

Abstract
Robinow syndrome is a genetically heterogeneous disorder characterized by mesomelic limb shortening, genital hypoplasia, and distinctive facial features and for which both autosomal-recessive and autosomal-dominant inheritance patterns have been described. Causative variants in the non-canonical signaling gene WNT5A underlie a subset of autosomal-dominant Robinow syndrome (DRS) cases, but most individuals with DRS remain without a molecular diagnosis. We performed whole-exome sequencing in four unrelated DRS-affected individuals without coding mutations in WNT5A and found heterozygous DVL1 exon 14 mutations in three of them. Targeted Sanger sequencing in additional subjects with DRS uncovered DVL1 exon 14 mutations in five individuals, including a pair of monozygotic twins. In total, six distinct frameshift mutations were found in eight subjects, and all were heterozygous truncating variants within the penultimate exon of DVL1. In five families in which samples from unaffected parents were available, the variants were demonstrated to represent de novo mutations. All variant alleles are predicted to result in a premature termination codon within the last exon, escape nonsense-mediated decay (NMD), and most likely generate a C-terminally truncated protein with a distinct −1 reading-frame terminus. Study of the transcripts extracted from affected subjects’ leukocytes confirmed expression of both wild-type and variant alleles, supporting the hypothesis that mutant mRNA escapes NMD. Genomic variants identified in our study suggest that truncation of the C-terminal domain of DVL1, a protein hypothesized to have a downstream role in the Wnt-5a non-canonical pathway, is a common cause of DRS.
Introduction
In 1969, Dr. Meinhard Robinow and colleagues described an “unrecognized dwarfing syndrome” in one non-consanguineous extended family with multiple affected individuals; analysis of the family pedigree revealed an autosomal-dominant segregation pattern with complete penetrance.1 Clinical description of autosomal-dominant Robinow syndrome (DRS [MIM 180700]) includes mesomelic shortening, genital hypoplasia, dental malalignment, normal intellect, and distinctive facial features resembling a “fetal face.” These features include a high and broad forehead, frontal bossing, marked hypertelorism with midfacial hypoplasia, prominent eyes, a depressed nasal bridge, and a short nose with anteverted nares and a broad tip1, 2, 3 and are collectively referred to as fetal face because the proportion of the neurocranium to the face is about equal in newborns, whereas in adults, the proportion of the head is about one-third neurocranium to two-thirds face. Heterozygous missense mutations in WNT5A (MIM 164975), which encodes a protein that participates in the non-canonical, β-catenin-independent signaling cascade, were later shown to segregate in the first described family.4 Such mutations most likely change Wnt-5a surface structure and affect interactions with other proteins in the same pathway.5 In that same study, an additional unrelated individual with sporadic DRS was also shown to be a carrier of a distinct heterozygous mutation in WNT5A.4 Wnt5a-null mice exhibit phenotypic features similar to those observed in subjects with DRS, including hypertelorism, a triangular mouth, micrognathia, and hypoplastic genitalia.4, 6, 7 Autosomal-recessive Robinow syndrome (RRS [MIM 268310]) is characterized by short stature and pronounced mesomelic shortening and vertebral anomalies3, 8, 9 and is associated with loss-of-function mutations affecting the extracellular or cytoplasmic region of the orphan tyrosine kinase receptor ROR2 (MIM 602337).10, 11 ROR2 is a putative receptor of Wnt-5a,12 and together they activate the non-canonical Wnt signaling cascade, which results in the establishment of planar cell polarity in Drosophila and in the equivalent convergent-extension movements during gastrulation in vertebrates.13, 14 Importantly, heterozygous mutations that truncate ROR2 around the tyrosine kinase domain cause a distinct disease known as autosomal-dominant brachydactyly type B1 (BDB1 [MIM 113000]), most likely as a result of a gain-of-function or dominant-negative effect of the truncating protein variant.15, 16
Here, we report eight individuals with six distinct frameshift mutations clustered in the same exon of DVL1 (MIM 601365). DVL1 encodes one out of three human homologs of the Drosophila segment polarity protein dishevelled (dsh). All six variants are predicted by conceptual translation to produce a truncated protein. This contention is supported by experimental detection of the expression of the premature termination codon (PTC)-containing mRNA in subject cells. Our data support the concept that mutations involving additional proteins in the Wnt-5a-ROR2 signaling pathway can cause Robinow syndrome.17 Furthermore, in contrast with the type of genetic alteration reported to cause WNT5A and ROR2 disease-associated mutations, variants in DVL1 seem to result in dominant-negative or gain-of-function proteins.
Subjects and Methods
Subjects
Phase 1 of our study consisted of candidate-gene discovery in four affected individuals (BAB4073, BAB4569, BAB4878, and BAB5264) with clinical diagnoses of Robinow syndrome and in both parents of each individual (except for BAB5264, for whom only maternal DNA was available for study); all affected subjects underwent personal genome studies using whole-exome sequencing (WES). Phase 2 of our study consisted of confirming and assessing the contribution of DVL1 variants in exon 14 to the disease phenotype. Phase 2 included 62 additional subjects, each with a clinical suspicion of Robinow syndrome. We did not pre-select for possible dominant or recessive inheritance. Targeted sequencing identified mutations in five individuals (016462, 016516, 016517, 017604, and 030526) from four families, including a monozygotic twin pair (016516 and 016517) about whom a clinical description was previously published.18 All five phase 2 subjects were included in the study by Radboud University Medical Center in Nijmegen, the Netherlands. The subjects’ countries of origin included Portugal (016516 and 016517), Turkey (017604), the United States (BAB4073, BAB4569, BAB4878, BAB5264, and 030526), and Denmark (016462). Clinical findings are shown in Table 1 (see Supplemental Data for clinical descriptions). DNA was obtained from the subjects and their families after they gave written informed consent. Selected family pedigrees and photographs of individuals who gave consent for these photos to be used are shown in Figure 1 and Figures S1 and S2. The study was approved by the Radboud University Medical Center review board and by the institutional review board at the Baylor College of Medicine (protocol no. H-29697) for all sequencing conducted at the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC).
Table 1.
