

Abstract
Background
Most amyotrophic lateral sclerosis (ALS) research has focused on mice, but there are distinct differences in the functional neuroanatomy of the corticospinal pathway in primates vs. rodents. A non-human primate model may be more sensitive and more predictive for therapeutic efficacy.
Methods
Rhesus macaques received recombinant adeno-associated virus (AAV9) encoding either the ALS-related pathological protein TDP-43 or a green fluorescent protein (GFP) control by intravenous administration. Motor function and electromyography were assessed over a nine-month expression interval followed by post-mortem analyses.
Results
Recombinant TDP-43 or GFP was stably expressed long term. Although the TDP-43 subjects did not manifest severe paralysis and atrophy, there were trends of a partial disease state in the TDP-43 subjects relative to the control.
Conclusions
These data indicate that a higher gene vector dose will likely be necessary for more robust effects, yet augur that a relevant primate model is feasible.
Keywords: adeno-associated virus, amyotrophic lateral sclerosis, frontotemporal lobar degeneration, gene therapy, gene transfer, TDP-43
Introduction
Therapeutic options for amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases are limited, potentially due to the lack of disease models that are predictive for efficacy in humans. The corticospinal tract in primates is distinct from rodents. The corticospinal tract in primates is mediolateral and more of a pure motor pathway compared to rodents [1]. Due to the greater anatomical similarities, the functional tests in non-human primates (NHP) are clinically relevant, for example, neurological examination and electromyography (EMG), which is significant for tests that are more sensitive and more predictive than rodent models. Enhancing the resolution of measuring neuromotor deficits will help establish a more sensitive and more relevant detection system compared to rodent models. A NHP model of ALS may permit the study of underlying disease mechanisms occurring in humans that are not effectively assayed for or are entirely missed in other models. The overall premise is that capture of human relevant processes coupled with high fidelity behavioral tests will unlock our ability to develop therapies that work in humans. This experiment is a preliminary dosing study for a non-human primate model with the neurological and neuropathological pattern of ALS. Due to the high cost and ethical issues related to primate research, only a small-scale, first-step experiment was attempted through a pilot grant program at the Tulane National Primate Research Center (TNPRC).
One of the main pathological proteins in ALS is transactive response DNA-binding protein 43 kDa (TDP-43) because over 90% of the ALS population harbors TDP-43-associated lesions, including both familial and idiopathic forms [2–5]. TDP-43 is thus a defining pathological marker for ALS and other diseases with TDP-43 neuropathology such as frontotemporal lobar degeneration (FTLD). TDP-43 is involved in binding and processing of RNA and DNA, RNA transport, and transcription [6, 7]. Normally, TDP-43 is found predominantly in the nucleus, but in ALS and other TDP-43 proteinopathy diseases, TDP-43 forms pathological aggregates in the cytoplasm [4, 5]. A variety of models have been developed to study TDP-43 in animals [8, 9], including focal intraparenchymal injections of adeno-associated virus (AAV) vectors to the brain or spinal cord [10, 11]. Notably, an intravenous route of AAV serotype 9 (AAV9) vector administration has enabled widespread central nervous system (CNS) gene delivery to rodents [12, 13]. When TDP-43 is expressed in this manner in rats, key features of ALS manifest including paralysis, loss of spinal motor neurons, and muscle atrophy [13]. We expressed several vector doses and several isoforms of TDP-43 in rats, which yielded consistent dose-dependent functional effects and genotype-specific effects [14]. A vector approach is more applicable to NHPs than making germline transgenics. Several laboratories have reported spinal cord gene delivery after administration of AAV9 to NHPs [15–19]. Based on these studies and the consistent TDP-43 neurotoxicity that we observed in rats, we hypothesized a clinical phenotype defined as motor and EMG deficits and paralysis when TDP-43 is overexpressed.
Materials and methods
Animal care
All subjects were Indian-origin rhesus macaques (Macaca mulatta) born in the TNPRC breeding colony. Three female subjects were used in this study. Dams were part of a specific pathogen-free colony, which is seronegative for simian immunodeficiency virus, simian T-cell lymphotropic virus type 1, type D retrovirus and B virus. All aspects of management and research use conformed to applicable United States federal regulations and the guidelines described in the Guide for the Care and Use of Laboratory Animals [20] and the United States Department of Agriculture's Animal Welfare regulations (1991) [21] and adhered to the study's protocol as approved by the TNPRC Institutional Animal Care and Use Committees.
Subjects were removed from dams at birth and placed in a dedicated primate nursery. The subjects were housed indoors in rooms maintained on a 12:12-h light: dark cycle and ambient temperature between 22 and 26°C with a relative humidity of 30–70%. Subjects were initially placed in incubators with soft toweling and a fleece object for clinging (termed a surrogate). Once examined and determined able to maintain body temperature, infants were moved to stainless steel mesh cages with a floor area of 0.46 square meters with a ceiling height of 0.51 meters. Adjacent cages interconnect to permit social housing, which commenced when subjects were between 13 and 41 days of age. These cages were equipped with toweling, a fleece surrogate, other soft toys, and climbing structures. As infants became destructive, towels and fleece surrogates were removed, generally by 120 days of age. Water was available ad libitum.
