

Abstract
By using a directed evolution approach, we have identified aminoacyl-tRNA synthetase variants with significantly enhanced activity for the incorporation of unnatural amino acids into proteins in response to the amber nonsense codon in bacteria. We demonstrated that the optimization of anticodon recognition of tRNA by aminoacyl-tRNA synthetase led to improved incorporation efficiency that is unnatural amino acid-specific. The findings will facilitate the creation of an optimized system for the genetic incorporation of unnatural amino acids in bacteria.
Keywords: genetic code expansion, anticodon recognition, unnatural amino acid, aminoacyl-tRNA synthetase engineering, amber suppression
Orthogonal tRNA-aminoacyl-tRNA synthetase pairs are widely used for the site-specific incorporation of nearly 80 unnatural amino acids (unAAs) in Escherichia coli, Saccharomyces cerevisiae, plant, and mammalian cells in response to unique nonsense (e.g., amber)1,2 and frameshift (e.g., quadruplet)3−5 codons. This methodology enables site-specific introductions of unique chemical or physical probes into proteins, which could facilitate the study of protein structure and function as well as the investigation of biological processes.1,6−8 Recently, nonsense codon suppression-mediated regulation of biological events were also explored in synthetic biology applications.9−11 Although unAAs are typically incorporated into proteins in response to nonsense codon with good efficiency and excellent fidelity, further system optimization to increase the incorporation efficiency is still highly desirable. This is because inefficient incorporation not only results in a low yield of the desired unAA-containing protein but also leads to an increased accumulation of truncated protein products that may negatively affect the fitness of host cells.
An engineered Methanocaldococcus jannaschii amber suppressor tyrosyl-tRNA (MjtRNACUATyr) and tyrosyl-tRNA-synthetase (MjTyrRS) pair is the most extensively used system for the evolution of aminoacyl-tRNA synthetase (aaRS) variants that incorporate unAAs with aromatic functional groups in E. coli. In a previous effort, optimization of the interaction between MjtRNACUA and the E. coli native translational machinery, such as EF-Tu,12 was explored in order to improve the incorporation efficiency. Here, we report an approach that focuses on fine-tuning anticodon recognition of MjtRNACUATyr by MjTyrRS variants, which led to additional improvement in unAA incorporation efficiency beyond the EF-Tu strategy. The observed improvement likely resulted from more efficient aminoacylation of MjtRNACUA by the evolved MjTyrRS mutants.
The evolved MjTyrRS variant catalyzes the aminoacylation reaction of MjtRNACUATyr with a tyrosine analogue (an unAA) at the expense of an ATP. The catalytic efficiency of this reaction, which contributes to the overall efficiency of unAA incorporation, is dictated by the substrates (MjtRNACUA and unAA) recognition of the enzyme. Previous work successfully altered the specificity of MjTyrRS toward unAAs by focusing on changing the enzyme’s amino acid recognition pocket. While the anticodon of the tRNA was changed from GUA (Figure 1) into CUA (MjtRNAGUATyr to MjtRNACUA) to enable amber suppression with unAA, the substrate promiscuity of the MjTyrRS allowed reasonable recognition of MjtRNACUATyr without adjusting the tRNA recognition elements of MjTyrRS to such change. In light of the crystal structure of MjtRNAGUA-MjTyrRS complex,13 the anticodon region of MjtRNAGUATyr is a major recognition element of MjTyrRS (Figure 1). Therefore, the G34C change in the tRNA would render the interaction between MjTyrRS and the resulting MjtRNACUA less optimal. Previous rational design work showed that a single mutation in the anticodon recognition pocket of MjTyrRS improved the aminoacylation rate13 and the amber suppression efficiency.14 However, no systematic effort was devoted to study or to optimize the recognition of the mutated MjtRNACUATyr by MjTyrRS. In this work, we report a systematic effort to improve the unAA incorporation efficiency through fine-tuning the recognition of the mutated MjtRNACUA by MjTyrRS variants. We used a directed evolution approach to identify MjTyrRS mutants that led to significantly higher amber suppression efficiency than both the parent MjTyrRS variants and the reported MjTyrRS mutant.13,14 While other beneficial factors may be picked up as well in our cell growth-based selection process, the major contributor to the observed improvement is likely due to better recognition of MjtRNACUATyr by MjTyrRS mutants. Our results also showed that the mutations for each unAA-specific MjTyrRS variant are different, which further strikes the notion that the catalytic efficiency of MjTyrRS is determined by recognitions of both substrates, the tRNA and the amino acid. We finally demonstrated that the MjTyrRS mutants obtained in this study, when paired with a previously evolved MjtRNACUA (MjtRNACUATyr-Nap1) that has optimized interaction with E. coli EF-Tu,12 further improved amber suppression efficiency.
