

Abstract
We investigated complex genomic rearrangements (CGRs) consisting of triplication copy-number variants (CNVs) that were accompanied by extended regions of copy-number-neutral absence of heterozygosity (AOH) in subjects with multiple congenital abnormalities. Molecular analyses provided observational evidence that in humans, post-zygotically generated CGRs can lead to regional uniparental disomy (UPD) due to template switches between homologs versus sister chromatids by using microhomology to prime DNA replication—a prediction of the replicative repair model, MMBIR. Our findings suggest that replication-based mechanisms might underlie the formation of diverse types of genomic alterations (CGRs and AOH) implicated in constitutional disorders.
Introduction
Absence of heterozygosity (AOH) in the developing embryo can result from uniparental disomy (UPD).1 UPD is an important non-Mendelian human disease-causing genetic mechanism resulting either from involvement of imprinted loci or from expression of a recessive trait when only one parent is a carrier.2,3 Segmental post-fertilization AOH can be observed in regions identical by descent or can occur because of recombination between homologous chromosomes such as mitotic crossing-over4 and gene conversion. Long tracts of AOH can also be generated during repair of one-ended, double-stranded DNA breaks (i.e., DSBs) by break-induced replication (BIR) if a homologous chromosome is used as a template to prime DNA replication.5,6 Microhomology-mediated break-induced replication (MMBIR), a mechanistic model that proposes a less stringent homology requirement for recombination than BIR with regards to length of homology, is predicted to produce segmental AOH as well.6 However, observational evidence for AOH in humans resulting from either replicative repair or MMBIR-mediated template switching between homologous chromosomes versus sister chromatids has been lacking.
Occurrence of complex genomic rearrangements (CGRs) associated with an extended segment of UPD has been recently reported,7–9 but the underlying molecular mechanism has remained elusive. We speculate that the previous lack of such CGR + AOH observations is due to the absence of genomic visualization of such potential events prior to the recent introduction of high-resolution microarrays with genome-wide coverage and the capacity to resolve CNVs and genotype SNPs in the same experimental assay.10
Intrachromosomal triplications in the human genome can originate from distinct molecular mechanisms generating two recognizably different CGR products, type I and II, that produce recurrent or nonrecurrent rearrangements,11 respectively. Type I triplication structures consist of three copies of a genomic segment in tandem with a head-to-tail orientation, separated and flanked by paralogous low-copy repeats (LCRs). Such triplications can result from two independent, intergenerational nonallelic homologous recombination (NAHR) events using flanking LCRs: the first event produces a duplication, followed by a second event producing the triplication. Such type I structures were reported in triplications leading to a severe form of Charcot-Marie-Tooth type 1A neuropathy,12 recognized as phenotypic outliers in families segregating duplications, as well as in individuals carrying Xp22.31 triplications.13 Type II triplications are more complex and share a unique structure consisting of a triplicated segment inserted in an inverted orientation between two copies of the duplicated segments, i.e., DUP-TRP/INV-DUP. Replication-based mechanisms with potential iterative template switches either coupled or not with non-homologous end joining (NHEJ) underlie the formation of these inverted triplications embedded in duplications, frequently consisting of, at least, two breakpoint junctions (jct1 and jct2) generated in one single event.13 DUP-TRP/INV-DUP was reported in 20% of the MECP2 CNV gains at Xq28,13 at the PLP1 locus at Xq22,14,15 at VIPR2 at 7q36,16 and at 15q13.3.17
We investigated the mechanism underlying CGRs found in association with segmental AOH in five families. By using a combined analysis of different high-resolution array platforms and breakpoint junction sequencing, we provide evidence that, in humans, complex rearrangements generated post-zygotically via MMBIR can lead to regional UPD as observed by copy-number-neutral segmental AOH. The model we propose can help explain formation of inverted triplications followed by AOH; this latter rearrangement product is posited to result from a template switch between homologs versus sister chromatids (Figure 1). Replication-based mechanisms might underlie the formation of diverse types of human genomic (CGR) and genetic (UPD) alterations, the latter with implications for transmission genetics.
Figure 1.
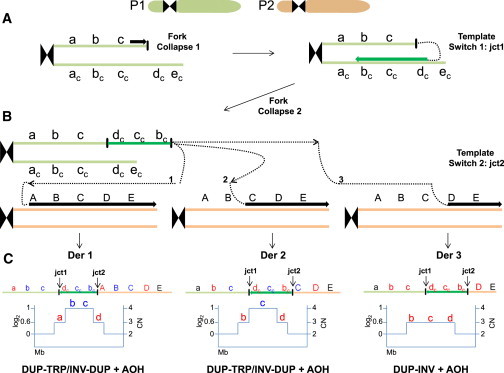
Replication-Based Mechanism Model for Generation of DUP-TRP/INV-DUP Rearrangements followed by AOH
(A) Event described probably occurred in a post-zygotic mitotic cell involving parental homolog chromosomes, P1 and P2. It might initiate due to a stalled or collapsed replication fork that uses a complementary strand to resume replication (template switch 1), generating jct1. Microhomology in the complementary strand at the annealing site (dc) is used to prime DNA synthesis, which will establish a unidirectional replication and will produce an inverted-oriented segment as compared to the reference genome.
(B) A new fork stalling or collapsing event releases a free 3′ end that can be resolved by a new cycle of template switching (template switch 2) and target annealing, this time using the homolog chromosome to prime and resume DNA synthesis. This second event can generate jct2 as well as AOH. It is possible that multiple cycles of fork collapse and target annealing occurs and produces additional complexities.
(C) Top: different genomic structures (Derivative [Der] 1, 2, 3) are expected to be generated depending on the location of the selected annealing site to prime DNA synthesis in the second template switch event. For instance, if the new annealing occurs at or before allele A to C, a triplication flanked by duplications will be produced (DER 1 and DER 2), whereas if annealing occurs at allele D, an inverted duplication will result (DER 3). Annealing at allele E will produce an inversion of segments b-c-d along with a partial duplication of b-c (not shown). AOH will result if unidirectional replication fork continues till the telomere. Bottom: expected segmental copy number (CN) variation in a simulated aCGH experiment. a, b, c: representative chromosome alleles; ac, bc, cc: complementary chromosome alleles; A, B, C, D, E: corresponding homolog chromosome alleles.