As neonates, subjects were bottle-fed formula every 6 hours. Soft foods were introduced at 30 days of age. Over time, standard monkey chow was introduced, until, by 150 days of age, infants were no longer fed formula but instead consumed nutritionally complete chow and a variety of foods distributed as feeding enrichment (e.g., produce, cereals) twice per day. Neonates were weighed one or two times per day, transitioning to once daily, once weekly, and once monthly as dictated by the veterinarian. Animals were observed daily, examined by a veterinarian monthly, and at the time of each study procedure. Animals did not receive vaccinations. The subjects were euthanized between 264 and 294 days of life.
Gene transfer vector injections
Anesthetized animals (ketamine, 10 mg/kg i.m. from Vedco, St. Joseph, MO, USA) received an intravenous injection into the saphenous vein of 1 ml of a solution of recombinant AAV9 vector diluted in lactated Ringer's buffer (Baxter Health Care, Deerfield, IL, USA). The injections were at post-natal day zero or post-natal day one which permitted wide-scale neuronal transduction in rodents [12, 13]. Two subjects received AAV9 TDP-43 and one AAV9 GFP. The first TDP-43 subject, referred to as TDP1, received AAV9 TDP-43 at a dose of 1.5 × 1014 vector genomes (vg)/kg. The other TDP-43 subject, TDP2, received AAV9 TDP-43 at a dose of 6.2 × 1013 vg/kg. The GFP subject received AAV9 GFP at a dose of 6.0 × 1013 vg/kg. The animals were studied for 8 months for behavior testing and EMG that should detect changes consistent with ALS [3]. Weights and blood draws were also collected periodically from birth to euthanasia over 9–10 months of age depending on the animal.
The expression cassette in the vectors included the hybrid cytomegalovirus/chicken beta-actin promoter and the woodchuck hepatitis virus post-transcriptional regulatory element and the terminal repeats from AAV serotype 2 as described [22]. The constructs were packaged into recombinant AAV9 capsid particles using plasmid DNAs provided by James Wilson (University of Pennsylvania, Philadelphia, PA, USA) as described in Klein et al. [23]. The AAV9 serotype was first described by Gao et al. [24].
Behavioral and EMG monitoring
The neuromotor tests employed to evaluate motor function were age-appropriate for the young subjects in this study. A modified version of the Bayley Scales of Infant Development based on Bayley [25], Champoux et al. [26], Isakova et al. [27], Borda et al. [28], and Gomez-Mancilla et al. [29] was run from 2 to 6 months. Composite ethogram motor scores encompass overall activity level, ability to reach and grasp objects, and the quality of small motor activity in the play cage. For limb movement scores, the parameters include tactile response (limb movement in response to stroking with a q-tip); placing reflex (animal positions its hand or foot appropriate to supporting itself when placed in contact with a surface); palmar/plantar grasp (closing the hand/foot in response to examiner placing finger on palm/sole); passive resistance (tester flexes and extends limbs); and active power (resistance when tester restrains limb movement). Another set of tests measures cognitive function, for example memory of where a food treat was baited, or overcoming obstacles to get to a food treat. The cognitive methods have a large motor component, selectively for the forelimbs and especially hand and digit manipulation. The cognitive measures thus include both cognitive and forelimb motor functions.
EMG was monitored to observe signs of spontaneous activity consistent with ALS-relevant denervation. Denervation patterns consist of positive waves and abnormal discharges such as fibrillation and fasciculation [30]. EMG was performed on anesthetized animals (isoflurane 1.5–2.5% from Lambert Vet Supply, Fairbury, NE, USA) using a bipolar needle (Carefusion, San Carlos, CA, USA) and a Nicolet Viking EMG system (Madison, WI, USA) and a standardized protocol. Muscles were observed at rest for the presence of spontaneous activity. Muscles analyzed included biceps, first dorsal interosseous (digits of the hand), vastus medialis (upper hind limb), and gastrocnemius. EMG was analyzed 5–6 times per animal at regular intervals over a 7-month period starting when the animals were 1–2 months old.