Figure 1.
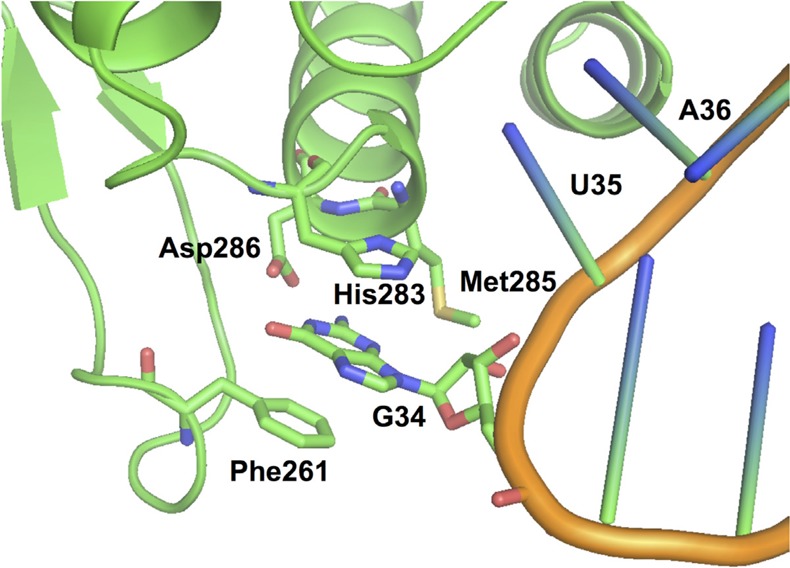
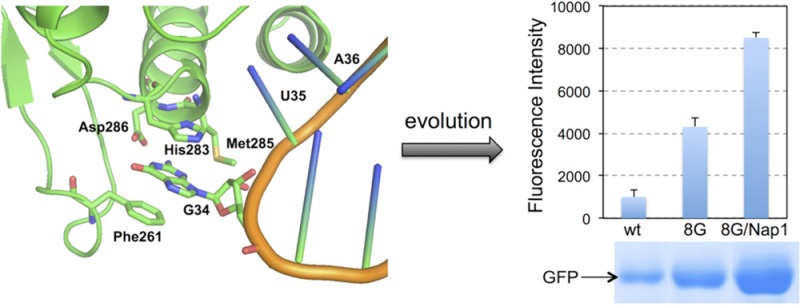
Recognition of G34 of MjtRNAGUATyr by MjTyrRS (PDB, 1J1U).
Examining the X-ray crystal structure of the MjtRNAGUATyr-MjTyrRS complex13 reveals that the anticodon of MjtRNAGUA is recognized by the C-terminal domain of MjTyrRS (Figure 1). Residues Phe261 and His283 engage in stacking interaction with the base of G34 in MjtRNAGUATyr. Residue Asp286 forms two hydrogen bonds with N1 and N2 of G34. Asp286 is well conserved among the archaeal and eukaryotic TyrRSs. It was reported that mutation of Asp286 into alanine led to a 10-fold reduction of the aminoacylation rate of MjtRNAGUA by MjTyrRS.15 On the other hand, the Asp286Arg mutation resulted in a more efficient aminoacylation of MjtRNACUATyr (an amber suppressor tRNA with G34C mutation).13,14 In addition to Phe261, His283, and Asp286, residue Met285 is in the close proximity to G34 and may provide additional interaction to fine-tune the anticodon recognition by MjTyrRS. We envisaged that a directed evolution approach involving mutagenesis of above residues within the anticodon recognition pocket of MjTyrRS could optimize the interaction between MjTyrRS and MjtRNACUA, and therefore improve the overall efficiency of unAA incorporation in response to amber nonsense codon.