Subjects and Methods
Subjects
Subjects carrying an unusual pattern of triplications associated with AOH were identified by Nijmegen Centre for Molecular Life Sciences, Radboud University Nijmegen Medical Centre (Nijmegen, the Netherlands) (BAB3922, BAB3923; Figure 2), by the Department of Clinical Genetics, Maastricht University Medical Center (BAB3924; Figure 2), by the Baylor College of Medicine Medical Genetics Laboratories (MGL) as part of their clinical diagnostic evaluation (BAB4539; Figures 2 and S1), and by the Wellcome Trust Deciphering Developmental Disorders (WTDDD) research program (DECIPHER_257814). Informed consent was obtained for participation in the research study.
Figure 2.
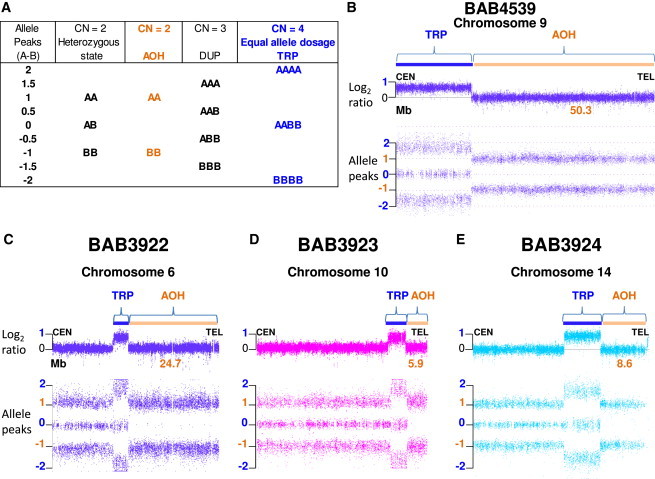
Affymetrix CytoScan Results
(A) Chromosome Analysis Suite (ChAS) software compares SNP probe intensities from index individual sample DNA with those of a sex-matched pooled reference DNA sample to calculate log2 ratios and infer copy number state (CN) that differ from the expected 0 value for CN = 2. Genotype calls and allele dosage normalization are performed as follows: the basic formula for allele peaks is A − B, where A is the signal of the A allele and B is the signal of the B allele. The allele peaks are normalized such that AA = 1.0, AB = 0.0, and BB = −1.0. Table shows allele dosage that differs from the heterozygous, diploid state.
(B–E) ChAS graphic results for chromosomes that presented triplication (TRP, blue rectangle) and absence of heterozygosity (AOH, orange rectangle) in subjects (B) BAB4539, (C) BAB3922, (D) BAB3923, and (E) BAB3924. Top, copy number log2 ratio; bottom, allele peaks (A − B). Alteration size is shown in Mb.
Determining Triplication Size and Absence of Heterozygosity Region
DNA from subjects BAB3922, BAB3923, and BAB3924 plus parental DNA samples were hybridized on an Affymetrix GeneChip 250k (NspI) SNP array platform or CytoScan array. Hybridizations were performed according to the manufacturer’s protocols (Affymetrix). Copy-number estimates were determined with the 2.0 version of the Copy Number Analyzer for Affymetrix GeneChip mapping (CNAG) software package (250k) or Chromosome Analysis Suite (CHAS) software (for CytoScan arrays) by comparing SNP probe intensities from index individual samples of DNA with those of a sex-matched pooled reference DNA sample (DNA from either ten healthy male or ten healthy female individuals). Subject BAB4539 was identified with V9.1 OLIGO, which is a custom-designed array with approximately 400,000 interrogating oligonucleotides that include 60,000 probes used for SNPs (Agilent Technologies).10 Subject DECIPHER_257814 was genotyped with an Illumina (Illumina) custom SNP genotyping chip, constituted by a backbone of 733,059 HumanOmniExpress-12v1_A-b37 positions and the addition of 94,840 selected positions. Genotypes were called by Illuminus genotyping software.18 Array comparative genomic hybridization was performed with a custom, exome-focused, two million probe Agilent aCGH array; CNVs were called via CNsolidate, a Sanger Centre in-house algorithm that integrates 12 change-point detection algorithms (T. Fitzpatrick, P. Vijayarangakannan, N. Carter, M.E.H., data not shown); uniparental disomy was detected with UPDio.19 To confirm triplication and AOH independently, samples were run on two different array platforms. For instance, BAB4539 plus parental samples were run on an Affymetrix CytoScan SNP array and samples BAB3922, BAB3923, and BAB3924 plus parental samples were analyzed with an Illumina array HumanOmniExpress-24 Beadchip at the Human Genome Sequencing Center of Baylor College of Medicine. Basic quality control and analysis of the genotyping data were performed on GenomeStudio software (Illumina).
The Affymetrix platform algorithm calls genotypes and normalizes allele dosage as follows: the basic formula for allele peaks is A − B, where A is the signal of the A allele and B is the signal of the B allele. The allele peaks are normalized such that AA = 1.0, AB = 0.0, and BB = −1.0; a hemizygous A maps to 0.5 and a hemizygous B maps to −0.5. On the Illumina platform the algorithm uses B-allele frequency (BAF) formula as follows: B / (A + B), where A is the signal of the A allele and B is the signal of the B allele. In BAFs, both AA and hemizygous A display as 0.0, AB displays as 0.5, and both BB and hemizygous B display as 1.0.
Fluorescence In Situ Hybridization Studies
Fluorescence in situ hybridization (FISH) for subject BAB4539 was performed with BAC probes for 9q21.13: RP11-655M14 (test, red) and RP11-338N12 (control, green).