Tissue preparation, PCR, Western blot, histology
For necropsy, the animals were deeply anesthetized with Telazol (8 mg/kg, i.m. from Zoetis, Florham Park, NJ, USA) and euthanized with pentobarbital (Beuthanasia, 100 mg/kg, intracardiac from Butler Schein, Fort Worth, TX, USA). The survival interval was 9 months for the GFP animal and 9–10 months for the TDP-43 animals. Tissues were collected and some were frozen on dry ice for RNA and protein analysis and some were post-fixated in 4% paraformaldehyde for histology. RNA was prepared using the STAT-60 reagent from Tel-test (Friendsville, TX, USA) and the RNA minElute Cleanup Kit from Qiagen (Germantown, MD, USA), and the quality was inspected by the Agilent (Santa Clara, CA, USA) 2100 Bioanalyzer system. The RNA was translated into cDNA for PCR with iScript Reverse Transcriptase Supermix reagents from Bio-Rad (Hercules, CA, USA). PCR was run on a Bio-Rad C1000 Thermal Cycler. One TDP-43 primer set (sense: CTGCTCTCCACGGTT; antisense: TGGATAGTTG-ACAACATAC) was used to amplify a 156-bp product within the N-terminal region of human TDP-43. This primer set incorporated four bases (2 on sense and 2 on antisense) that are mismatched between human and macaque TDP-43 to selectively amplify the human sequence. Importantly, each primer had a human base not present in the macaque sequence on its 3′ terminus to inhibit amplification of endogenous macaque TDP-43. Another TDP-43 primer set (sense: GCA-GAACCAGTCAGGCC; antisense: GCACAGTCGA-GGCTGATC) was used to amplify a 915-bp product from the C-terminal region of the human TDP-43 to the bovine growth hormone polyadenylation signal sequence in the recombinant expression cassette. This primer set should specifically detect only the recombinant TDP-43 mRNA but not macaque TDP-43 mRNA. A GFP primer set (sense: ATGGCGATGTGAA-TGGGC; antisense: AGATGGTTCTCTCCTGCA) was used to amplify a 240-bp product. Primers were ordered from Eurofins Operon (Huntsville, AL, USA).
For protein, tissues were homogenized in RIPA buffer with a protease inhibitor cocktail from Pierce (Rock-ford, IL, USA) and measured for protein content using Bio-Rad (Hercules, CA, USA) Protein Assay Dye. Equal amounts of protein were loaded on 12% gels from Bio-Rad. Secondary antibodies and ECL reagents were from Santa Cruz (Dallas, TX, USA) and GE Health Care (Pittsburgh, PA, USA). Primary antibodies for Western blots were TDP-43 antibodies from Abnova (Taipei City, Taiwan) and ProteinTech (Chicago, IL, USA), GFP from Chemicon (Merck, Damstadt, Germany), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from Ambion (Austin, TX, USA) at 1:1000.
Some of the fixated tissues were prepared for thin paraffin sections (5 μm) and some for thicker frozen sections (either 20 or 50 μm) by embedding in OCT compound from Tissue-Tek (Elkhart, IN, USA) and sectioning on either a cryostat or a sliding microtome. Several regions of the brain and spinal cord were analyzed as well as gastrocnemius muscle, heart, liver, kidney, ovaries, and others. Sections were stained by hematoxylin and eosin or by cresyl violet (Nissl substance) from Sigma (St. Louis, MO, USA) by standard methods. Primary antibodies included TDP-43 from Abnova, GFP from Invitrogen (Carlsbad, CA, USA), and NeuN from Chemicon at 1:1000 to 1:2000. Fluorescent DNA counterstains were used (TO-PRO3 stain from LifeTechnologies, Grand Island, NY, USA or DAPI from Sigma). Secondary antibodies were from DAKO (Glastrop, Denmark), Sigma, Invitrogen, or Jackson Immuno Research (Westgrove, PA, USA) and used at 1:300 to 1:500. Because relatively efficient expression was observed in the cerebellum Purkinje cells and a trend of more Purkinje cells with cytoplasmic TDP-43 immunoreactivity in the TDP-43 subjects was noted, the number of cells in the Purkinje cell layer of the cerebellum with dark staining of TDP-43 in the cytoplasm was estimated by a counting method. Two sagittal sections of the cerebellum stained for TDP-43 immunohistochemistry (paraffin sections and Abnova TDP-43 antibody) were counted for each animal, and the number was normalized per unit area of cerebellum as measured by a Cavalieri method using the Micro-brightfield (Williston, VT, USA) StereoInvestigator software and motorized stage. A blinded observer counted the samples.
Results
In previous studies in the rat, intravenous gene delivery with AAV9 provided sufficient expression of TDP-43 to yield a model with the major features of ALS [13].
Our i.v. vector doses per body mass were 2- to 5-fold lower in the macaques than in the previous rat studies [13]. There was widespread GFP expression in the brain and spinal cord in the macaque infused with the GFP vector (Fig. 1), although overall, the numbers of neurons transduced were clearly fewer compared to rats, likely due at least in part to the lower dose. Similarly, the marked paralysis and atrophy found in rats [13] were not reproduced in the TDP-43 macaques, so a readily observable, clinically relevant phenotype did not manifest in the two TDP-43 animals, which was again attributed primarily to the lower vector dose than used in rats.
Fig. 1.