To test the hypothesis, we first examined an MjTyrRS variant, AcPheRS (referred as AcPheRS-wt hereafter),16 that was evolved previously for the incorporation of p-acetyl-l-phenylalanine (AcPhe, Figure 2A) in response to amber nonsense codon. We created an AcPheRS library in which residues Phe261, His283, Met285, and Asp286 were completely randomized. Overlapping polymerase chain reaction (PCR) was performed with synthetic oligonucleotide primers in which the randomized residues were encoded as NNK (N = A, C, T, or G; K = T or G) to generate a library with a theoretical diversity of 1.05 × 106. The quality of the library (>99% coverage) was validated by DNA sequencing. The resulting AcPheRS library was subjected to a positive selection to identify functional AcPheRS variants followed by a negative selection to remove AcPheRS variants that could charge MjtRNACUATyr with natural amino acid as previously described.16 Briefly, the positive selection is based on resistance to chloramphenicol (Cm), which is conferred by the suppression of an amber mutation at a permissive site (Asp112) in the chloramphenicol acetyltransferase-encoding gene in the presence of MjtRNACUA, AcPhe, and functional AcPheRS mutants. The negative selection uses the toxic barnase gene with amber mutations at permissive sites (Gln2TAG and Asp44TAG) and was carried out in the absence of AcPhe. The surviving AcPheRS variants from two positive and one negative rounds of selection were subsequently screened for chloramphenicol resistance level in the presence and absence of AcPhe. A few clones that survived on 150 μg/mL chloramphenicol in the presence of AcPhe and did not grow on 75 μg/mL chloramphenicol in the absence of AcPhe were identified. Among these clones, AcPheRS-8G and AcPheRS-12B displayed the fastest growth rate in the presence of chloramphenicol and the brightest GFP fluorescence (The selection plasmid, pREP,17 contains a T7 RNA polymerase gene with amber mutation at permissive site. The synthesis of full-length T7 RNA polymerase with amber suppression drives the expression of a green fluorescent protein). Another clone, AcPheRS-2B, showed higher amber suppression efficiency than that of AcPheRS-wt, but the efficiency is lower than that of AcPheRS-8G and AcPheRS-12B. We next examined the relative protein expression level of AcPheRS-wt and AcPheRS-8G (Supporting Information Figure S3) by Western blot. We did not detect any notable difference between the two, suggesting that the observed improvement in AcPhe incorporation was not a result of higher expression level of the evolved AcPheRS-8G mutant. We also conducted cell growth experiments and observed similar growth rates of strains harboring different AcPheRS variants (Supporting Information Figure S4A). It is therefore unlikely that the observed improvement was due to lower toxicity of the evolved AcPheRS mutants.
Figure 2.
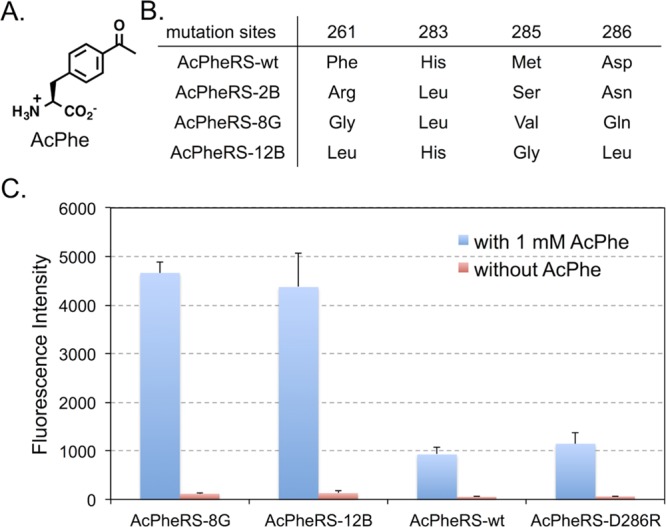
Evolution of anticodon recognition region of AcPheRS. (A) The structure of p-acetyl-l-phenylalanine (AcPhe). (B) Mutations in the anticodon recognition region of the evolved AcPheRS variants. (C) GFP fluorescence assays of cells expressing AcPheRS variants. Fluorescence readings of E. coli GeneHogs cells expressing wild type (AcPheRS-wt) or the evolved mutants, each coexpressed with MjtRNACUATyr, in the presence (blue column) or the absence (red column) of 1 mM AcPhe. Fluorescence intensity was normalized to cell growth. Each data point is the average of duplicate measurements with standard deviation.
DNA sequencing results revealed mutation convergence at positions Phe261 and Asp286 of all three hits (Figure 2B). The Asp286 residue was mutated into neutral residues (Asn, Gln, and Leu) that have similar side chain size to that of the Asp residue in AcPheRS-wt. Residue Phe261 in all three hits was mutated into nonaromatic amino acids, which apparently reduce the stacking interaction between the synthetase and the pyrimidine base of C34 in MjtRNACUATyr. This observation indicates that the favorable stacking interaction in MjTyrRS-MjtRNAGUA may not be essential for AcPheRS-MjtRNACUATyr when the G34C mutation leads to the replacement of a purine base with a pyrimidine base in MjtRNACUA. In addition to mutations at positions Phe261 and Asp286, Met285 was mutated to smaller amino acids (Figure 2B), which is inconsistent with the notion that cytosine is a smaller base than guanine and a larger amino acid may be needed to restore the lost interaction.13,18 The possible explanation is that the amino acid residue at position 285 does not directly interact with the nucleotide but rather affect anticodon recognition through fine-tuning the local conformation of the anticodon recognition region of AcPheRS.