Refining Triplication Size and Gene Content
To refine the breakpoint junctions of triplications in subjects BAB3922, BAB3923, BAB3924, and BAB4539, we designed a high-density custom tiling-path oligonucleotide microarray spanning each of the chromosomes involved (6, 9, 10, and 14) via the earray website. The average coverage was 1 probe per 1,000 bp, spanning the following genomic regions (UCSC Browser build hg19): chr6: 140,000,000–150,000,000; chr9: 65,000,000–141,213,430; chr10: 120,000,000–135,000,000; chr14: 80,000,000–105,000,000. Probe labeling and hybridization were performed according to the manufacturer’s protocols with modifications.
Long-Range PCR Amplification
Reverse and forward primer pairs (relative to the reference genome) were designed at the apparent boundaries of the copy-number gain segments as defined by high-density aCGH analysis.14 Long-range PCR was performed with TaKaRa LA Taq (Clontech). PCR sample-specific products were sequenced by the Sanger sequencing methodology. PCR and sequencing results were independently repeated to confirm results. Any additional potential point mutation observed in the rearrangement breakpoint junction of the index subject was also studied in parental samples to examine the ancestral chromosome status for those specific alterations and determine whether they occurred de novo.
Results
Intrachromosomal Triplications Followed by Extensive Regions of Absence of Heterozygosity
In total, five triplications associated with adjacent copy-number-neutral AOH were studied. The five complex genomic rearrangements (CGRs) studied were from four independent diagnostic and research laboratories and each represented a de novo rare variant. These five subjects were found to carry de novo genomic triplications, that is, copy number (CN) equal to four, followed by AOH that extended from the end of the triplication to the telomere. Both log2 ratio and allele peaks (A − B) in the triplicated region presented distinct patterns (log2 ratio = 1, AAAA = 2, AABB = 0, BBBB = −2) that could be readily differentiated from the diploid, heterozygous state (log2 ratio = 0, AA = 1, AB = 0, BB = −1) (Figure 2). The observed allele dosage values indicate that, within the triplicated segment, each parent contributed equally with two alleles (either AA or BB). AOH segments presented with log2 ratio = 0 and allele peaks of AA = 1 or BB = −1. Triplication sizes varied from 3.8 Mb to 21.7 Mb whereas accompanying AOH genomic intervals ranged from 5.9 Mb to 50.3 Mb (Figure 2, Table 1), involving different chromosomes: 1, 6, 9, 10, and 14.
Table 1.
Summary of Results for Subjects with Triplication Followed by AOH
| Subject | Chromosome Sub-band |
DUP |
TRP |
DUP |
AOH Region |
|
|---|---|---|---|---|---|---|
|
a |
b-c |
d |
e |
Uniparental |
||
| Mb | Mb | Mb | Mb | disomy | ||
| BAB3922 | 6q24.1 | 0.258 | 3.8 | 0.004 | 24.7 | maternal |
| BAB3923 | 10q26.13 | 0.022 | 3.8a | unknown | 5.9 | paternal |
| BAB3924 | 14q32.12 | 0.031 | 5.9 | 0.002 | 8.6 | paternal |
| BAB4539 | 9q12–q21.11 | LCR | 21.7 + 0.055a | LCR | 50.3 | paternal |
| DECIPHER_257814 | 1p36.12 | 0.001 | 8.7 | 0.001 | 11.8 | maternal |
Size of CNV segments was defined by inspection of high-resolution aCGH data coupled to breakpoint junction sequencing unless otherwise stated. Size of AOH region was inferred from SNP array data (Affymetrix platform: BAB3922, BAB3923, BAB3924, BAB4539; Illumina platform: DECIPHER_257814). Segments a, b, c, d, and e are defined in Figure 1. Abbreviations are as follows: LCR, non-paralogous low-copy repeat; Mb, Megabase.
Estimated size based on Affymetrix platform data.
To study the structure of these distinct genomic alterations further, we first asked whether the triplicated segments were located on the same chromosome or inserted into homologous or heterologous chromosomes. FISH assays were performed on samples that had biological material available. For samples BAB3922, BAB3924, and BAB4539, FISH studies were consistent with the triplicated genomic segments being located at an adjacent position, in the same chromosome homolog (Figure S2 for BAB4539; data not shown for BAB3922 and BAB3924).
Triplications Are Flanked by Small Duplications
We fine-mapped the breakpoint junctions of rearrangements in individuals BAB3922, BAB3923, BAB3924, and DECIPHER_257814 to the nucleotide resolution level. For four cases (BAB3922, BAB3923, BAB3924, and BAB4539), we designed chromosome-specific, high-resolution, custom-tiled oligonucleotide CGH arrays to examine the CGRs in subjects and families. Remarkably, in two out of four cases (BAB3922 and BAB3924, Figure 3A), high-density aCGH revealed small duplications flanking both proximal and distal junctions of the triplications. Triplication observed in BAB3923 also presented a small, proximal, centromeric duplication (Figure 3B) but there was no apparent flanking duplication at the distal, telomeric side.
Figure 3.
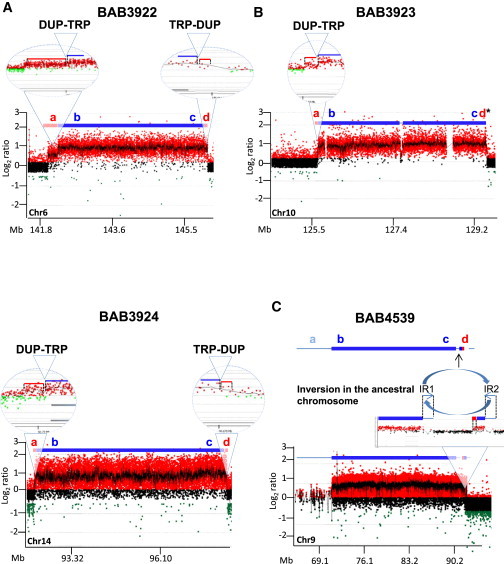
Agilent Tiling High-Density aCGH Revealed Triplications Flanked by Duplications at Both Centromeric and Telomeric Junctions
(A and B) In BAB3922 and BAB3924 (A), the flanking duplications were experimentally documented by aCGH and breakpoint junction sequencing, whereas in BAB3923 (B), a flanking duplication was observed at the centromeric junction and a duplication at the telomeric junction is predicted (d∗), but that breakpoint junction was not identified. Red rectangles represent duplicated segments (a, d); blue rectangles represent triplicated segments (b, c) named according to Figure 1 scheme. Chromosome-specific coordinates (hg19) shown on x axis; mean normalized log2 (Cy5/Cy3) ratio shown on y axis: approximate values of 0.6 indicates DUP, 1.0 indicates TRP. DUP-TRP junctions are shown in enlarged detailed view.
