.1158/0008-5472.CAN-10-2787. [DOI] [PubMed] [Google Scholar]
- 28.Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Chen D, Ringler J, Chen W, Cui QC, Ethier SP, et al. Disulfiram treatment facilitates phosphoinositide 3-kinase inhibition in human breast cancer cells in vitro and in vivo. Cancer Res. 2010;70:3996–4004. doi: 10.1158/0008-5472.CAN-09-3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang H, Liu G, Dziubinski M, Yang Z, Ethier SP, Wu G. Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast Cancer Res Treat. 2008;112:217–27. doi: 10.1007/s10549-007-9847-6. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Meng F, Wu S, Kreike B, Sethi S, Chen W, et al. Engagement of I-branching {beta}-1, 6-N-acetylglucosaminyltransferase 2 in breast cancer metastasis and TGF-{beta} signaling. Cancer Res. 2011;71:4846–56. doi: 10.1158/0008-5472.CAN-11-0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sleeman KE, Kendrick H, Ashworth A, Isacke CM, Smalley MJ. CD24 staining of mouse mammary gland cells defines luminal epithelial, myo-epithelial/basal and non-epithelial cells. Breast Cancer Res. 2006;8:R7. doi: 10.1186/bcr1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones AV, Cross NC. Oncogenic derivatives of platelet-derived growth factor receptors. Cell Mol Life Sci. 2004;61:2912–23. doi: 10.1007/s00018-004-4272-z. [DOI] [PubMed] [Google Scholar]
- 36.Kim Y, Kim E, Wu Q, Guryanova O, Hitomi M, Lathia JD, et al. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev. 2012;26:1247–62. doi: 10.1101/gad.193565.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laimer D, Dolznig H, Kollmann K, Vesely PW, Schlederer M, Merkel O, et al. PDGFR blockade is a rational and effective therapy for NPM-ALK-driven lymphomas. Nat Med. 2012;18:1699–704. doi: 10.1038/nm.2966. [DOI] [PubMed] [Google Scholar]
- 38.Ostman A, Heldin CH. PDGF receptors as targets in tumor treatment. Adv Cancer Res. 2007;97:247–74. doi: 10.1016/S0065-230X(06)97011-0. [DOI] [PubMed] [Google Scholar]
- 39.Pardanani A, Tefferi A. Imatinib targets other than bcr/abl and their clinical relevance in myeloid disorders. Blood. 2004;104:1931–9. doi: 10.1182/blood-2004-01-0246. [DOI] [PubMed] [Google Scholar]
- 40.Patel BB, He YA, Li XM, Frolov A, Vanderveer L, Slater C, et al. Molecular mechanisms of action of imatinib mesylate in human ovarian cancer: a proteomic analysis. Cancer Genomics Proteomics. 2008;5:137–49. [PubMed] [Google Scholar]
- 41.Rink L, Skorobogatko Y, Kossenkov AV, Belinsky MG, Pajak T, Heinrich MC, et al. Gene expression signatures and response to imatinib mesylate in gastrointestinal stromal tumor. Mol Cancer Ther. 2009;8:2172–82. doi: 10.1158/1535-7163.MCT-09-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jechlinger M, Sommer A, Moriggl R, Seither P, Kraut N, Capodiecci P, et al. Autocrine PDGFR signaling promotes mammary cancer metastasis. J Clin Invest. 2006;116:1561–70. doi: 10.1172/JCI24652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strawn LM, Mann E, Elliger SS, Chu LM, Germain LL, Niederfellner G, et al. Inhibition of glioma cell growth by a truncated platelet-derived growth factor-beta receptor. J Biol Chem. 1994;269:21215–22. [PubMed] [Google Scholar]
- 44.Uehara H, Kim SJ, Karashima T, Shepherd DL, Fan D, Tsan R, et al. Effects of blocking platelet-derived growth factor-receptor signaling in a mousemodel of experimental prostate cancer bone metastases. J Natl Cancer Inst. 2003;95:458–70. doi: 10.1093/jnci/95.6.458. [DOI] [PubMed] [Google Scholar]
- 45.Heldin CH, Ostman A, Eriksson A, Siegbahn A, Claesson-Welsh L, Wester-mark B. Platelet-derived growth factor: isoform-specific signalling via heterodimeric or homodimeric receptor complexes. Kidney Int. 1992;41:571–4. doi: 10.1038/ki.1992.84. [DOI] [PubMed] [Google Scholar]
- 46.Eriksson A, Siegbahn A, Westermark B, Heldin CH, Claesson-Welsh L. PDGF alpha- and beta-receptors activate unique and common signal transduction pathways. EMBO J. 1992;11:543–50. doi: 10.1002/j.1460-2075.1992.tb05085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, et al. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19:372–86. doi: 10.1016/j.ccr.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klinghoffer RA, Mueting-Nelsen PF, Faerman A, Shani M, Soriano P. The two PDGF receptors maintain conserved signaling in vivo despite divergent embryological functions. Mol Cell. 2001;7:343–54. doi: 10.1016/s1097-2765(01)00182-4. [DOI] [PubMed] [Google Scholar]
- 49.Pietras K, Sjoblom T, Rubin K, Heldin CH, Ostman A. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3:439–43. doi: 10.1016/s1535-6108(03)00089-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.