To determine the efficiency and the fidelity of AcPhe incorporation into proteins in E. coli, an amber mutation (TAG) was introduced at position Asp149 in a C-terminal His-tagged GFP variant (GFP149TAG). Protein expression experiments using the two most promising hits (AcPheRS-8G and AcPheRS-12B) as well as two controls (AcPheRS-wt and AcPheRS-D286R) were carried out in LB medium supplemented with and without 1 mM AcPhe. Fluorescence analysis of E. coli cultures showed that significant amount of full-length GFP protein was produced only in the presence of AcPhe for the two evolved AcPheRS variants (Figure 2C). This result indicates that the evolved AcPheRS mutants are not cross-active with any endogenous amino acids in E. coli. The incorporation fidelity of the evolved AcPheRS mutants is comparable to that of the AcPheRS-wt. Fluorescence intensities of GFP also showed that the evolved AcPheRS-8G and AcPheRS-12B mutants had significantly higher amber suppression efficiency (Figure 2C) than that of AcPheRS-wt and AcPheRS-D286R, an AcPheRS variant with the previously reported beneficial mutation (Asp286Arg).13,14 Comparing to the rational designed AcPheRS-D286R mutant, better recognition of MjtRNACUATyr by AcPheRS-8G and AcPheRS-12B was likely achieved by exploiting a much larger conformational space using the directed evolution approach. Between the two best hits, AcPheRS-8G displayed better reproducibility and lower background in AcPhe incorporation (Figure 2C). We, therefore, focused on the AcPheRS-8G hit in following studies.
We next investigated if the beneficial mutations in the evolved AcPheRS-8G could be functionally transferred to other unAA-specific MjTyrRS variants to achieve general improvements in the incorporation efficiency. We reasoned that the beneficial mutations within the anticodon recognition region are away from the amino acid-binding pocket of MjTyrRS variants and might not affect the unAA recognition by MjTyrRSs. To this end, we focused on two other MjTyrRS variants, BpaRS19 and sTyrRS,20 which were evolved previously to recognize p-benzoyl-l-phenylalanine (Bpa, Figure 3A) and sulfotyrosine (sTyr, Figure 4A), respectively. The Bpa is a useful cross-linking amino acid for the study of protein–protein interactions. The sTyr, which is a product of post-translational modification,21 is found in many secreted and membrane-bound proteins. The direct and more efficient incorporation of sTyr into proteins is useful for the investigation of its biological functions.22 Side chain structures of these two unAAs are significantly different from that of AcPhe. We constructed BpaRS-8G and sTyrRS-8G, each of which contained beneficial mutations (Phe261Gly, His283Leu, Met285Val, and Asp286Gln) from AcPheRS-8G. Based on the expression of GFP149TAG (Supporting Information Figure S1), no expected improvements were observed. Two possible explanations could be proposed from the above result: (1) the observed improvement of unAA incorporation via the optimization of anticodon recognition by MjTyrRS is an isolated case for AcPheRS, and (2) the anticodon and unAA recognition by MjTyrRS are mutually dependent.
Figure 3.
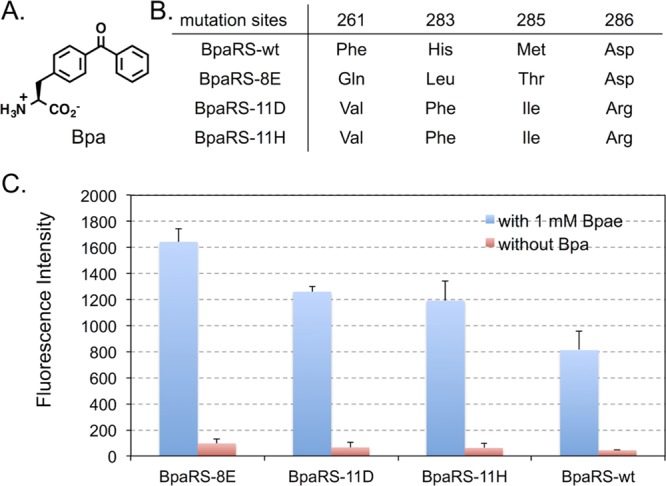
Evolution of anticodon recognition region of BpaRS. (A) The structure of p-benzoyl-l-phenylalanine (Bpa). (B) Mutations in the anticodon recognition region of the evolved BpaRS mutants. (C) GFP fluorescence assays of cells expressing BpaRS variants. Fluorescence readings of E. coli GeneHogs cells expressing wild type (BpaRS-wt) or the evolved mutants, each coexpressed with MjtRNACUATyr, in the presence (blue column) or the absence (red column) of 1 mM Bpa. Fluorescence intensity was normalized to cell growth. Each data point is the average of duplicate measurements with standard deviation.