(C) BAB4539: high-density aCGH revealed an apparent complex rearrangement consisting of a large TRP separated by a copy-number-neutral region from a smaller DUP-TRP segment. Both TRPs are flanked by inverted repeats (IR1 and IR2), which make this segment susceptible to inversion through NAHR. Such an inversion present in an ancestral chromosome would produce an apparent interrupted triplication. Low-copy repeats (LCRs) are represented by thin light blue bar.
Interestingly, BAB4539 presented an apparent complex rearrangement consisting of two triplications separated by a ∼163 kb segment without copy-number alteration. LCRs flanking the centromeric and telomeric side of the large triplication hampered our ability to map the breakpoint junctions (Figure 3C), whereas the smaller triplication was flanked by a small duplication (Figure 3C) at the centromeric side. Importantly, the distal telomeric breakpoint junctions of both large and small triplications map within 30 kb of an inverted repeat pair, IR1 and IR2, that each share 99% nucleotide identity (Figure 3C). The high nucleotide similarity and the relative short genomic distance between IR1 and IR2 suggest that they can act as substrates for an intrachromosomal ectopic recombination that would result in an inversion of the genomic segment in between them. In fact, the region between IR1 and IR2 seems to be inverted in seven out of nine individuals that had their genomes subcloned to fosmid libraries in the Human Genome Structural Variation Project (HGSV) (Figure S3),20 suggesting that a frequent polymorphic inversion of such a region is present in the population. If the ancestral chromosome that underwent the rearrangement in subject BAB4539 carried such an inversion, then the apparent complex rearrangement formed by a triplication and a duplication/triplication can be, in fact, a single triplication flanked by a duplication as observed in the other cases reported herein (Figure 3C). The observation of inverted haplotypes, with respect to the haploid human reference genome, producing apparent structural complexities have been reported and reveal the limitations of a haploid reference for interpreting structural variation of the diploid human genome.21,22
Breakpoint Junction Sequencing Supports a DUP-TRP/INV-DUP Pattern
Duplications flanking triplications have been observed and characterized as type II triplications with a particular DUP-TRP/INV-DUP structure.11,14 Thus, in these AOH-associated triplications with flanking duplications, we anticipated the possibility that the triplicated segment might be inverted, which would be consistent with our proposed model in Figure 1. To test this hypothesis, we designed outward-facing sets of primer pairs for long-range PCR in which the amplification was predicted to span the transitions from an unchanged copy-number state to gains of genomic sequence based on the breakpoint mapping results of the high-density aCGH for each subject described above.
Breakpoint junctions of four out of five cases (BAB3922, BAB3923, BAB3924, and DECIPHER_257814) were sequenced, and we confirmed that there is an inversion of the triplicated segment, consistent with a pattern of DUP-TRP/INV-DUP. Sequences obtained from each junction are depicted in Figure S4. Junctions 1 and 2 (jct1 and jct2) were precisely characterized, revealing that the size of centromeric duplications, a, varied from 964 bp to 258 kb, whereas the size of the telomeric duplication, d, varied from 612 bp to 4.0 kb (Table 1). Importantly, the small size of the telomeric duplications might evade array detection of these latter CNV gains; also a likely scenario for the CGR in BAB3923 for whom we were not able either to detect telomeric duplication or to obtain sequencing of jct1. BAB4539 had no breakpoint junction sequenced despite multiple attempts, probably because they map within LCRs. The BAB4539 CGR jct1 maps within a flanking inverted repeat (IR1, IR2) whereas jct2 mapped somewhere within the pericentromeric region of chromosome 9 (9q12/qh) and 9q21, which is rich in LCRs and repetitive sequences (Figure 3C). This cytogenetic interval is known to show frequent polymorphic structural variation.
Microhomology from 1 to 10 bp was observed in all seven breakpoint junctions studied (Table 2, Figure S4). Insertion of sequences were observed in three junctions: BAB3922 presented with four nucleotide insertions at the junction; BAB3923 presented with an insertion of 12 nt of which 10 nt (CACCCAGATG) might represent a duplication of the sequence found adjacent to the breakpoint; and DECIPHER_257814 presents with a single nucleotide insertion (C) flanking the junction. In addition, an A to T transversion was observed in subject BAB3922. Remarkably, all insertions and point mutations seem to have occurred at jct2. None of the parents carry these single-nucleotide variants (SNVs), as tested by PCR and Sanger sequencing of these loci in parental DNA, so we infer that they arose during the same event as the de novo CGR/AOH formation. We have previously observed novel insertions, deletions, and point mutations near to the breakpoint junctions of CGR—a potential signature feature of replication-based mechanisms.22
Table 2.
Breakpoint Junction Features in Four Subjects
| Subjects |
jct1 |
jct2 |
||||
|---|---|---|---|---|---|---|
| Microhomology | Ins/Dela | Point Mutationa | Microhomology | Ins/Dela | Point Mutationa | |
| BAB3922 | AAAA | 0 | 0 | A | Ins CC + Del C, Del T, Del G, Del CA | A>T |
| BAB3923 | NA | NA | NA | CA | Ins CATCTGGGTGCA + Ins T | 0 |
| BAB3924 | TTT | 0 | 0 | AGGCTG | 0 | 0 |
| DECIPHER_257814 | GGTGGCAGGC | 0 | 0 | CT | Ins C | 0 |
Abbreviations are as follows: jct, breakpoint junction; Ins, insertion; Del, deletion; NA, not available.
Flanking 50 bp sequence analyzed for de novo mutations.