Summary of Clinical Features of DRS-Affected Individuals in This Study
|
Individual |
||||||||
|---|---|---|---|---|---|---|---|---|
| BAB4073 | BAB4878 | BAB5264 | 016462 | 016516 | 016517 | 017604 | 030526 | |
| Current age | 11 years | 22 years | 6 years | 20 years | 21 years | 21 years | 15 years, 6 months | 42 years |
| Age at last examination | 9 years | 20 years | 28 months | 20 years | 21 years | 21 years | 15 years, 6 months | 18 years |
| Genotype | c.1570_1571delins | c.1505_1517del | c.1519del | c.1505_1517del | c.1508del | c.1508del | c.1615del | c.1529del |
| De novo | + | + | ND | + | + | + | + | ND |
| Gender | F | F | M | F | M | M | M | F |
| Growth | ||||||||
| Height percentile | 10% | 17% | <3% | 70% | 90% | 75% | 80% | <3% |
| Occipitofrontal circumference SD | +4 SDs | +2.5 SDs | >+2 SDs | >+4 SDs | >+4 SDs | >+4 SDs | >+4SDs | >+6 SDs |
| Facial Features | ||||||||
| Macrocephaly | + | + | + | + | + | + | + | + |
| Frontal bossing | + | + | + | + | + | + | + | + |
| High forehead | + | + | + | + | + | + | + | + |
| Midface hypoplasia | + | + | + | + | + | + | + | + |
| Hypertelorism | + | + | + | + | + | + | + | + |
| Upslanting palpebral fissures | + | + | − | ND | − | − | − | − |
| Long eyelashes | + | + | + | ND | − | − | − | + |
| Prominent eyes | + | − | + | ND | + | + | − | + |
| Blue sclerae | + | − | + | ND | − | − | − | − |
| Epicanthal folds | − | + | + | + | − | − | + | ND |
| Anteverted nares | + | + | + | + | + | + | − | ND |
| Wide, low nasal bridge | + | + | + | ND | + | + | + | + |
| Short nose | + | + | + | + | + | + | + | + |
| Long philtrum | − | + | + | + | − | − | + | − |
| Triangular mouth | + | + | + | + | + | + | + | ND |
| Thin upper lip | + | + | + | + | − | − | − | ND |
| Gingival hyperplasia | + | + | + | + | + | + | + | − |
| Absent uvula | − | − | − | ND | − | − | + | ND |
| Cleft soft palate | + | − | − | ND | − | − | − | − |
| Bilobed tongue | + | + | + | − | − | − | + | − |
| Dental anomalies | + | + | + | + | + | + | + | + |
| Short neck | + | − | − | − | − | − | + | |
| Micrognathia | + | − | + | + | − | − | − | − |
| Abnormal ear shape and position | + | + | + | + | + | + | + | − |
| Skeletal Features | ||||||||
| Mesomelia | + | + | + | + | + | + | + | + |
| Brachydactyly | + | + | + | + | + | + | − | + |
| Clinodactyly | + | + | + | + | − | − | + | + |
| Camptodactyly | + | − | ND | ND | − | − | + | − |
| Broad thumb | + | − | + | + | + | + | − | − |
| Nail dysplasia | + | − | − | + | − | − | − | − |
| Bifid first and/or second phalanges | − | − | − | ND | + | + | ND | − |
| Hypoplastic phalanges | + | + | + | ND | + | + | + | + |
| Fetal finger and/or toe pads | − | − | − | ND | − | − | + | − |
| Broad first toe | + | + | + | + | + | + | + | + |
| Scoliosis and/or kyphosis | − | − | − | + | − | − | + | + |
| Pectus anomaly | + | + | − | ND | + | + | + | − |
| Urogenital Features | ||||||||
| Sacral dimple | + | − | ND | ND | − | − | − | − |
| Cryptorchidism | NA | NA | + | NA | + | + | + | NA |
| Hypospadias | NA | NA | ND | NA | − | − | + | NA |
| Micropenis | NA | NA | + | NA | + | + | + | NA |
| Agenesis of the labia minora | ND | − | NA | + | NA | NA | NA | ND |
| Small clitoris | ND | − | NA | + | NA | NA | NA | ND |
| Renal anomalies | − | − | − | ND | + | − | + | − |
| Inguinal hernia | − | − | − | ND | − | − | − | − |
| Other Features | ||||||||
| Heart defects | + | + | − | ND | − | − | − | − |
| Umbilical hernia | + | + | + | ND | − | − | + | − |
| Seizures | − | − | − | ND | − | − | + | − |
| Hearing loss | − | − | + | + | − | − | − | + |
| Obstructive sleep apnea | ND | + | ND | + | − | − | − | − |
| Absent anterior nasal spine | − | − | − | + | − | − | − | + |
| Increased bone density (skull) | ND | + | ND | + | − | − | + | ND |
| Omphalocele | − | − | − | ND | + | − | − | − |
| Hepatomegaly | − | − | − | ND | − | − | − | + |
Additional clinical case reports are available in the Supplemental Data. Abbreviations are as follows: +, present; −, absent; F, female; M, male; NA, not applicable; ND, no data; OFC, occipitofrontal circumference.
Figure 1.
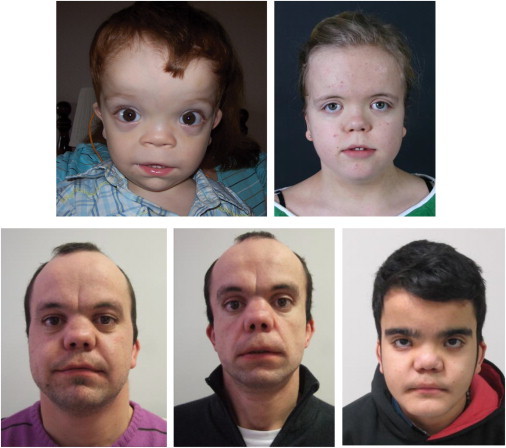
Clinical Presentation of the Individuals with DRS in This Study
Subjects (clockwise from the top left) BAB5264, 016462, 016516, 016517, and 017604 have DRS due to mutations in DLV1 and show characteristic facial features of frontal bossing with a high, broad forehead, hypertelorism, a broad nasal tip, and low-set ears.
WES, Variant Calling, and Selection of De Novo Variants
DNAs from individuals BAB4073, BAB4569, BAB4878, and BAB5264 were subjected to WES at the BCM-HGSC through the Baylor-Hopkins Center for Mendelian Genomics initiative. With 1 μg of DNA, an Illumina paired-end pre-capture library was constructed according to the manufacturer’s protocol (Illumina Multiplexing_SamplePrep_Guide_1005361_D) with modifications as described in the BCM-HGSC Illumina Barcoded Paired-End Capture Library Preparation protocol. Four pre-captured libraries were pooled and then hybridized in solution according to the BCM-HGSC in-house CORE design19 (52 Mb, NimbleGen) and the manufacturer’s protocol (NimbleGen SeqCap EZ Exome Library SR User’s Guide version 2.2) with minor revisions. The sequencing run was performed in paired-end mode with the Illumina HiSeq 2000 platform; sequencing-by-synthesis reactions were extended for 101 cycles from each end and an additional seven cycles for the index read. With a sequencing yield of 9.9 Gb on average, all samples achieved 89% of the targeted exome bases covered to a depth of 20× or greater. Illumina sequence analysis was performed with the BCM-HGSC Mercury analysis pipeline,20, 21 which moves data from the initial sequence generation on the instrument to annotated variant calls (SNPs and intra-read indels) via various analysis tools. We identified de novo mutations in silico by subtracting variants observed in either parent. Candidate variants were filtered against exome data in publicly available databases, including the 1000 Genomes Project, the NHBLI Exome Sequencing Project (ESP) Exome Variant Server, the Atherosclerosis Risk in Communities Study (ARIC) database, and our in-house-generated database of more than 4,282 exomes.