Green fluorescent protein (GFP) expression in the macaque, 9 months after gene transfer. Detection of GFP used an immunofluorescent method. (A) Cross-section of the lumbar spinal cord. (B) Ventral horn of the lumbar spinal cord. Cells with neuronal (arrow) and non-neuronal (arrowhead) morphologies were expressing GFP. (C) GFP fibers in the dorsal columns of the spinal cord. (D) Motor cortex, cells with neuronal and non-neuronal morphologies. (E) Brainstem, non-neuronal morphologies. (F) Hippocampus, neuronal (arrow) and non-neuronal morphologies (arrowhead). (G) Sagittal section of the cerebellum, efficient transduction of Purkinje neurons. (H) The gray matter of the cerebellum is counterstained in red for the neuronal marker, NeuN. (I) Efficient transduction of cells in the heart. (J–M) Confocal images from cerebellum showing GFP-positive Purkinje neurons, brainstem showing GFP in cells with non-neuronal morphologies, and gastrocnemius muscle showing GFP-positive muscle fibers. Counterstains: J, L: TO-PRO3 DNA in blue; K: TDP-43 in red. Bar in A = 536 μm, same magnification in G, H. Bar in B = 134 μm, same magnification in C–E. Bar in F = 67 μm. Bar in I = 268 μm. Bar in J = 150 μm. Bar in K = 38 μm. Bar in L = 75 μm, same magnification in M.
Two animals received the AAV9 TDP-43 and one the AAV9 GFP. The GFP animal was unusually small at birth prior to treatment. However, she outweighed the two TDP-43 females from 3 to 7 months (Fig. 2A). The trend of lower weights in the TDP-43 macaques is consistent with a partial effect relative to the significantly deficient weight gain previously observed in TDP-43 rats [13]. Monthly blood analysis suggested neither general morbidity nor infection (WBCs) in the TDP-43 subjects (Table S1). The main histopathological alterations in ALS are loss of motor neurons and muscle atrophy [3]. However, histological examination of the motor neurons in the anterior horn of the spinal cord and the gastrocnemius muscle was negative for any indications of ALS-relevant neuronal loss or atrophy, respectively (Fig. 3), which is consistent with the overall lack of clinical phenotype that was found.
Fig. 2.
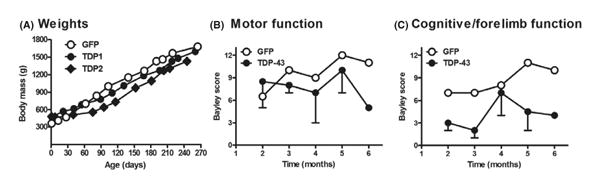
Functional trends in the TDP-43 subjects consistent with a disease state. Neonatal macaques were injected with AAV9 vectors i.v. (A) Weight gain curves: lower weights in the two TDP-43 subjects between 90 and 240 days. (B) Bayley composite ethogram score of motor function (GFP, N = 1; TDP-43, N = 2 from 2 to 5 months and N = 1 at 6 months due to technical issues; bars = SEM shown in the minus direction). (C) Bayley scale of cognitive functions, which also involves forelimb motor function: trend of impairment in the TDP-43 subjects.
Fig. 3.

Lumbar spinal cord and gastrocnemius muscle: no loss of motor neurons or muscle atrophy in the TDP-43 subjects relative to control. (A–C) Spinal cord was stained for Nissl substance to visualize large motor neurons in the anterior horn. (D–F) Gastrocnemius was stained with hematoxylin and eosin (H & E). Bar in C = 134 μm, same magnification in A–C. Bar in F = 67 μm, same magnification in D–F.
However, behavioral analyses suggested trends of impairment in measures of motor and cognitive performance (Fig. 2B, C). Relative to the age-matched control GFP animal, the composite ethograms suggested a slight tendency for motor impairment in the TDP-43 subjects (Fig. 2B). The trend was stronger for cognitive/forelimb function (Fig. 2C), in which the subjects performed memory tasks and used their hands to solve the puzzle or retrieve the food reward. In combining all of the motor and cognitive data, TDP1 showed lower scores than the GFP control on 80% of all testing intervals, and TDP2 showed lower scores than the control on 75% of all the testing time points, so the TDP-43 subjects trended lower scores than the control. We attempted to record positive waves and complex repetitive discharges in muscles, that is, the abnormal discharges that accompany acute and chronic neurogenic processes [30, 31]. Overall, there was little evidence of abnormal discharges in the muscles of either the TDP-43 or GFP subjects. However, at an age of 5 months for the TDP1 subject, positive waves and complex repetitive discharges were unequivocally present in the left first dorsal interosseous muscle, but these abnormal findings were not found upon subsequent testing 7 weeks later.