Change in Allele Dosage Ratio Suggest a Third Breakpoint Junction within the Triplicated Segment
Individual DECIPHER_257814 was genotyped with a customized Illumina SNP array and analyzed with in-house developed software, CNsolidate and UPDio, that detected an 8.7 Mb triplication and an 11.8 Mb segmental isodisomy, respectively, involving 1p36.12 (Figure 4). Importantly, inspection of B-allele frequencies in the raw SNP data of the triplicated region was significant for two patterns. One large region of ∼8.7 Mb showed equal allele dosage (AABB), consistent with data obtained in the four above-mentioned CGR + AOH genomes from individuals genotyped by Affymetrix arrays. Surprisingly, though, in the DECIPHER subject, a small ∼148.5 kb region within the triplicated interval consistently presented SNPs with unequal parental contribution, i.e., ABBB or BAAA (Figures 4B and 4C). Genotyping inspection of the unequal allele dosage region indicated that this segment was inherited from the same parent that contributed the UPD segment (maternal inheritance). The most likely explanation for this observation is that a new junction upstream of jct2 is present in this rearrangement; more importantly, such a new junction is revealed as defined by changing from equal to unequal allele dosage via a SNP platform for the genomic assay without a clear accompanying change in copy number (Figure 4B).
Figure 4.
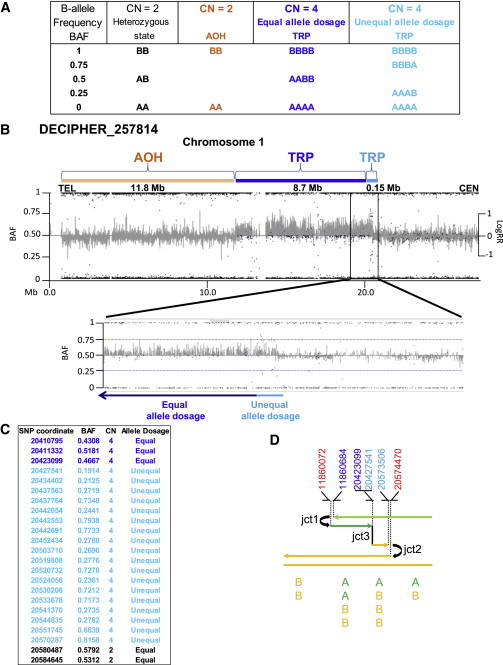
Subject DECIPHER_257814 Presents a Third Breakpoint Junction
Analysis of the Illumina SNP array revealed a triplicated segment with unequal allele dosage indicating dissimilar genotype inheritance (unequal allele dosage, light blue) followed by a region containing similar genotype inheritance (equal allele dosage, dark blue).
(A) Table shows some of the possible genotype calls performed by the allele dosage normalization Illumina’s platform algorithm. B-allele frequency (BAF) formula is calculated as: B / (A + B), where A is the signal of the A allele and B is the signal of the B allele. In BAFs, both AA and hemizygous A display as 0.0, AB displays as 0.5, and both B and hemizygous B display as 1.0.
(B) Graphic view of telomeric 20 Mb spanning 1p showing BAF and LogRR for subject DECIPHER_257814 indicating an 11.8 Mb region of absence of heterozygosity (AOH, orange rectangle) and triplication (TRP, dark and light blue rectangles). BAF data revealed that the centromeric TRP breakpoint segment of 148.5 kb (enlarged detailed view) presents with unequal allele dosage (light blue) of the parental alleles that is followed by a TRP segment with equal allele dosage (8.7 Mb, dark blue). LogRR indicates Log R ratio used to calculate copy number state that differ from the expected 0 value for CN = 2.
(C) Informative Illumina SNP BAF data spanning the region containing jct3 (triplication with an equal parental allele dosage to triplication with an unequal parental allele dosage) and jct2.
(D) Color-matched schematic representation of the 1p36.12 CGR formation. DECIPHER_257814 is hypothesized to present at least three breakpoint junctions generated by template switches during replication-based repair (refer to model described in Figure 1). Top: genomic coordinates (hg19) of breakpoint junctions in chromosome 1 inferred from multiple technical approaches. For sequencing data at jct1 and jct2, refer to Figure S4. Red indicates duplicated segment; light blue indicates triplicated segment harboring unequal allele dosage; dark blue indicates triplicated segment harboring equal allele dosage. Bottom: representation of the SNP allele dosage in each segment involved in this CGR. A, B: SNP alleles. Note that the different orientation of the rearrangement in DECIPHER_257814 compared to that shown in Figure 1, in this case involving the p-arm.
Intrigued by the apparent template switch without copy-number change, we further analyzed Affymetrix data to search for evidence of unequal allele dosage in all four individuals, BAB3922, BAB3923, BAB3924, and BAB4539. No evidence for an unequal allele dosage inheritance for the triplicated alleles could be observed for individuals BAB3922 and BAB3924 but analysis of subject BAB3923 data revealed two SNPs that seemed to be inherited from a single parent (rs7073245 and rs9422252, paternal inheritance), both of which map near to jct2. Sequencing of both alleles by Sanger in samples from the family trio revealed that the maternal allele was being sequenced but it was barely detectable (Figure S5), supporting the contention that paternal alleles were being amplified in higher quantity compared to maternal alleles as would be expected in an unequal allele dosage pattern biased toward the paternal alleles. In addition, we also tested two informative SNPs present on each side of jct2 (rs11517442 and rs4132312) by Sanger sequencing that allowed us to confirm exclusive inheritance of both segments from the father (Figure S6). A summary of alterations observed in BAB3923 is shown in Figure S7.