Figure 4.
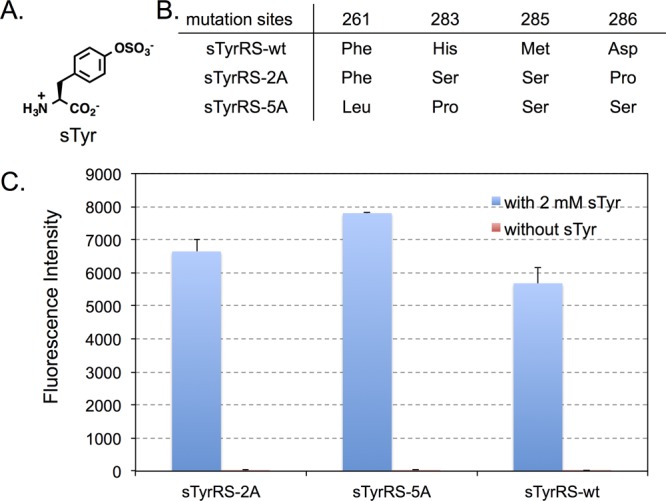
Evolution of anticodon recognition region of sTyrRS. (A) The structure of sulfotyrosine (sTyr). (B) Mutations in the anticodon recognition region of evolved sTyrRS mutants. (C) GFP fluorescence assays of cells expressing sTyrRS variants. Fluorescence readings of E. coli GeneHogs cells expressing wild type (sTyrRS-wt) or the evolved mutants, each coexpressed with MjtRNACUATyr, in the presence (blue column) or the absence (red column) of 1 mM sTyr. Fluorescence intensity was normalized to cell growth. Each data point is the average of duplicate measurements with standard deviation.
To investigate above possibilities, we examined if BpaRS and sTyrRS variants with improved incorporation efficiency can be obtained through the evolution of anticodon recognition pocket. Using the same approach in AcPheRS evolution, we created a BpaRS library and a sTyrRS library, where residues Phe261, His283, Met285, and Asp286 were randomized in each library. After consecutive rounds of positive and negative selections, three BpaRS hits (BpaRS-8E, BpaRS-11D, and BpaRS-11H) and two sTyrRS hits (sTyrRS-2A and sTyrRS-5A) were identified to display higher amber suppression efficiency over their parents (Figures 3 and 4). Among these, BpaRS-8E and sTyrRS-5A are the best ones for the incorporation of Bpa and sTyr, respectively. It is worth of noticing that the degree of chloramphenicol resistance (amber suppression at position 112 of chloramphenicol acetyl transferase) does not correlate very well with the intensity of GFP fluorescence (amber suppression at position 149 of GFP). While cells containing BpaRS hits can survive much higher concentrations of chloramphenicol (>250 μg/mL) than sTyrRS hits (75 μg/mL), greater fluorescence intensities were observed for sTyrRS hits (∼6000–8000 au/OD600 nm; Figure 4C) relative to those of the BpaRS hits (∼1200–1600 au/OD600 nm; Figure 3C). In addition, the fold improvement of sTyrRS hits over the sTyrRS-wt is apparently greater when the chloramphenicol resistance level was used to evaluate the improvement. The observation may be due to the structure/function changes of reporter proteins caused by the incorporation of a given unAA with unique physical and chemical properties. Nevertheless, the general trend of improvement is obvious regardless which data set is used for evaluation.
As shown in Figure 3B, BpaRS-11D and BpaRS-11H have converged protein sequence, which is different from that of BpaRS-8E. The two sTyrRS hits, sTyrRS-2A and sTyrRS-5A, have different sequences (Figures 4B). None of the BpaRS and sTyrRS hits has the same mutation combinations as the AcPheRS hits. The most significant difference was observed at position 286 of BpaRS hits. While all the AcPheRS and sTyrRS hits contain a neutral amino acid residue at position 286, the BpaRS hits either retained a negatively charged Asp residue or changed into a positively charged Arg residue. On the other hand, all the evolved BpaRS, sTyrRS, and AcPheRS variants have mutations at either positions 261 or 283 or both, which apparently led to reduced stacking interactions with the C34 of MjtRNACUATyr. In addition, the Met285 residues in all evolved mutants were mutated into amino acids with shorter side chains. These analyses implicate that MjTyrRS variants (i.e., AcPheRS, BpaRS, and sTyrRS) interact with the anticodon of MjtRNACUA in a similar fashion but with subtle differences. The aminoacylation of MjtRNACUATyr by MjTyrRS is likely collaboratively affected by both MjTyrRS-unAA and MjTyrRS-anticodon interactions, which leads to unAA-specific beneficial mutations in the anticodon recognition region of different MjTyrRS variants.