The weights of several organs were recorded at necropsy and expressed as percent of body weight (Table S2). The TDP-43 subjects suggested lower brain to body weight and heart to body weight ratios than the GFP control. Regarding GFP expression in the CNS, there was relatively stronger expression in the spinal cord and the cerebellum compared to other areas (Fig. 1). In the spinal cord, there was GFP labeling of cells with both neuronal and non-neuronal morphologies (Fig. 1A, B). In the cerebellum, GFP-positive Purkinje neurons were clearly observed (Fig. 1G, H, J, K), although other cell types were also more faintly positive for GFP in this area. Throughout most of the brainstem, midbrain, hippocampus, and motor cortex, there was relatively diffuse and sporadic labeling in cells with both neu-ronal and non-neuronal morphologies (depicted in Fig. 1D–F, L) with a relative preponderance for the non-neuronal astroglial morphology. This mainly astroglial transduction pattern in the brain was observed by Foust et al. [12] and Bevan et al. [15] when injecting adult mice or primates. Outside the CNS, pronounced GFP expression was observed in the heart (Fig. 1I), relatively lower levels of expression in the gastrocnemius muscle (Fig. 1M), and nearly no expression in the liver (Fig. S1A). The kidney (Fig. S1B) and ovaries (Fig. S1C) were also stained with the GFP antibody and showed sporadic low level expression in comparison to the spinal cord, cerebellum, and heart.
We were able to demonstrate an upregulation of TDP-43 levels in the TDP-43 subjects as expected. Two different PCR primer sets were used to detect the recombinant human TDP-43 mRNA, one targeting differences in the human and macaque sequences and one that only amplified a region from the recombinant vector sequence (Fig. 4A). On Western blots, elevated levels of TDP-43 in the TDP-43 subjects relative to the control GFP subject were detected in the dorsal root ganglia and the spinal cord. From analysis of the blots, after normalizing to a housekeeping protein, averaging the two TDP-43 bands and comparing to the band from the control GFP macaque, we estimated an 81% and a 64% increase in the dorsal root ganglia and spinal cord, respectively (Fig. 4B). However, in the cerebellum, TDP-43 levels were similar in all of the subjects. This was surprising, given the observation of GFP expression in Purkinje neurons in the GFP animal as well as the evidence of recombinant TDP-43 mRNA in the cerebellum in the TDP-43 subjects. The lack of change on the Western blot from the cerebellum samples may be due to a technical issue such as a higher fraction of protein from non-transduced cells.
Fig. 4.

(A) Detection of recombinant human TDP-43 mRNA or recombinant GFP mRNA in primates. The cerebellum was analyzed because of relatively efficient gene transfer in this area. TDP primer set 1 amplified a portion of human TDP-43 with mismatches between the human and macaque TDP-43 sequences, which demonstrated upregulated expression in the two TDP-43 subjects compared to the GFP control. TDP primer set 2 amplified recombinant TDP-43 only by specifically recognizing a downstream sequence in the expression cassette. GFP mRNA was specifically present in only the GFP subject as expected. (B) TDP-43 protein levels in primates. Western blots of dorsal root ganglia, lumbar spinal cord, and cerebellum at 9–10 months post-injection. There was evidence of elevated expression in the TDP-43 subjects in the dorsal root ganglia and spinal cord but not the cerebellum. The density of the TDP-43 bands (T) was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression (G).
Evidence of induced TDP-43 neuropathology was inconclusive overall despite trends. Although the human TDP-43 sequence was expressed, specific labeling of the recombinant human TDP-43 protein by immunohisto-chemistry was unsuccessful because the TDP-43 antibody from Abnova that is directed against human TDP-43 also labeled macaque TDP-43. TDP-43 immunoreactivity was observed in cell nuclei (small circular staining in Fig. 5A, B, arrows). While TDP-43 expression in the nucleus is the normal pattern, the main diagnostic lesion in ALS and FTLD-TDP is abnormal deposition and aggregation of TDP-43 in the cytoplasm [4, 5]. Surprisingly, TDP-43 immunoreactivity was also observed in the cytoplasm of some spinal cord anterior horn neurons in the control animal (Fig. 5B). However in the TDP-43 subjects, there was a pattern supporting a greater frequency of dark immunoreactivity in the cytoplasm, with dark staining of neuronal processes (Fig. 5D–F, arrowheads). Some of these cells displayed granular TDP-43 immunoreactivity (Fig. 5D–F). In the cerebellum of TDP-43 subjects, we noticed greater frequency of cytoplasmic TDP-43 immunoreactivity in the Purkinje cell layer (Fig. 5G, H). We scored the tissue containing these cells in a blind manner and normalized per unit area to estimate the density of cells with cyto-plasmic TDP-43 immunoreactivity. On average, counts of cells with cytoplasmic TDP-43 immunoreactivity in the TDP-43 subjects were increased by 75% relative to the control (TDP1/GFP = 1.98; TDP2/GFP = 1.52). Spinal cord tissues were probed with antibodies for the diagnostic marker proteins ubiquitin and p62 [32], but we did not observe specific pathological aggregates in the TDP-43 subjects. Positive findings included GFP expression in the control, elevated TDP-43 in the TDP-43 subjects evidenced for RNA and protein, trends of subtle behavioral and EMG impairments, and increased cytoplasmic TDP-43 in the TDP-43 subjects.