Variant Confirmation
We verified and evaluated potential disease-associated variants identified via WES for co-segregation with the phenotype by using standard PCR amplification. The primers we used to amplify exon 14 of DVL1 were as follows: 5′-GGGGAAGGGCAGGTAGGG-3′ (forward) and 5′-CAGTGAGTGGGGGCTTCG-3′ (reverse). PCR products were purified with ExoSAP-IT (Affymetrix) and sequenced with di-deoxy nucleotide Sanger sequencing at the DNA Sequencing and Gene Vector Core at Baylor College of Medicine. To confirm each frameshift individually, we manually cloned both alleles into a standard TOPO TA cloning vector (Life Technologies) and then transformed the recombinant clones into chemically competent E. coli to be grown overnight; individual clonal colonies were sequenced by a standard Sanger capillary protocol.
RNA Analysis
We extracted total RNA from peripheral blood from BAB5265, BAB5264, BAB4073, BAB4074, and BAB4075 by using the PAXgene Blood RNA System (PreAnalytiX). Blood was collected and transported in PAXgene Blood RNA Tubes. RNA was manually extracted according to the PAXgene Blood RNA purification protocol. cDNA was synthesized with 1 μg of RNA and the qScript cDNA SuperMix (Quanta Biosciences). 2 μl of cDNA was used for amplifying the mRNA fragment with primers in DVL1 exon 13 (5′-TGCTACTACGTCTTCGGGGA-3′ [forward]) and exon 15 (5′-TGTGATCCGATTCACTGCCA-3′ [reverse]). To quantify DVL1 expression, we used a qPCR assay on a reaction mixture consisting of 200 ng of cDNA with TaqMan probes (Life Technologies) located within the exon boundaries of exons 4 and 5 (Hs00182896_m1) and exons 14 and 15 (Hs00737028_m1). The endogenous control probe was specific to TBP (MIM 600075), encoding TATA box binding protein. Expression levels were analyzed with the SDS RQ software via the ΔΔ cycle-threshold (CT) method (Applied Biosystems).
Results
Genomic and Molecular Analysis
DVL1 maps to the 1p36.3 chromosomal sub-band; its canonical transcript contains 15 coding exons, which together comprise 2,941 nt (RefSeq accession number NM_004421.2), and encodes a 670-aa protein (RefSeq NP_004412) (Figure 2A). WES identified frameshift mutations in DVL1 exon 14 (RefSeq NM_004421.2) in individuals BAB4878 (c.1505_1517del [p.His502Profs∗143] on chromosome 1) and BAB5264 (c.1519del [p.Trp507Glyfs∗142] on chromosome 1). Sanger sequencing of exon 14 from the PCR product and from PCR cloning confirmed the presence of mutated and variant alleles in both subjects (Figure 2B). In subject BAB4073, a missense variant (chr1:1273426A>G) was identified by WES, but Sanger sequencing of exon 14 failed to confirm that mutation and instead unveiled an apparent frameshift in the subject’s electropherogram. Further allele-specific cloning confirmed the presence of such a frameshift involving the missense variant detected by WES, now interpreted as c.1570_1571delins (p.Phe524Profs∗125) (Figure 2B). This mutation was missed during the WES variant calling most likely as a result of poor mappability. Indel calling remains a challenge for massively parallel next-generation sequencing and might reflect errors in both library preparation and algorithm artifacts.17 Complex indels, such as the one identified in subject BAB4073 (c.1570_1571delins), can be challenging to identify in exome sequencing and can be misinterpreted as a simple base-pair substitution. Therefore, our data suggest that some frameshift mutations might need to be confirmed with allele-specific cloning and sequencing in addition to sequencing directly from mixed wild-type and mutant PCR products.
Figure 2.
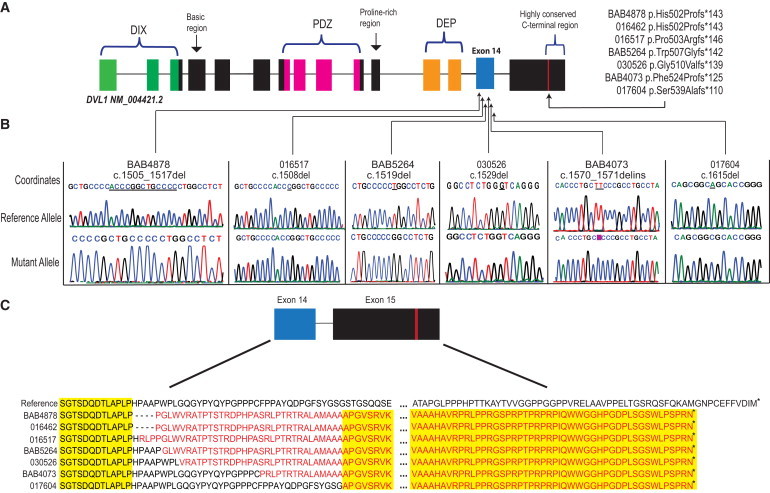
DVL1 Variants Identified in This Study
(A) Position of known functional domains and their distribution within the DVL1 transcript. The N-terminal DIX domain is most likely involved in regulating canonical Wnt signaling, leading to β-catenin stability;22 the PDZ domain is required for DVL1-mediated microtubule stabilization23 and for regulation of signal transduction. The C-terminal DEP domain is predicted to be important for protein interaction and most likely has a role in signal transduction; it can be found in other proteins involved in G protein signaling.24 The approximate locations of variants identified for each affected individual all cluster within exon 14 and display the same PTC within exon 15.
(B) Sanger sequencing of allele-specific PCR-product clones confirmed the presence of frameshift mutations in each affected individual and revealed a C insertion in subject BAB4073. Electropherograms displaying the cloning results of both alleles for six affected individuals are shown. Electropherograms for individual 016462, who carries the recurrent deletion c.1505_1517, are displayed in Figure S3. Underlined bases are deleted in the mutant allele, and the highlighted pink base (for subject BAB4073) in the mutant allele represents an insertion that did not exist in the reference allele.