To further investigate above conclusion, we examined the substrate specificity of the evolved AcPheRS-8G toward different unAAs. We found that the AcPheRS-8G showed significantly improved recognition toward p-azido-l-phenylalanine (AzPhe),23 a close analogue of AcPhe (Supporting Information Figure S2). The results further confirm the theory that the MjTyrRS-anticodon interaction and the MjTyrRS-unAA interaction mutually affect each other.
In order to further improve unAA incorporation efficiency, we next examined if a previously reported MjtRNACUATyr variant (MjtRNACUA-Nap1)12 with beneficial T-stem mutations can be efficiently recognized by the evolved MjTyrRS variants from the present work. The T-stem mutations in MjtRNACUATyr-Nap1 (the “general” hit that gave the best overall yield improvements with all unAAs tested)12 was predicted to increase its binding energy with EF-Tu, which compensates for weaker binding of unAA in the EF-Tu binding pocket. Since the T-stem of MjtRNACUA-Nap1 does not interact with MjTyrRS, we envisaged that MjtRNACUATyr-Nap1 could still be recognized by the evolved MjTyrRS variants. To test this hypothesis, amber suppression efficiency was examined using GFP-149TAG. As shown in Figure 5A, both AcPheRS-8G and AcPheRS-12B worked very well with MjtRNACUA-Nap1 and resulted in significantly improved amber suppression efficiency relative to that when MjtRNACUATyr was used. We also verified the fluorescence readings by conducting large-scale protein expression, partial purification, and SDS-PAGE analysis (Figure 5B).
Figure 5.

GFP expression with AcPheRS and MjtRNACUATyr variants. (A) GFP fluorescence assays of AcPhe incorporation by using different combinations of AcPheRS and MjtRNACUA variants. Fluorescence intensity was normalized to cell growth; (B) GFP expression yield and SDS-PAGE analysis.
In conclusion, by using a directed evolution approach, we were able to identify MjTyrRS variants with significantly enhanced efficiency for the incorporation of unAA into proteins in response to amber nonsense codon. We also found that the optimal efficiencies of MjTyrRS variants-MjtRNACUATyr pairs are unAA-specific. In addition, the apparent improvement in anticodon recognition (or other unknown beneficial factors picked up during the selection) can be combined with previous improvement in tRNA-EF-Tu interaction to achieve further increase in unAA incorporation efficiency. Therefore, the evolved MjTyrRS variants from this work will facilitate the creation of an optimized and standardized system for the genetic incorporation of unAA into proteins in E. coli. The same strategy should be generally applicable to the evolution of other orthogonal tRNA–aaRS pairs for highly efficient unAA incorporations in living cells.
Methods
Materials and General Methods
Bpa and AzPhe were purchased from Bachem. The synthesis of AcPhe16 and sTyr24 followed previously reported procedures. Restriction enzymes, antarctic phosphatase (AP), and T4 DNA ligase were purchased from New England Biolabs. KOD hot start DNA polymerase was purchased from EMD Millipore. Primers were ordered from Eurofins MWG Operon. Standard molecular biology techniques were used throughout.25 Site-directed mutagenesis was carried out using either overlapping PCR or the QuikChange II site-directed mutagenesis kit by following the manufacturer’s protocol. E. coli DH5α and GeneHogs were used for routine cloning and DNA propagation. All solutions were prepared in deionized water further treated by Barnstead Nanopure ultrapure water purification system. Antibiotics were added where appropriate to following final concentrations: ampicillin, 100 μg/mL; kanamycin, 50 μg/mL; tetracycline, 12.5 μg/mL, and chloramphenicol (varied from 34 to 250 μg/mL).
Library Construction
Mutants of MjTyrRS were obtained26 by overlapping PCR using AcPheRS,16 BpaRS,19 or sTyrRS20 as template. Digestion of PCR products with NdeI and PstI followed by ligation between NdeI and PstI sites of pBK vector27 resulted in MjTyrRS libraries. Primers that were used for library construction are listed in the Supporting Information.