Fig. 5.

TDP-43 immunoreactivity in the lumbar spinal cord and cerebellum. (A, B) Anterior horn and motor neurons in the control GFP subject. Arrows, TDP-43 staining in cell nuclei, found in both GFP and TDP-43 subjects. (C–F) Anterior horn and motor neurons in the TDP-43 subjects with examples of dense cytoplasmic immunoreactivity and staining in neuronal processes (arrowheads). (G, H) Purkinje cell layer in cerebellum. Cells with cytoplasmic immunoreactivity were relatively more frequent in the TDP-43 subjects. Bar in A = 134 μm, same magnification in C. Bar in B = 13 μm, same magnification in D–F. Bar in G = 21 μm, same magnification in H.
Discussion
The cohort was small and clear-cut mimicry of clinical features of ALS, that is, muscle weakness and paralysis were not observed in contrast to earlier findings in rats using this gene transfer method to express TDP-43, one of the main pathological proteins in ALS and FTLD. The lack of effects in the primates was probably due to the lower AAV9 vector dose that was used but could have potentially involved immunological differences between primates and rodents, for example, neutralizing antibodies to AAV which could affect the degree of transduction [19, 33]. However, we demonstrated sustained 9-month expression of recombinant transgenes in the CNS, and there was an overall trend of impairment in the TDP-43 subjects by EMG and the cognitive and forelimb tests. Furthermore, there was a trend for greater cytoplasmic and neuritic TDP-43 staining in the TDP-43 subjects. We estimated a less than twofold increase in TDP-43 levels, which was similar to the estimated fold increase in cells with cytoplasmic expression. While the effects were only subtle, the expression of either GFP or TDP-43 was unequivocal. However, in the previous rat studies [13], there was more neuronal transduction in the brain than in this study, so greater expression levels would likely be needed to achieve the same behavioral effects. If the vector dose is increased to yield greater expression or if a more efficient mode of expression is used, that is, stronger promoter or self-complementary double-stranded AAV [34], the approach may be feasible based on the results in this study. A NHP model could be valuable to therapeutic development by virtue of the similarity to neurological testing in ALS. EMG and motor and cognitive function were assessed in this study. The greater anatomical similarity among primates allows for highly sensitive and clinically relevant tests that may allow for accurate predictions on efficacy. The opportunity for meaningful clinical testing may be missed in other less relevant systems along with the underlying molecular mechanisms relevant to disease that are shared among primates. For these reasons, refinement of this preliminary work could lead to a more human-like model of ALS, which may provide passage for uncovering efficacious compounds by effectively capturing mechanisms that occur in humans. A disadvantage of this system is the high cost and ethical issues associated with primates, but it could yield a unique opportunity for predicting efficacy of potential ALS therapeutics. As ALS is an age-related disease usually affecting individuals in their 50s and 60s, it would be more relevant to induce the disease state in older subjects. In this regard, it would have been interesting to monitor these subjects to older age. We conducted gene transfer to neonates because widespread CNS gene transfer can be achieved from a treatment in the neonatal period [12, 13, 16, 18]. Another practical issue is that larger subjects will require larger per kg vector doses, which could be cost-prohibitive in terms of dose escalation.
In terms of the gene transfer expression pattern in this study, there was relatively strong expression in the spinal cord and the cerebellum in the CNS. In addition, heart and muscle cells were also transduced outside of the CNS. However, we believe expression in the spinal cord is essential to the disease state. When we expressed TDP-43 in rats with a promoter that did not express in the spinal cord but did in the liver and the heart, paralysis did not manifest as when TDP-43 was expressed in the spinal cord [14]. Interestingly, the liver did not express GFP in this study, which could have been due to the long survival interval and vector DNA washout during hepatocyte cell division. In studying ALS, it would be advantageous to make restricted tissue expressions, for example, either neurons vs. glia or neurons vs. muscle, in order to study the site of the functional effects. Given the apparent shift from more neuronal expression to more of an astroglial pattern upon vector administration to adults [12, 15], this developmental difference could potentially be used to study the requirement of neuronal expression. Administration directly into the cerebrospinal fluid is effective in selectively transducing the CNS and preventing the peripheral expression that occurs after systemic vector delivery as used in Bevan et al. [15] and Samaranch et al. [19]. Administration within the thecal sac of the spinal cord may offer particular relevance to ALS by targeting the spinal cord as used by Federici et al. [35] in swine and Wang et al. [36] in primates. This would also overcome the need to use neonatal subjects and provide an adult onset model. Additionally, an intraspinal approach could reduce the amount of vector needed to produce the effect [15, 19]. While the data are preliminary and only a first step, given the high cost of primate studies, they may aid investigators attempting a similar approach in terms of vector dosing and methodology. With refinement, a more relevant primate model could advance therapeutic progress. The models could be used either to validate a therapeutic lead from more reductionistic systems or to uncover one that would only be detected in a highly sensitive, clinically relevant, and predictive system.