(C) Predicted amino acid sequence with a change in the reading frame for part of exon 14 and exon 15. All variants in affected individuals have an identical PTC within exon 15. Identical amino acids are highlighted in yellow. Asterisks indicate termination codons.
Trio analysis revealed that frameshift mutations are de novo in subjects BAB4073 and BAB4878. The mother of BAB5264 does not carry the mutation, and the father is unavailable. We did not detect pathogenic or predicted pathogenic mutations in subject BAB4569 in the coding regions of ROR2, WNT5A, or DVL1 by WES (data not shown).
Further targeted gene studies identified three unique and one recurrent frameshift in five additional subjects. All of these frameshifts affect exon 14 of DVL1 and are as follows: c.1505_1517del (p.His502Profs∗143) in subject 016462 (subject BAB4878 harbors a similar frameshift), c.1508del (p.Pro503Argfs∗146) on chromosome 1 in identical twins 016516 and 016517, c.1615del (p.Ser539Alafs∗110) in subject 017604, and c.1529del (p.Gly510Valfs∗139) in subject 030526 (Figure 2B and Figure S3). Sanger sequencing of parental DNAs revealed a de novo origin for the DVL1 mutations identified in the personal genomes from the twin pair (016516 and 016517) and from individuals 016462 and 017604. Thus, de novo occurrence was verified in five unrelated families for which parental DNA was available. PCR fragments were cloned and sequenced for confirmation of reading frames.
All the variants observed in this group of subjects with DRS are predicted to result in a PTC located within the last exon (15) of DVL1 (Figure 2C). Because of the location of the predicted PTC, we hypothesized that the resulting mutant transcript could potentially escape nonsense-mediated decay (NMD).25 To test this hypothesis, we performed PCR of exons 13 to 15 by using the cDNA synthesized from isolated RNA extracted from subjects BAB4073 and BAB5264, as well as parental samples as wild-type controls when available. Sequencing of the RT-PCR products revealed the presence of both wild-type and mutated transcripts in affected individuals only (Figures 3A and 3B). Additionally, we measured the expression levels of DVL1 by using two independent TaqMan probes to target junctional exons 4 and 5 and exons 14 and 15 and observed no changes in the DVL1 expression between affected individuals and their parents (Figure 3C). The expression data indicate that both alleles are expressed in affected individuals’ leukocytes and support the hypothesis that these PTC-containing transcripts are not degraded by NMD.
Figure 3.
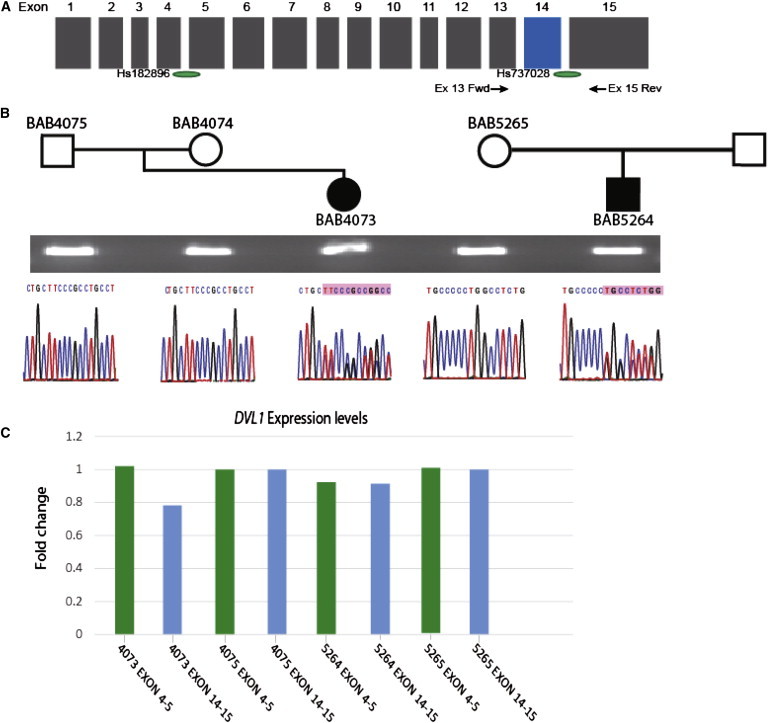
mRNA Expression of Mutant Alleles
(A) Schematic representation of the DVL1 coding region. Green ovals display approximate locations of TaqMan probes Hs00182896_m1 and Hs00737028_m1. Arrows show approximate locations of primers 5′-TGCTACTACGTCTTCGGGGA-3′ (exon 13 fwd) and 5′-TGTGATCCGATTCACTGCCA-3′ (exon 15 rev), which were used to amplify cDNA of mRNA extracted from subjects’ and parents’ leukocytes.
(B) Pedigrees of subjects BAB4073 and BAB5264 and available parents for RNA isolation. Agarose gel shows a 343-bp RT-PCR band, which was amplified with primers 5′-TGCTACTACGTCTTCGGGGA-3′ (exon 13 fwd) and 5′-TGTGATCCGATTCACTGCCA-3′ (exon 15 rev); below, electropherograms display sequencing results of amplified cDNA. Affected individuals harbor frameshift mutations identical to the genomic mutations identified. The variants are present in the cDNA of affected individuals but are not present in unaffected parents.
(C) Levels of DVL1 expression in leukocytes were measured by qPCR with TaqMan probes Hs00182896_m1 and Hs00737028_m1. For evaluating relative expression of DVL1, the ΔΔCT method was used with TBP as the endogenous control and one unaffected parent per family as the relative control. This experiment was replicated once and showed similar results.
The identified mutations were not observed in publicly available databases or in our in-house database. Analysis of the Exome Aggregation Consortium (ExAC) Browser (release 0.20), a public database that provides access to more than 61,486 unrelated subjects sequenced by WES without confirmation by an orthogonal sequencing technology as part of distinct disease-specific and population genetic studies, did reveal four frameshift alleles among 651 total coding mutations in DVL1. We do not predict that any of the four suggested frameshift mutations cause DRS because none are in exon 14. Notably, the subjects in the ExAC collection were not generally phenotyped, nor is it known from which disease population the study sample was obtained. Furthermore, the ExAC Browser includes data of heterogeneous quality, which requires that WES-identified mutations be confirmed with an orthogonal DNA sequencing technique that currently is not permitted.