We then investigated samples BAB3922, BAB3923, and BAB3924 plus parents in an Illumina SNP array aiming to confirm the Affymetrix results independently. BAB3922 and BAB3924 indeed present a triplication segment with an equivalent contribution from both parents, and therefore there was no evidence for jct3 formation in those subjects. For BAB3923, Illumina BAF values support existence of an unequal allele dosage region within the triplicated segment spanning a maximum of 226 kb including jct2 (data not shown). Importantly, as in DECIPHER_257814, the small unequal allele dosage segment was inherited from the same parent that contributed to the UPD segment, in this case a paternal inheritance. A model for formation of the CGR in BAB3923 is shown in Figure S7. We could not confirm whether jct3 was formed without an accompanying change in the CNV status as observed in DECIPHER_257814 because in BAB3923, jct3 seems to have occurred between genomic coordinates chr10: 125,868,868 and 125,946,185 where a gap in the reference genome remains (therefore, none of the arrays provide CNV information for that region).
Discussion
The main molecular mechanism invoked to explain large regions of AOH not inherited as identical by descent is mitotic recombination,23 first described by Stern in 1936 to explain somatic crossovers (CO) observed in Drosophila experiments.4 DSB-induced COs between homologs followed by segregation of crossover chromatids to separate daughter cells is a very rare event in mitotic cells and might lead to AOH within part of the chromosome arm distal from the point of CO.24,25 The second mechanism that can also generate very large regions of AOH is BIR. BIR acts on one-ended, double-stranded DNA breaks; for instance, those that result from replication fork collapse. In the case that an homologous chromosome is used as template to re-establish a unidirectional replication fork, long tracts of AOH of all markers located distal to the DSB can be observed and might proceed several hundred kilobases to the telomere.5,6
Experiments performed here unveiled multiple layers of complexity, many of which were shared by all individual CGRs studied (Figure S8). In three cases (BAB3922, BAB3924, and DECIPHER_257814), rearrangements could be consistently classified as DUP-TRP/INV-DUP, or type II triplications, and in two cases (BAB3923 and BAB4539), preliminary data also support such a structure: BAB3923 presents with a DUP-TRP/INV structure and BAB4539 presents with a DUP-TRP structure. Furthermore, in all cases equal allele dosage is observed within the majority of the triplicated segments, which indicates that each parent had contributed equally to the copy-number gain in each subject. This observation is relevant for two reasons: first, it implies that at least two events, one intrachromosomal and another one interchromosomal in nature, must have occurred to produce triplications with AABB genotyping patterns; and second, the interchromosomal event indicates that there was at least one post-zygotic event. Therefore, triplications followed by AOH might, in fact, present with a similar genomic structure, which allows us to hypothesize a general model for their formation.
Importantly, mitotic crossing-over alone cannot account for our observations including the presence of segmental copy-number gain associated with AOH and a consistent pattern of DUP-TRP/INV-DUP rearrangement formed by a mixture of paternal and maternal segments originating from homologous chromosomes. BIR would be another possible mechanism to explain our findings; BIR in human cells under replication stress was recently reported in vitro.26 Nonetheless, BIR is RAD51 dependent and homology dependent,27 which cannot account for the observations of microhomology present in both jct1 and jct2. Because the underlying mechanism for formation is not driven by sequence homology (e.g., inverted repeats), we therefore propose that junctions are a result of multiple template-switches during MMBIR (Figure 1). Events might have occurred either all at once in a post-zygotic mitotic cell or in two steps: first one in a pre-meiotic cell followed by resolution in a post-zygotic cell; current data do not allow distinguishing between these alternatives. At least two template switches might have occurred triggered by stalled or collapsed replication forks. An initial template switch is predicted to use a sister chromatid to resume replication (Figure 1A). Microhomology at annealing site (dc) in the complementary strand is used to prime DNA synthesis relatively close to the breakpoint c (612 bp to 4.0 kb). Therefore, a unidirectional replication resumes in an inverted orientation and will form an inverted partially duplicated segment consisting of dc (no copy-number gain), cc to bc (duplication) (Figure 1A). This is unlikely to result in a healthy viable cell unless a second compensating inversion occurs, which might happen if the fork again collapses. A new fork stalling or collapsing event releases a free 3′ end that can be resolved by a second template switch to the homologous chromosome; this will result in a jct2 (Figure 1B). There are at least three possible target annealing sites to resume replication, represented here by different alleles A, C, D (annealing site 3). Each one will result in derivative chromosomes Der 1, 2, or 3; two of them will result in a DUP-TRP/INV-DUP structure (Der 1 and Der 2), whereas an inverted duplication will result in Der 3 (Figure 1C).
The resolution of the second break might be accomplished by other mechanisms that cannot be ruled out at this point. All of them would be capable of inducing the formation of AOH to the telomere: (1) a fully processive replication fork is established and can continue to the end of the chromosome or replicon; (2) repair might be accomplished by non-homologous end joining;28 or (3) repair is finished by formation of a half-crossover.29,30 Half-crossovers were shown to result from aberrant processing of BIR intermediates in mutants deficient in DNA synthesis in S. cerevisiae that are analogous to mammalian nonreciprocal translocations.31 One of the predicted products of a half-crossover would be a template chromosome carrying a large deletion including the telomere that can just segregate away, leading to cell death or triggering further instability. None of those by-products have yet been detected but this could be due to the few individual cases studied thus far.
Interestingly, DUP-TRP/INV-DUP rearrangements observed on autosomes seem to contrast somewhat with our previous observations regarding DUP-TRP/INV-DUP formation involving the MECP2 locus on the X chromosome. In the latter case an inverted repeat mediates jct1 formation probably through BIR coupled with MMBIR or NHEJ.14 In the cases described here, BIR cannot account for the observations of microhomology present in jct1 and jct2, and we speculate that MMBIR might be involved. The reason underlying such differences needs further investigation, but one possibility is that the genomic architecture of chromosome X plays a role. For example, we have recently constructed a genome-wide map of inverted repeats larger than 800 bp with at least 98% sequence identity (named DTIP-LCRs32) that could facilitate duplication/triplication formation. We observed that: (1) chromosome X has the largest chromosomal percent coverage of DTIP-LCRs (∼1.2%); and (2) chromosome X has a higher chromosomal percent coverage of genomic regions flanked by such DTIP-LCRs (>40%), which implies that nearly 2/5 of that chromosome is at risk of undergoing DUP-TRP/INV-DUP formation.32 This contrasts sharply with chromosome 14, involved in the DUP-TRP/INV-DUP rearrangement in subject BAB3924, which shows less than 10% of genomic regions flanked by DTIP-LCRs and therefore presents with a lower number of DTIP-LCRs substrates.