Supplementary Material
Acknowledgments
This work was supported by a Pilot Grant from the Tulane National Primate Research Center [NIH NCRR RR00164] and the Fidelity Biosciences Research Initiative. We thank Christopher Jackson, Mychal Grames, and Daniel Daunis (Louisiana State University Health Sciences Center in Shreveport) for technical assistance, Kerith Luchins and Lara Doyle-Meyers for veterinary care, Michelle Weedman and Michelle Muniz for conducting the behavioral tests, Cynthia Trygg for administrative assistance (Tulane National Primate Research Center), and Kevin Foust (Ohio State College of Medicine) for discussion.
Footnotes
Supporting Information: Additional Supporting Information may be found in the online version of this article:
Table S1. Average blood counts. Blood draws were taken periodically throughout the 8 month study.
Table S2. Tissue weights at necropsy at 8 months expressed as percent of body weight.
Figure S1. Green fluorescent protein immunolabeling in non-CNS tissues.
References
- 1.Watson C, Paxinos G, Kayalioglu G. Chapter 7: Localization of motoneurons in the spinal cord. In: McHanwell S, Watson C, editors. The Spinal Cord: A Christopher and Dana Reeve Foundation Text and Atlas. 1st. Sydney: Academic Press; 2009. pp. 94–114. [Google Scholar]
- 2.Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6:211–20. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–55. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 4.Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM, Trojanowski JQ. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–34. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 5.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McClus-key LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 6.Baralle M, Buratti E, Baralle FE. The role of TDP-43 in the patho-genesis of ALS and FTLD. Biochem Soc Trans. 2013;41:1536–40. doi: 10.1042/BST20130186. [DOI] [PubMed] [Google Scholar]
- 7.Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14:459–68. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dayton RD, Wang DB, Klein RL. The advent of AAV9 expands applications for brain and spinal cord gene delivery. Expert Opin Biol Ther. 2012;12:757–66. doi: 10.1517/14712598.2012.681463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang DB, Gitcho MA, Kraemer BC, Klein RL. Genetic strategies to study TDP-43 in rodents and to develop preclinical therapeutics for amyotrophic lateral sclerosis. Eur J Neurosci. 2011;34:1179–88. doi: 10.1111/j.1460-9568.2011.07803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tatom JB, Wang DB, Dayton RD, Skalli O, Hutton ML, Dickson DW, Klein RL. Mimicking aspects of frontotemporal lobar degeneration and Lou Gehrig's disease in rats via TDP-43 overexpression. Mol Ther. 2009;17:607–13. doi: 10.1038/mt.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchida A, Sasaguri H, Kimura N, Tajiri M, Ohkubo T, Ono F, Sakaue F, Kanai K, Hirai T, Sano T, Shibuya K, Kobayashi M, Yamamoto M, Yokota S, Kubodera T, Tomori M, Sakaki K, Enomoto M, Hirai Y, Kumagai J, Yasutomi Y, Mochizuki H, Kuwabara S, Uchihara T, Mizusawa H, Yokota T. Non-human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP-43. Brain. 2012;135:833–46. doi: 10.1093/brain/awr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang DB, Dayton RD, Henning PP, Cain CD, Zhao LR, Schrott LM, Orchard EA, Knight DS, Klein RL. Expansive gene transfer to the rat CNS and amyotrophic lateral sclerosis relevant sequelae when TDP-43 is overexpressed. Mol Ther. 2010;18:2064–74. doi: 10.1038/mt.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dayton RD, Gitcho MA, Orchard EA, Wilson JD, Wang DB, Cain CD, Johnson JA, Zhang YJ, Petrucelli L, Mathis JM, Klein RL. Selective forelimb impairment in rats expressing a pathological TDP-43 25 kDa C-terminal fragment to mimic amyotrophic lateral sclerosis. Mol Ther. 2013;21:1324–34. doi: 10.1038/mt.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bevan AK, Duque S, Foust KD, Morales PR, Braun L, Schmelzer L, Chan CM, McCrate M, Chicoine LG, Coley BD, Porensky PN, Kolb SJ, Mendell JR, Burghes AH, Kaspar BK. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther. 2011;19:1971–80. doi: 10.1038/mt.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, Le TT, Morales PR, Rich MM, Burghes AH, Kaspar BK. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;28:271–4. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Gray SJ, Nagabhushan Kalburgi S, McCown TJ, Samulski RJ. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20:450–9. doi: 10.1038/gt.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattar CN, Waddington SN, Biswas A, Johana N, Ng XW, Fisk NM, Tan LG, Rahim AA, Buckley SM, Tan MH, Lu J, Choolani M, Chan JK. Systemic delivery of scAAV9 in fetal macaques facilitates neuronal transduction of the central and peripheral nervous systems. Gene Ther. 2013;20:69–83. doi: 10.1038/gt.2011.216. [DOI] [PubMed] [Google Scholar]