Clinical Observations of Subjects with DVL1 Mutations
In general, the phenotype of individuals with DVL1 mutations is concordant with earlier clinical descriptions of DRS (Table 1 and Figure 1; see Supplemental Data).2, 3 Typical facial features, dental anomalies, and mesomelia were noted in 100% of DVL1-mutation-affected individuals in our study. In contrast to the DRS-affected individuals with WNT5A mutations thus far reported in the literature, 75% of DRS-affected individuals with DVL1 mutations were reported to have normal stature (above the tenth percentile). Another possible distinguishing feature between the two types of DRS is macrocephaly, which was observed in 100% of individuals with DVL1 mutations in this cohort. An occipitofrontal circumference above +4 SDs was noted in six out of the eight individuals with DVL1 mutations. Except for mild intellectual disability in one individual, intellectual function was normal in all other DVL1-mutation carriers. The majority of individuals had additional congenital anomalies, including genital hypoplasia (5/6 individuals), umbilical hernias (4/7 individuals), heart defects (2/7 individuals), renal anomalies (2/7 individuals), and hearing loss (3/7 individuals) (Table 1). Increased skull bone density was noted in three individuals. In addition, two subjects presented with severe obstructive breathing problems during sleep, and these necessitated long-term treatment with continuous positive airway pressure and eventually craniofacial surgery for correction of airway obstruction (Figures S1 and S2).
Discussion
Wingless (Wg)/Wnt signaling is a highly conserved pathway involved in cell-fate determination, including cell proliferation, migration, and establishment of planar cell polarity, in eukaryotic organisms. In humans, hypomorphic alleles and loss-of-function mutations of WNT5A and ROR2, respectively, both of which encode proteins involved in the non-canonical, β-catenin-independent signaling cascade, lead to DRS and RRS, respectively. Disturbing the non-canonical Wnt-5a pathway in diverse model organisms, including mice, causes morphogenic defects during embryonic development.6, 26, 27 DRS is hypothesized to display genetic heterogeneity given that mutations affecting WNT5A have been previously reported to account for only 1 of 24 cases.4 Recently, a report of three new unrelated subjects with de novo mutations in WNT5A5 confirmed that hypomorphic mutations in this gene are an important cause of DRS, but most subjects clinically diagnosed with Robinow syndrome do not have mutations in genes with a known association with the disease. Therefore, it is possible that the majority of the affected subjects will have mutations in distinct genes potentially involved in the same signaling pathway.3 In agreement with this hypothesis, we report eight DRS-affected subjects carrying frameshift mutations in exon 14 of DVL1.
In general, the phenotype of the herein-studied individuals with DVL1 mutations is concordant with earlier clinical descriptions of DRS (Table 1 and Figure 1; see Supplemental Data).2, 3 However, we suggest macrocephaly and absence of short stature as potential genotype-phenotype correlations for subjects with DVL1-associated Robinow syndrome. Individuals with WNT5A mutations can have macrocephaly at birth, but head circumference generally undergoes a postnatal progression to normocephaly.1, 4, 5 Even though Robinow syndrome is generally considered a “typical gestalt” diagnosis, facial features can become less clear during puberty and adulthood, and the diagnosis can be less obvious in individuals with normal stature18 (Figures S1 and S2). Therefore, the availability of another gene test for DRS could help confirm this clinical diagnosis. Furthermore, given the mappability challenges of the frameshift mutations identified herein, future clinical application might benefit from the use of traditional Sanger sequencing as a first approach for clinical diagnosis in individuals suspected of having DRS. Moreover, if a clinical diagnosis is suspected in a subject who previously had WES testing, then it might be prudent to examine the raw data obtained at the DVL1 locus.
DVL is a segment polarity gene (dsh) originally discovered in Drosophila, where it mediates Wg signaling transduction of patterning information in multiple tissues during the developmental processes, including axial patterning of legs and wings in adults.28, 29 In the developing Drosophila embryo, lack of dsh results in functional defects similar to those observed in wg mutants; further experiments revealed that Dsh functions in the reception or interpretation of the Wg signal.29, 30 Three dsh homologs, DVL1, DVL2 (MIM 602151), and DVL3 (MIM 601368), have been identified in mammals, including humans.31 Experimental data indicate that there could be functional redundancy among Dvl proteins,32, 33 but individual specific roles for each of these proteins are also being proposed.34 These observations are in agreement with a recently published mosaic genetic screen of lethal mutations on the Drosophila X chromosome; the screen revealed that fly genes with more than one human homolog are more likely to be associated with human syndromes than are fly genes with a single homolog, potentially indicating duplication and further specialization during evolution.35
Human DVL proteins, DVL1, DVL2, and DVL3, share 59%–67% of amino acid sequence identity, and the corresponding mouse orthologs show >95% of amino acid sequence identity.31 Mouse Dvl1 was shown to be ubiquitously expressed in adult tissues, particularly in the brain and testes.36 However, Dvl1-null mice do not present with developmental defects; instead, they show abnormal social behavior, such as a lack of whisker trimming, poor nest building, and decreased huddling during sleep, in addition to diminished sensorimotor gating.37 These data suggest that Dvl1 might contribute to the modulation of social behaviors in mammals; however, it is unclear what might constitute human “homologous” instinctual behaviors that one might screen for in our subjects. Furthermore, there is no evidence of altered behavior traits associated with DRS; in fact, most individuals from our cohort perform well in terms of social integration and functioning (see Supplemental Data), supporting the contention that the mechanism of DRS caused by mutations in DVL1 is not due to loss of function.
In our study, we observed a total of six frameshift mutations, five of which were confirmed to be de novo. Analysis of the ExAC Browser revealed the presence of four frameshift variants affecting DVL1, but the relevance of such findings for DRS is unknown because (1) we cannot confirm the ExAC Browser mutations by Sanger sequencing, (2) we do not have phenotypic information on those particular subjects, and (3) none of these mutations affect exon 14 of DVL1, which might suggest that frameshift mutations elsewhere in DVL1 might not cause DRS.
Remarkably, all variants observed in this cohort are predicted to produce a PTC that maps within the last exon of DVL1 and are predicted to escape NMD; this hypothesis is supported by our mRNA studies. NMD is a conserved post-transcriptional surveillance system that is intended to degrade transcripts that contain a PTC and thus eliminate abnormal transcripts (reviewed in Conti et al.38 and Popp et al.39); evidence of such NMD was not observed in our subjects. In the resulting predicted altered proteins, 22 amino acids from the C terminus will be removed, but other highly conserved functional domains, i.e., DIX, PDZ, and DEP, will be preserved. The exact function of the DVL1 C terminus is unknown, but the last 35 amino acids have been shown to be conserved among all vertebrate Dvl proteins.31 Intriguingly, in all six mutations reported here, the frameshift leads to the same open reading frame, which is predicted to generate a PTC within the coding region of exon 15. This most likely produces a novel DVL1 C-terminal truncation that retains 109 amino acids shared by all eight subjects (Figure 2C). Further studies are required to investigate whether DRS-associated mutations in exon 14 of DVL1 produce a protein with residual activity that competes with the wild-type protein and disturbs its function (antemorph) or whether the novel C-terminal domain performs a new, unpredicted biological role (neomorph). In any case, a dominant-negative or gain-of-function altered protein is predicted to contribute to the dominant disease trait.