Positive Selection
Library DNAs were transformed into GeneHogs electrocompetent cells containing plasmid pREP27 that harbors MjtRNACUATyr and a chloramphenicol acetyltransferase-encoding gene with an amber mutation at position Asp112. Transformants were cultivated in LB media containing kanamycin and tetracycline. After 12 h of cultivation, cells were harvested. Based on calculation, a certain number of cells (>4.6× the size of the library) were plated on LB agar containing kanamycin (to maintain the pBK-MjTyrRS plasmid), tetracycline (to maintain the pREP plasmid), 1 mM unAA (e.g., AcPhe), and chloramphenicol (varied from 50 to 250 μg/mL). The selection plates were incubated at 37 °C for 24 h. Survived cells were pooled and the pBK-MjTyrRS plasmids were isolated.
Negative Selection Assay
E. coli GeneHogs was cotransformed with plasmids pNEG27 (containing MjtRNACUATyr and a barnase-encoding gene with two amber mutations at permissive sites, Gln2 and Asp44) and pBK-MjTyrRS plasmids isolated from the positive selection. Transformants were plated on LB agar containing ampicillin (to maintain the pNEG plasmid), kanamycin, and 0.2% l-arabinose (to activate the transcription of mutant barnase gene). The selection plates were incubated at 37 °C for 12 h. Survived cells were then pooled and the pBK-MjTyrRS plasmids were isolated.
Hit Verification
Selected numbers of single colonies from the last round of positive selection were screened by replication onto plates with varied concentrations of chloramphenicol (34, 50, 75, 100, 150, or 250 μg/mL) in the presence and the absence of the appropriate unAA. Only the ones that grew in the presence of unAA and did not grow in the absence of unAA were selected for further evaluation.
Fluorescence Analysis of Bacterial Culture
E. coli GeneHogs strain harboring plasmids pBK-MjTyrRS variant and pLei-GFP-N149TAG12 was cultured in 5 mL LB media containing kanamycin and chloramphenicol at 37 °C. The protein expression was induced at the OD600 nm of 0.6 by additions of IPTG (0.1 mM) and appropriate unAA (1 mM). Following cultivation at 37 °C for an additional 16 h, 1 mL of cell culture were collected, washed, resuspended in 1 mL of potassium phosphate buffer (50 mM, pH 7.4). The processed cell suspensions were directly used for fluorescence and cell density measurements using a Synergy H1 Hybrid plate reader (BioTek Instruments). The fluorescence of GFP (GFPUV) was monitored at λEx = 390 nm and λEm = 510 nm. The cell density was estimated by measuring the absorbance at 600 nm. Values of fluorescence intensity were normalized to cell growth. Reported data are the average of two or more measurements with standard deviations.
Protein Expression and Purification
Similar cell cultivation procedure for fluorescence analysis was applied to preparing 25 mL of E. coli culture for protein purification. Cells were collected by centrifugation at 5000g and 4 °C for 15 min. Harvested cells were resuspended in lysis buffer containing potassium phosphate (20 mM, pH 7.4), NaCl (300 mM), and imidazole (10 mM). Cells were subsequently disrupted by sonication. Cellular debris was removed by centrifugation (21 000g, 30 min, 4 °C). The cell-free lysate was applied to Ni Sepharose 6 Fast Flow resin (GE Healthcare). Protein purification followed manufacture’s instructions. Protein concentrations were determined by Bradford assay (Bio-Rad). Purified proteins were analyzed by SDS-PAGE.
Acknowledgments
This work was funded by the New Faculty Startup Fund (to J.G.) from the University of Nebraska—Lincoln and by Grant 1R01AI111862 (to J.G.) from the National Institutes of Health, DHHS-NIH-NIAID.