- 19.Samaranch L, Salegio EA, San Sebastian W, Kells AP, Foust KD, Bringas JR, Lamarre C, Forsayeth J, Kaspar BK, Bankiewicz KS. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum Gene Ther. 2012;23:382–9. doi: 10.1089/hum.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.National Research Council. Institute of Laboratory Animal Resources: Guide for the Care and Use of Laboratory Animals. Washington: National Academy Press; 1996. [Google Scholar]
- 21.United States Department of Agriculture: Final rules: code of federal regulations, title 9, part 3. Fed Regist. 1991;55:6426–505. [Google Scholar]
- 22.Klein RL, Hamby ME, Hirko AC, Wang S, Hughes JA, King MA, Meyer EM. Dose and promoter effects of adeno-associated viral vector for green fluorescent protein expression in the rat brain. Exp Neurol. 2002;176:66–74. doi: 10.1006/exnr.2002.7942. [DOI] [PubMed] [Google Scholar]
- 23.Klein RL, Dayton RD, Tatom JB, Henderson KM, Henning PP. AAV 8, 9, Rh10, Rh43 vector gene transfer in the rat brain: effects of serotype, promoter and purification method. Mol Ther. 2008;16:89–96. doi: 10.1038/sj.mt.6300331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–8. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayley N. Bayley Scales of Infant Development: Birth to Two Years. New York: The Psychological Corporation; 1969. [Google Scholar]
- 26.Champoux M, DiGregorio G, Schneider ML, Suomi SJ. Inanimate environmental enrichment for group-housed rhesus macaque infants. Am J Primatol. 1990;22:61–7. doi: 10.1002/ajp.1350220104. [DOI] [PubMed] [Google Scholar]
- 27.Isakova IA, Baker K, DuTreil M, Dufour J, Gaupp D, Phinney DG. Age and dose-related effects on MSC engraftment levels and anatomical distribution in the CNS of non-human primates: identification of novel MSC subpopulations that respond to guidance cues in brain. Stem Cells. 2007;25:3261–70. doi: 10.1634/stemcells.2007-0543. [DOI] [PubMed] [Google Scholar]
- 28.Borda JT, Alvarez X, Mohan M, Ratterree MS, Phillippi-Falkenstein K, Lackner AA, Bunnell BA. Clinical and immunopathologic alterations in rhesus macaques affected with Globoid Cell Leukodystrophy. Am J Pathol. 2008;172:98–111. doi: 10.2353/ajpath.2008.070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gomez-Mancilla B, Boucher R, Ga-gnon C, Di Paolo T, Markstein R, Bedard PJ. Effect of adding the D1 agonist CY 208–243 to chronic bromocriptine treatment. I: evaluation of motor parameters in relation to striatal catecholamine content and dopamine receptors. Mov Disord. 1993;8:144–50. doi: 10.1002/mds.870080205. [DOI] [PubMed] [Google Scholar]
- 30.Katirji B. Electro-myography in Clinical Practice. 2nd. United States of America: Mosby Elsevier; 2007. Chapter 2: Routine clinical electromyography; pp. 29–31. [Google Scholar]
- 31.England JD, Bohm RP, Jr, Roberts ED, Phillip MT. Mononeuropathy multiplex in rhesus monkeys with chronic lyme disease. Ann Neurol. 1997;41:375–84. doi: 10.1002/ana.410410313. [DOI] [PubMed] [Google Scholar]
- 32.Fecto F, Siddique T. UBQLN2/P62 cellular recycling pathways in amyotrophic lateral sclerosis and frontotemporal dementia. Muscle Nerve. 2012;45:157–62. doi: 10.1002/mus.23278. [DOI] [PubMed] [Google Scholar]
- 33.Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intra-vascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther. 2012;19:1058–69. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8:1248–54. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- 35.Federici T, Taub JS, Baum GR, Gray SJ, Grieger JC, Matthews KA, Handy CR, Passini MA, Samulski RJ, Boulis NM. Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. 2012;19:852–9. doi: 10.1038/gt.2011.130. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Yang B, Qiu L, Yang C, Kramer J, Su Q, Guo Y, Brown RH, Jr, Gao G, Xu Z. Widespread spinal cord transduction by intra-thecal injection of rAAV delivers efficacious RNAi therapy for amyotrophic lateral sclerosis. Hum Mol Genet. 2014;23:668–81. doi: 10.1093/hmg/ddt454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.