Nonsense mutations within the last 50 base pairs of the penultimate exon and within the last exon are hypothesized to escape NMD, leading to the synthesis of C-terminally truncated proteins. These truncated proteins can result in diseases as a result of dominant-negative or gain-of-function mechanisms.40 Importantly, a mutant transcript being subjected to NMD or escaping NMD can lead to distinct disease-inheritance patterns and variable clinical severity,15, 25 examples of which include mutations in ROR2: recessive ROR2 mutations cause RRS, whereas dominant ROR2 mutations cause BDB1. SOX10 (MIM 602229) mutations also cause one of two distinct dominant diseases, Waardenburg syndrome (MIM 602229) or PCWH (peripheral demyelinating neuropathy, central dysmyelination, Waardenburg syndrome, and Hirschsprung disease [MIM 609136]), depending on whether the transcript containing the PTC is subjected to or escapes NMD, respectively.41 Recently, de novo truncation in the single-exon gene AHDC1 (MIM 615790) was found in subjects with a new neurological disorder, Xia-Gibbs syndrome (MIM 615829), consisting of developmental delay, hypotonia, mild dysmorphic features, sleep apnea, and other symptoms. Single-exon genes all escape NMD. In aggregate, these observations suggest that the interpretation of the biological effects of nonsense and frameshift alleles can be difficult to predict and most certainly do not always cause loss of function, but rather can be null (haploinsufficiency), hypomorphic, antemorphic, or neomorphic. The presence of frameshift or nonsense mutations that allow transcripts to escape NMD can provide an improved set of tools for identifying mutations associated with human disease.41, 42, 43
Both canonical and non-canonical Wnt signaling, including via Wnt-5a, have been demonstrated to be important effectors of bone mass in humans and animal models.44 Wnt signaling increases osteoblastogenesis, which increases bone mass. As examples, loss-of-function mutations in AMER1 (MIM 300647), a gene that represses canonical Wnt signaling, results in the increased-bone-mass phenotype of osteopathia striata with cranial sclerosis (MIM 300373),45 whereas loss-of-function mutations in LRP5 (MIM 603506), which normally transduces canonical Wnt signaling, results in the decreased-bone-mass phenotype of osteoporosis-pseudoglioma syndrome (MIM 259770).46 Given the increased cranial density seen in a few of our subjects, further investigation for any potential role of DVL1 in regulating bone density is warranted.
No constitutional diseases have yet been associated with specific mutations in human DVL genes. Copy-number alteration spanning 17p13.1, specifically deletion copy-number variation (CNV) that includes DVL2, can lead to intellectual disability and microcephaly.47 DVL1 has been proposed as a candidate gene for the cardiovascular malformations and cardiomyopathy phenotypes observed in individuals with the 1p36 deletion syndrome (MIM 607872).48 Nonetheless, this deletion-CNV syndrome does not include the typical facial and skeletal features observed in Robinow syndrome, supporting the contention that haploinsufficiency is not the mechanism for disease in DRS caused by DVL1 mutations. Loss-of-function mutations in Dvl1−/− mice cause a relatively mild phenotype consisting of behavioral problems. This contrasts sharply with the severe morphological defects observed in mice with null mutations in Wnt5a or Ror2. These observations in mice support our hypothesis that truncated DVL1 in subjects with DRS is probably not due to loss-of-function mutations. Our data suggest that mutations that lead to the formation of truncated DVL1, which has a distinct terminus due to specific −1 frameshifting variants in the penultimate exon, can disrupt the Wnt-5a-ROR2 pathway and cause overlapping clinical phenotypes. We therefore hypothesize that WNT5A, ROR2, and DVL1, similar to their orthologs in other organisms, act in the same pathway in humans.
Acknowledgments
We thank the families and the Robinow Syndrome Foundation for their participation in the study. The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about. This work was supported by the Baylor Hopkins Center for Mendelian Genomics (U54HG006542), jointly funded by the National Human Genome Research Institute (NHGRI) and NHBLI, a grant (U54HG003273) to R.A.G., the Fundação de Amparo à Pesquisa do Estado de Minas Gerais (CBB-APQ-01456-13), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (476217/2013-0), a Science without Borders program Young Investigator fellowship grant (402520/2012-2) to C.M.B.C., the Netherlands Organization for Scientific Research ZonMW Veni grant (916-12-095) to A.H., and a Scientific and Technological Research Council of Turkey (TUBITAK) grant (SBAG-112S398, CRANIRARE-2, ERA-NET project) to H.K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHGRI, NHBLI, or NIH. J.R.L. holds stock ownership in 23andMe and Lasergen, is a paid consultant for Regeneron Pharmaceuticals, and is a co-inventor of multiple United States and European patents related to molecular diagnostics. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered in the Baylor-Miraca Medical Genetics Laboratories (https://www.bcm.edu/geneticlabs/).
Published: March 26, 2015
Footnotes
Supplemental Data include detailed clinical case reports of the DRS-affected individuals and three figures and can be found with this article online at http://dx.doi.org/10.1016/j.ajhg.2015.02.015.