Supporting Information Available
Additional figures and data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Liu C. C.; Schultz P. G. (2010) Adding new chemistries to the genetic code. Annu. Rev. Biochem. 79, 413–444. [DOI] [PubMed] [Google Scholar]
- Hohsaka T.; Sisido M. (2002) Incorporation of non-natural amino acids into proteins. Curr. Opin. Chem. Biol. 6, 809–815. [DOI] [PubMed] [Google Scholar]
- Niu W.; Schultz P. G.; Guo J. (2013) An expanded genetic code in mammalian cells with a functional quadruplet codon. ACS Chem. Biol. 8, 1640–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. C.; Wu N.; Santoro S. W.; Lakshman V.; King D. S.; Schultz P. G. (2004) An expanded genetic code with a functional quadruplet codon. Proc. Natl. Acad. Sci. U.S.A. 101, 7566–7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H.; Wang K.; Davis L.; Garcia-Alai M.; Chin J. W. (2010) Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 464, 441–444. [DOI] [PubMed] [Google Scholar]
- Wu X.; Schultz P. G. (2009) Synthesis at the interface of chemistry and biology. J. Am. Chem. Soc. 131, 12497–12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W.; Guo J. (2013) Expanding the chemistry of fluorescent protein biosensors through genetic incorporation of unnatural amino acids. Mol. BioSyst. 9, 2961–2970. [DOI] [PubMed] [Google Scholar]
- Davis L.; Chin J. W. (2012) Designer proteins: applications of genetic code expansion in cell biology. Nat. Rev. Mol. Cell Biol. 13, 168–182. [DOI] [PubMed] [Google Scholar]
- Liu C. C.; Qi L.; Yanofsky C.; Arkin A. P. (2011) Regulation of transcription by unnatural amino acids. Nat. Biotechnol. 29, 164–168. [DOI] [PubMed] [Google Scholar]
- Anderson J. C.; Voigt C. A.; Arkin A. P. (2007) Environmental signal integration by a modular AND gate. Mol. Syst. Biol. 3, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Li Y.; Niu W.; Sun M.; Cerny R.; Li Q.; Guo J. (2014) Construction of a live-attenuated HIV-1 vaccine through genetic code expansion. Angew. Chem., Int. Ed. 53, 4867–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.; Melancon C. E. III; Lee H. S.; Groff D.; Schultz P. G. (2009) Evolution of amber suppressor tRNAs for efficient bacterial production of proteins containing nonnatural amino acids. Angew. Chem., Int. Ed. 48, 9148–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T.; Nureki O.; Ishitani R.; Yaremchuk A.; Tukalo M.; Cusack S.; Sakamoto K.; Yokoyama S. (2003) Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat. Struct. Biol. 10, 425–432. [DOI] [PubMed] [Google Scholar]
- Young T. S.; Ahmad I.; Yin J. A.; Schultz P. G. (2010) An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374. [DOI] [PubMed] [Google Scholar]
- Steer B. A.; Schimmel P. (1999) Domain–domain communication in a miniature archaebacterial tRNA synthetase. Proc. Natl. Acad. Sci. U.S.A. 96, 13644–13649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Zhang Z.; Brock A.; Schultz P. G. (2003) Addition of the keto functional group to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 100, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro S. W.; Wang L.; Herberich B.; King D. S.; Schultz P. G. (2002) An efficient system for the evolution of aminoacyl-tRNA synthetase specificity. Nat. Biotechnol. 20, 1044–1048. [DOI] [PubMed] [Google Scholar]
- Takimoto J. K.; Adams K. L.; Xiang Z.; Wang L. (2009) Improving orthogonal tRNA-synthetase recognition for efficient unnatural amino acid incorporation and application in mammalian cells. Mol. BioSyst. 5, 931–934. [DOI] [PubMed] [Google Scholar]
- Chin J. W.; Martin A. B.; King D. S.; Wang L.; Schultz P. G. (2002) Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 99, 11020–11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C.; Schultz P. G. (2006) Recombinant expression of selectively sulfated proteins in Escherichia coli. Nat. Biotechnol. 24, 1436–1440. [DOI] [PubMed] [Google Scholar]
- Stone M. J.; Chuang S.; Hou X.; Shoham M.; Zhu J. Z. (2009) Tyrosine sulfation: An increasingly recognized post-translational modification of secreted proteins. New Biotechnol. 25, 299–317. [DOI] [PubMed] [Google Scholar]
- Ju T.; Niu W.; Cerny R.; Bollman J.; Roy A.; Guo J. (2013) Molecular recognition of sulfotyrosine and phosphotyrosine by the Src homology 2 domain. Mol. Biosyst. 9, 1829–1832. [DOI] [PubMed] [Google Scholar]
- Chin J. W.; Santoro S. W.; Martin A. B.; King D. S.; Wang L.; Schultz P. G. (2002) Addition of p-azido-l-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 124, 9026–9027. [DOI] [PubMed] [Google Scholar]
- Liu C. C.; Choe H.; Farzan M.; Smider V. V.; Schultz P. G. (2009) Mutagenesis and evolution of sulfated antibodies using an expanded genetic code. Biochemistry 48, 8891–8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J. F. and Russell D. W. (2000) Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Wang L.; Brock A.; Herberich B.; Schultz P. G. (2001) Expanding the genetic code of Escherichia coli. Science 292, 498–500. [DOI] [PubMed] [Google Scholar]
- Wang L.; Xie J.; Schultz P. G. (2006) Expanding the genetic code. Annu. Rev. Biophys. Biomol. Struct. 35, 225–249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.