Remarkably, subjects DECIPHER_257814 and BAB3923 present with a potential third junction (jct3) not observed in the other three subjects described here. Existence of such a junction was inferred upon careful analysis of the SNP array data that revealed a triplicated segment consisting of two regions, each one presenting with different segregating patterns regarding parental contribution (equal allele dosage region or unequal allele dosage region). A third junction is required to explain the transition between those two patterns. Interestingly, in both cases (DECIPHER_257814 and BAB3923), jct3 seemed to have occurred a hundred kilobases from jct2 and without evidence of an accompanying change of the copy-number status. One possible explanation is that homology could have driven the formation of jct3 by template switching between homologs and, therefore, regular BIR might be involved rather than MMBIR. Coupled-homology and microhomology-mediated BIR was previously implicated in the generation of triplications at the chromosome X loci, MECP2 and PLP1,14 and might also be involved in generating triplications in autosomes. Alternatively, an accompanying CNV below the level of resolution afforded by our multiple array platforms used here is also potentially possible.
In conclusion, we present a model for formation of a recently described disease-causing mechanism: segmental AOH associated with complex rearrangements produced by long-range template switching via a RAD51-independent BIR. Replicative mechanisms to explain human CGRs have all incorporated the concept of long-distance template switching of replication forks, including fork stalling and template switching (FoSTeS),33,34 recombination restarted replication forks (RRRF or 3RF),35 and MMBIR,6 which are thought to proceed independently of RAD51.36 The molecular features of the rearrangements described here, including triplication formation and de novo SNVs observed at breakpoint junctions, support the involvement of replicative mechanisms underlying its formation and implicate an expected level of mutational load that can be generated in a unique event (i.e., copy-number variation or CNV, inversion, point mutations or SNV, and UPD), which has important implications for human disease and evolution.
Acknowledgments
We thank the families for their participation in the study. This work was supported in part by US National Institute of Neurological Disorders and Stroke (R01NS058529 to J.R.L.), the National Institute of General Medical Sciences (RO1GM106373 to P.J.H. and J.R.L. and R01GM080600 to G.I.), the National Human Genome Research Institute/National Heart Blood Lung Institute jointly funded Baylor Hopkins Center for Mendelian Genomics (U54HG006542 to J.R.L.), and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) through the Young Investigator fellowship (Science without Borders Program) (grant 402520/2012-2 to C.M.B.C.). The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NINDS, NHGRI/NHBLI, NIH, the Wellcome Trust, or the Department of Health. J.R.L. holds stock ownership in 23andMe, Inc., and Lasergen, Inc., is a paid consultant for Regeneron Pharmaceuticals, and is a co-inventor on multiple United States and European patents related to molecular diagnostics. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered in the Medical Genetics Laboratories.
Accession Numbers
Arrays have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession numbers GSE65379 and GSE65113. DECIPHER data have been deposited in the European Genome-phenome Archive (EGA) through the following accession numbers: dataset EGAD00010000702 and sample EGAN01000271322.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Agilent eArray, https://earray.chem.agilent.com
Baylor Miraca Genetics Laboratories, https://www.bcm.edu/research/medical-genetics-labs/
dbSNP, v.137, http://www.ncbi.nlm.nih.gov/projects/SNP/
Deciphering Developmental Disorders (DDD) Project, http://www.ddduk.org/
European Genome-phenome Archive (EGA), https://www.ebi.ac.uk/ega
Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/
UCSC Human Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Engel E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am. J. Med. Genet. 1980;6:137–143. doi: 10.1002/ajmg.1320060207. [DOI] [PubMed] [Google Scholar]
- 2.Spence J.E., Perciaccante R.G., Greig G.M., Willard H.F., Ledbetter D.H., Hejtmancik J.F., Pollack M.S., O’Brien W.E., Beaudet A.L. Uniparental disomy as a mechanism for human genetic disease. Am. J. Hum. Genet. 1988;42:217–226. [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y., Muzny D.M., Xia F., Niu Z., Person R., Ding Y., Ward P., Braxton A., Wang M., Buhay C. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stern C. Somatic crossing over and segregation in Drosophila melanogaster. Genetics. 1936;21:625–730. doi: 10.1093/genetics/21.6.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith C.E., Llorente B., Symington L.S. Template switching during break-induced replication. Nature. 2007;447:102–105. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]
- 6.Hastings P.J., Ira G., Lupski J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujita A., Suzumura H., Nakashima M., Tsurusaki Y., Saitsu H., Harada N., Matsumoto N., Miyake N. A unique case of de novo 5q33.3-q34 triplication with uniparental isodisomy of 5q34-qter. Am. J. Med. Genet. A. 2013;161A:1904–1909. doi: 10.1002/ajmg.a.36026. [DOI] [PubMed] [Google Scholar]