Accession Numbers
The dbGaP accession number for the WES-study DNA sequences reported in this paper is phs000711 under the Baylor Hopkins Center for Mendelian Genomics (BHCMG) study. Sample identifiers are SAMN03361066, SAMN03361124, SAMN03360998, SAMN03361122, SAMN03361069, SAMN03361195, SAMN03361120, SAMN03361178, SAMN03361186, SAMN03361014, and SAMN03361076.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Atherosclerosis Risk in Communities Study (ARIC) database, http://www2.cscc.unc.edu/aric/
BCM-HGSC Mercury analysis pipeline, https://www.hgsc.bcm.edu/software/mercury
ExAC Browser, http://exac.broadinstitute.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
Supplemental Data
References
- 1.Robinow M., Silverman F.N., Smith H.D. A newly recognized dwarfing syndrome. Am. J. Dis. Child. 1969;117:645–651. doi: 10.1001/archpedi.1969.02100030647005. [DOI] [PubMed] [Google Scholar]
- 2.Mazzeu J.F., Pardono E., Vianna-Morgante A.M., Richieri-Costa A., Ae Kim C., Brunoni D., Martelli L., de Andrade C.E., Colin G., Otto P.A. Clinical characterization of autosomal dominant and recessive variants of Robinow syndrome. Am. J. Med. Genet. A. 2007;143:320–325. doi: 10.1002/ajmg.a.31592. [DOI] [PubMed] [Google Scholar]
- 3.Patton M.A., Afzal A.R. Robinow syndrome. J. Med. Genet. 2002;39:305–310. doi: 10.1136/jmg.39.5.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Person A.D., Beiraghi S., Sieben C.M., Hermanson S., Neumann A.N., Robu M.E., Schleiffarth J.R., Billington C.J., Jr., van Bokhoven H., Hoogeboom J.M. WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev. Dyn. 2010;239:327–337. doi: 10.1002/dvdy.22156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roifman M., Marcelis C.L., Paton T., Marshall C., Silver R., Lohr J.L., Yntema H.G., Venselaar H., Kayserili H., van Bon B., FORGE Canada Consortium De novo WNT5A-associated autosomal dominant Robinow syndrome suggests specificity of genotype and phenotype. Clin. Genet. 2015;87:34–41. doi: 10.1111/cge.12401. [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi T.P., Bradley A., McMahon A.P., Jones S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–1223. doi: 10.1242/dev.126.6.1211. [DOI] [PubMed] [Google Scholar]
- 7.Chawengsaksophak K., Svingen T., Ng E.T., Epp T., Spiller C.M., Clark C., Cooper H., Koopman P. Loss of Wnt5a disrupts primordial germ cell migration and male sexual development in mice. Biol. Reprod. 2012;86:1–12. doi: 10.1095/biolreprod.111.095232. [DOI] [PubMed] [Google Scholar]
- 8.Wadia R.S. Covesdem syndrome. J. Med. Genet. 1979;16:162. doi: 10.1136/jmg.16.2.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wadia R.S., Shirole D.B., Dikshit M.S. Recessively inherited costovertebral segmentation defect with mesomelia and peculiar facies (Covesdem syndrome): A new genetic entity? J. Med. Genet. 1978;15:123–127. doi: 10.1136/jmg.15.2.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Afzal A.R., Rajab A., Fenske C.D., Oldridge M., Elanko N., Ternes-Pereira E., Tüysüz B., Murday V.A., Patton M.A., Wilkie A.O., Jeffery S. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat. Genet. 2000;25:419–422. doi: 10.1038/78107. [DOI] [PubMed] [Google Scholar]
- 11.van Bokhoven H., Celli J., Kayserili H., van Beusekom E., Balci S., Brussel W., Skovby F., Kerr B., Percin E.F., Akarsu N., Brunner H.G. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat. Genet. 2000;25:423–426. doi: 10.1038/78113. [DOI] [PubMed] [Google Scholar]
- 12.Oishi I., Suzuki H., Onishi N., Takada R., Kani S., Ohkawara B., Koshida I., Suzuki K., Yamada G., Schwabe G.C. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells. 2003;8:645–654. doi: 10.1046/j.1365-2443.2003.00662.x. [DOI] [PubMed] [Google Scholar]
- 13.Sokol S. A role for Wnts in morpho-genesis and tissue polarity. Nat. Cell Biol. 2000;2:E124–E125. doi: 10.1038/35017136. [DOI] [PubMed] [Google Scholar]
- 14.Yamanaka H., Moriguchi T., Masuyama N., Kusakabe M., Hanafusa H., Takada R., Takada S., Nishida E. JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements in vertebrates. EMBO Rep. 2002;3:69–75. doi: 10.1093/embo-reports/kvf008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben-Shachar S., Khajavi M., Withers M.A., Shaw C.A., van Bokhoven H., Brunner H.G., Lupski J.R. Dominant versus recessive traits conveyed by allelic mutations - to what extent is nonsense-mediated decay involved? Clin. Genet. 2009;75:394–400. doi: 10.1111/j.1399-0004.2008.01114.x. [DOI] [PubMed] [Google Scholar]
- 16.Oldridge M., Fortuna A.M., Maringa M., Propping P., Mansour S., Pollitt C., DeChiara T.M., Kimble R.B., Valenzuela D.M., Yancopoulos G.D., Wilkie A.O. Dominant mutations in ROR2, encoding an orphan receptor tyrosine kinase, cause brachydactyly type B. Nat. Genet. 2000;24:275–278. doi: 10.1038/73495. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z., Liu X., Yang B.Z., Gelernter J. The role and challenges of exome sequencing in studies of human diseases. Front. Genet. 2013;4:160. doi: 10.3389/fgene.2013.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saraiva J.M., Cordeiro I., Santos H.G. Robinow syndrome in monozygotic twins with normal stature. Clin. Dysmorphol. 1999;8:147–150. [PubMed] [Google Scholar]
- 19.Bainbridge M.N., Wang M., Wu Y., Newsham I., Muzny D.M., Jefferies J.L., Albert T.J., Burgess D.L., Gibbs R.A. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol. 2011;12:R68. doi: 10.1186/gb-2011-12-7-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Challis D., Yu J., Evani U.S., Jackson A.R., Paithankar S., Coarfa C., Milosavljevic A., Gibbs R.A., Yu F. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics. 2012;13:8. doi: 10.1186/1471-2105-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reid J.G., Carroll A., Veeraraghavan N., Dahdouli M., Sundquist A., English A., Bainbridge M., White S., Salerno W., Buhay C. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics. 2014;15:30. doi: 10.1186/1471-2105-15-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kishida S., Yamamoto H., Hino S., Ikeda S., Kishida M., Kikuchi A. DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta-catenin stability. Mol. Cell. Biol. 1999;19:4414–4422. doi: 10.1128/mcb.19.6.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krylova O., Messenger M.J., Salinas P.C. Dishevelled-1 regulates microtubule stability: a new function mediated by glycogen synthase kinase-3beta. J. Cell Biol. 2000;151:83–94. doi: 10.1083/jcb.151.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponting C.P., Bork P. Pleckstrin’s repeat performance: a novel domain in G-protein signaling? Trends Biochem. Sci. 1996;21:245–246. [PubMed] [Google Scholar]
- 25.Khajavi M., Inoue K., Lupski J.R. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 2006;14:1074–1081. doi: 10.1038/sj.ejhg.5201649. [DOI] [PubMed] [Google Scholar]
- 26.Moon R.T., Campbell R.M., Christian J.L., McGrew L.L., Shih J., Fraser S. Xwnt-5A: a maternal Wnt that affects morphogenetic movements after overexpression in embryos of Xenopus laevis. Development. 1993;119:97–111. doi: 10.1242/dev.119.1.97. [DOI] [PubMed] [Google Scholar]
- 27.Rauch G.J., Hammerschmidt M., Blader P., Schauerte H.E., Strähle U., Ingham P.W., McMahon A.P., Haffter P. Wnt5 is required for tail formation in the zebrafish embryo. Cold Spring Harb. Symp. Quant. Biol. 1997;62:227–234. [PubMed] [Google Scholar]
- 28.Noordermeer J., Klingensmith J., Perrimon N., Nusse R. dishevelled and armadillo act in the wingless signalling pathway in Drosophila. Nature. 1994;367:80–83. doi: 10.1038/367080a0. [DOI] [PubMed] [Google Scholar]
- 29.Klingensmith J., Nusse R., Perrimon N. The Drosophila segment polarity gene dishevelled encodes a novel protein required for response to the wingless signal. Genes Dev. 1994;8:118–130. doi: 10.1101/gad.8.1.118. [DOI] [PubMed] [Google Scholar]
- 30.Gao C., Chen Y.G. Dishevelled: The hub of Wnt signaling. Cell. Signal. 2010;22:717–727. doi: 10.1016/j.cellsig.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 31.Semënov M.V., Snyder M. Human dishevelled genes constitute a DHR-containing multigene family. Genomics. 1997;42:302–310. doi: 10.1006/geno.1997.4713. [DOI] [PubMed] [Google Scholar]
- 32.Hamblet N.S., Lijam N., Ruiz-Lozano P., Wang J., Yang Y., Luo Z., Mei L., Chien K.R., Sussman D.J., Wynshaw-Boris A. Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development. 2002;129:5827–5838. doi: 10.1242/dev.00164. [DOI] [PubMed] [Google Scholar]
- 33.Etheridge S.L., Ray S., Li S., Hamblet N.S., Lijam N., Tsang M., Greer J., Kardos N., Wang J., Sussman D.J. Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 2008;4:e1000259. doi: 10.1371/journal.pgen.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee Y.N., Gao Y., Wang H.Y. Differential mediation of the Wnt canonical pathway by mammalian Dishevelleds-1, -2, and -3. Cell. Signal. 2008;20:443–452. doi: 10.1016/j.cellsig.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto S., Jaiswal M., Charng W.L., Gambin T., Karaca E., Mirzaa G., Wiszniewski W., Sandoval H., Haelterman N.A., Xiong B. A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell. 2014;159:200–214. doi: 10.1016/j.cell.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sussman D.J., Klingensmith J., Salinas P., Adams P.S., Nusse R., Perrimon N. Isolation and characterization of a mouse homolog of the Drosophila segment polarity gene dishevelled. Dev. Biol. 1994;166:73–86. doi: 10.1006/dbio.1994.1297. [DOI] [PubMed] [Google Scholar]
- 37.Lijam N., Paylor R., McDonald M.P., Crawley J.N., Deng C.X., Herrup K., Stevens K.E., Maccaferri G., McBain C.J., Sussman D.J., Wynshaw-Boris A. Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell. 1997;90:895–905. doi: 10.1016/s0092-8674(00)80354-2. [DOI] [PubMed] [Google Scholar]
- 38.Conti E., Izaurralde E. Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr. Opin. Cell Biol. 2005;17:316–325. doi: 10.1016/j.ceb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Popp M.W., Maquat L.E. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu. Rev. Genet. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng H.X., Jiang H., Fu R., Zhai H., Shi Y., Liu E., Hirano M., Dal Canto M.C., Siddique T. Molecular dissection of ALS-associated toxicity of SOD1 in transgenic mice using an exon-fusion approach. Hum. Mol. Genet. 2008;17:2310–2319. doi: 10.1093/hmg/ddn131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia F., Bainbridge M.N., Tan T.Y., Wangler M.F., Scheuerle A.E., Zackai E.H., Harr M.H., Sutton V.R., Nalam R.L., Zhu W. De novo truncating mutations in AHDC1 in individuals with syndromic expressive language delay, hypotonia, and sleep apnea. Am. J. Hum. Genet. 2014;94:784–789. doi: 10.1016/j.ajhg.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simpson M.A., Irving M.D., Asilmaz E., Gray M.J., Dafou D., Elmslie F.V., Mansour S., Holder S.E., Brain C.E., Burton B.K. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 2011;43:303–305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- 43.Isidor B., Lindenbaum P., Pichon O., Bézieau S., Dina C., Jacquemont S., Martin-Coignard D., Thauvin-Robinet C., Le Merrer M., Mandel J.L. Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat. Genet. 2011;43:306–308. doi: 10.1038/ng.778. [DOI] [PubMed] [Google Scholar]
- 44.Okamoto M., Udagawa N., Uehara S., Maeda K., Yamashita T., Nakamichi Y., Kato H., Saito N., Minami Y., Takahashi N., Kobayashi Y. Noncanonical Wnt5a enhances Wnt/β-catenin signaling during osteoblastogenesis. Sci. Rep. 2014;4:4493. doi: 10.1038/srep04493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jenkins Z.A., van Kogelenberg M., Morgan T., Jeffs A., Fukuzawa R., Pearl E., Thaller C., Hing A.V., Porteous M.E., Garcia-Miñaur S. Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat. Genet. 2009;41:95–100. doi: 10.1038/ng.270. [DOI] [PubMed] [Google Scholar]
- 46.Gong Y., Slee R.B., Fukai N., Rawadi G., Roman-Roman S., Reginato A.M., Wang H., Cundy T., Glorieux F.H., Lev D., Osteoporosis-Pseudoglioma Syndrome Collaborative Group LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 47.Carvalho C.M., Vasanth S., Shinawi M., Russell C., Ramocki M.B., Brown C.W., Graakjaer J., Skytte A.B., Vianna-Morgante A.M., Krepischi A.C. Dosage changes of a segment at 17p13.1 lead to intellectual disability and microcephaly as a result of complex genetic interaction of multiple genes. Am. J. Hum. Genet. 2014;95:565–578. doi: 10.1016/j.ajhg.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zaveri H.P., Beck T.F., Hernández-García A., Shelly K.E., Montgomery T., van Haeringen A., Anderlid B.M., Patel C., Goel H., Houge G. Identification of critical regions and candidate genes for cardiovascular malformations and cardiomyopathy associated with deletions of chromosome 1p36. PLoS ONE. 2014;9:e85600. doi: 10.1371/journal.pone.0085600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.