- 8.Rodríguez-Santiago B., Malats N., Rothman N., Armengol L., Garcia-Closas M., Kogevinas M., Villa O., Hutchinson A., Earl J., Marenne G. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am. J. Hum. Genet. 2010;87:129–138. doi: 10.1016/j.ajhg.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sahoo T., Wang J.C., Elnaggar M.M., Sanchez-Lara P., Ross L.P., Mahon L.W., Hafezi K., Deming A., Hinman L., Bruno Y. Concurrent triplication and uniparental isodisomy: evidence for microhomology-mediated break-induced replication model for genomic rearrangements. Eur. J. Hum. Genet. 2015;23:61–66. doi: 10.1038/ejhg.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiszniewska J., Bi W., Shaw C., Stankiewicz P., Kang S.H., Pursley A.N., Lalani S., Hixson P., Gambin T., Tsai C.H. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur. J. Hum. Genet. 2014;22:79–87. doi: 10.1038/ejhg.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu P., Carvalho C.M., Hastings P.J., Lupski J.R. Mechanisms for recurrent and complex human genomic rearrangements. Curr. Opin. Genet. Dev. 2012;22:211–220. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu P., Gelowani V., Zhang F., Drory V.E., Ben-Shachar S., Roney E., Medeiros A.C., Moore R.J., DiVincenzo C., Burnette W.B. Mechanism, prevalence, and more severe neuropathy phenotype of the Charcot-Marie-Tooth type 1A triplication. Am. J. Hum. Genet. 2014;94:462–469. doi: 10.1016/j.ajhg.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu P., Erez A., Nagamani S.C., Bi W., Carvalho C.M., Simmons A.D., Wiszniewska J., Fang P., Eng P.A., Cooper M.L. Copy number gain at Xp22.31 includes complex duplication rearrangements and recurrent triplications. Hum. Mol. Genet. 2011;20:1975–1988. doi: 10.1093/hmg/ddr078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvalho C.M., Ramocki M.B., Pehlivan D., Franco L.M., Gonzaga-Jauregui C., Fang P., McCall A., Pivnick E.K., Hines-Dowell S., Seaver L.H. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat. Genet. 2011;43:1074–1081. doi: 10.1038/ng.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimojima K., Mano T., Kashiwagi M., Tanabe T., Sugawara M., Okamoto N., Arai H., Yamamoto T. Pelizaeus-Merzbacher disease caused by a duplication-inverted triplication-duplication in chromosomal segments including the PLP1 region. Eur. J. Med. Genet. 2012;55:400–403. doi: 10.1016/j.ejmg.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Beri S., Bonaglia M.C., Giorda R. Low-copy repeats at the human VIPR2 gene predispose to recurrent and nonrecurrent rearrangements. Eur. J. Hum. Genet. 2013;21:757–761. doi: 10.1038/ejhg.2012.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soler-Alfonso C., Carvalho C.M., Ge J., Roney E.K., Bader P.I., Kolodziejska K.E., Miller R.M., Lupski J.R., Stankiewicz P., Cheung S.W. CHRNA7 triplication associated with cognitive impairment and neuropsychiatric phenotypes in a three-generation pedigree. Eur. J. Hum. Genet. 2014;22:1071–1076. doi: 10.1038/ejhg.2013.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teo Y.Y., Inouye M., Small K.S., Gwilliam R., Deloukas P., Kwiatkowski D.P., Clark T.G. A genotype calling algorithm for the Illumina BeadArray platform. Bioinformatics. 2007;23:2741–2746. doi: 10.1093/bioinformatics/btm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King D.A., Fitzgerald T.W., Miller R., Canham N., Clayton-Smith J., Johnson D., Mansour S., Stewart F., Vasudevan P., Hurles M.E., DDD Study A novel method for detecting uniparental disomy from trio genotypes identifies a significant excess in children with developmental disorders. Genome Res. 2014;24:673–687. doi: 10.1101/gr.160465.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang F., Khajavi M., Connolly A.M., Towne C.F., Batish S.D., Lupski J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carvalho C.M., Pehlivan D., Ramocki M.B., Fang P., Alleva B., Franco L.M., Belmont J.W., Hastings P.J., Lupski J.R. Replicative mechanisms for CNV formation are error prone. Nat. Genet. 2013;45:1319–1326. doi: 10.1038/ng.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagstrom S.A., Dryja T.P. Mitotic recombination map of 13cen-13q14 derived from an investigation of loss of heterozygosity in retinoblastomas. Proc. Natl. Acad. Sci. USA. 1999;96:2952–2957. doi: 10.1073/pnas.96.6.2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee P.S., Greenwell P.W., Dominska M., Gawel M., Hamilton M., Petes T.D. A fine-structure map of spontaneous mitotic crossovers in the yeast Saccharomyces cerevisiae. PLoS Genet. 2009;5:e1000410. doi: 10.1371/journal.pgen.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaRocque J.R., Stark J.M., Oh J., Bojilova E., Yusa K., Horie K., Takeda J., Jasin M. Interhomolog recombination and loss of heterozygosity in wild-type and Bloom syndrome helicase (BLM)-deficient mammalian cells. Proc. Natl. Acad. Sci. USA. 2011;108:11971–11976. doi: 10.1073/pnas.1104421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costantino L., Sotiriou S.K., Rantala J.K., Magin S., Mladenov E., Helleday T., Haber J.E., Iliakis G., Kallioniemi O.P., Halazonetis T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science. 2014;343:88–91. doi: 10.1126/science.1243211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis A.P., Symington L.S. RAD51-dependent break-induced replication in yeast. Mol. Cell. Biol. 2004;24:2344–2351. doi: 10.1128/MCB.24.6.2344-2351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lieber M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 29.Malkova A., Ira G. Break-induced replication: functions and molecular mechanism. Curr. Opin. Genet. Dev. 2013;23:271–279. doi: 10.1016/j.gde.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson M.A., Kwon Y., Xu Y., Chung W.H., Chi P., Niu H., Mayle R., Chen X., Malkova A., Sung P., Ira G. Pif1 helicase and Polδ promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013;502:393–396. doi: 10.1038/nature12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deem A., Barker K., Vanhulle K., Downing B., Vayl A., Malkova A. Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics. 2008;179:1845–1860. doi: 10.1534/genetics.108.087940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dittwald P., Gambin T., Gonzaga-Jauregui C., Carvalho C.M., Lupski J.R., Stankiewicz P., Gambin A. Inverted low-copy repeats and genome instability—a genome-wide analysis. Hum. Mutat. 2013;34:210–220. doi: 10.1002/humu.22217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 34.Slack A., Thornton P.C., Magner D.B., Rosenberg S.M., Hastings P.J. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet. 2006;2:e48. doi: 10.1371/journal.pgen.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mizuno K., Miyabe I., Schalbetter S.A., Carr A.M., Murray J.M. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature. 2013;493:246–249. doi: 10.1038/nature11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malkova A., Ivanov E.L., Haber J.E. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc. Natl. Acad. Sci. USA. 1996;93:7131–7136. doi: 10.1073/pnas.93.14.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.