

Abstract
We have identified TUBGCP4 variants in individuals with autosomal-recessive microcephaly and chorioretinopathy. Whole-exome sequencing performed on one family with two affected siblings and independently on another family with one affected child revealed compound-heterozygous mutations in TUBGCP4. Subsequent Sanger sequencing was performed on a panel of individuals from 12 French families affected by microcephaly and ophthalmic manifestations, and one other individual was identified with compound-heterozygous mutations in TUBGCP4. One synonymous variant was common to all three families and was shown to induce exon skipping; the other mutations were frameshift mutations and a deletion. TUBGCP4 encodes γ-tubulin complex protein 4, a component belonging to the γ-tubulin ring complex (γ-TuRC) and known to regulate the nucleation and organization of microtubules. Functional analysis of individual fibroblasts disclosed reduced levels of the γ-TuRC, altered nucleation and organization of microtubules, abnormal nuclear shape, and aneuploidy. Moreover, zebrafish treated with morpholinos against tubgcp4 were found to have reduced head volume and eye developmental anomalies with chorioretinal dysplasia. In summary, the identification of TUBGCP4 mutations in individuals with microcephaly and a spectrum of anomalies in eye development, particularly photoreceptor anomalies, provides evidence of an important role for the γ-TuRC in brain and eye development.
Main Text
A rare subset of syndromic inherited congenital microcephalies is associated with chorioretinal dysplasia. Inheritance has been reported to be either autosomal dominant or autosomal recessive. Autosomal-dominant microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR [MIM 152950]), also known as Alzial syndrome,1 has recently been attributed to mutations in KIF11 (MIM 148760), the gene encoding kinesin motor protein Eg5, a homotetramer kinesin motor.2 Autosomal-recessive microcephaly and chorioretinopathy (MCCRP [MIM 251270]) is a very rare entity. It was reported by McKusick in 1966 in an inbred Mennonite family and then reported in a dozen families worldwide.3 In MCCRP, microcephaly ranges from mild to severe and has variable impact on cognitive performance, ranging from moderate developmental delay to normal intelligence.4 The MCCRP phenotypic eye spectrum has also been reported as variable, but the chorioretinopathy is a constant feature and includes typical punched-out lesions that can severely impair vision and occasional retinal folds that can progress to retinal detachment. To date, the original MCCRP-affected family described by McKusick has been shown to carry a read-through variant in TUBGCP65 (tubulin, gamma complex associated protein 6 [MIM 610053]), and more recently, four additional individuals (in three families) with primordial dwarfism, microcephaly, and retinopathy have been reported to have TUBGCP6 mutations.6
We used whole-exome sequencing to investigate families affected by autosomal-recessive microcephaly and developmental eye disease fitting the MCCRP phenotype. Subjects were recruited from genetic and ophthalmic clinics in France and the UK. Informed consent was obtained for all subjects. Ethical approval was obtained from the local ethics committees at Hôpitaux Universitaires de Strasbourg (Strasbourg University Hospital), Moorfields Eye Hospital, and Robert Debré Hospital. Whole-exome sequencing was performed independently in the French and the British families, and systemic and ophthalmological examinations were performed for all families affected by mutations in TUBGCP4 (tubulin, gamma complex associated protein 4 [MIM 609610]) at the respective clinics (Table 1).
Table 1.
Summary of the Clinical and Molecular Data of Individuals with TUBGCP4 Mutations
|
Individuals |
||||
|---|---|---|---|---|
| AII-1 | AII-3 | BII-2 | CII-1 | |
| Mutations in TUBGCP4 | c.[579dupT];[1746G>T] | c.[579dupT];[1746G>T] | c.[1732-?_∗544+?del];[1746G>T] | c.[298delT];[1746G>T] |
| Predicted protein changes in TUBGCP4 | p.[Gly194Trpfs∗8];[=] | p.[Gly194Trpfs∗8];[=] | p.[?];[=] | p.[Tyr100Ilefs∗27];[=] |
| Congenital microcephaly (OFC in SD) | + (<−3) | + (<−4) | + (<−3) | + (<−4) |
| Learning difficulties | lower range of intellectual ability | + (mild) | + | + |
| Cerebral MRI (age at evaluation) | thin corpus callosum (3 months old) | normal limits (3 months old) | normal limits (5 years old) | normal limits (7 months old) |
| Ocular Features at Last Examination (Age) | 9 Years Old | 3 Years Old | 5 Years Old | 8 Years Old |
| Fundus examination | ||||
| Multiple punched-out lesions | + | + | + | + |
| Chorioretinal fold | + | − | + | + |
| Other | retinal detachment | − | − | optic nerve hypoplasia |
| Microphthalmia | + | + | − | + |
| Hyperopia | R: +1.75D L: +1.75D |
R: +8.25D L: +8D |
R: +4.5D L: +6.5D |
R: +9D L: +9D |
| Visual acuity (logMAR value) | R: +1 L: NL |
R: +0.6 L: +0.6 |
R: +1.2 at 1 month L: +1.3 at 1 month |
R: +1.3 L: +1.3 |
| ERG | NA | NA | residual response | no response |
Abbreviations are as follows: D, diopter; L, left; NA, not available; NL, no light perception; OFC, occipital frontal circumference; and R, right.
Family A consists of two affected French siblings (one boy and one girl) and one healthy brother born to non-consanguineous parents. The eldest affected brother (AII-1) was referred to the Strasbourg national center for genetic eye diseases (Centre de Référence pour les Affections Rares en Génétique Ophtalmologique) because of chorioretinal dysplasia associated with congenital microcephaly. His fundus examination revealed multiple punched-out retinal lesions on the right and a large retinal fold on the left, which was later complicated by total retinal detachment leading to blindness in that eye at the age of 7 years (Table 1; Figures 1A and 1B). He has normal growth and development. He walked at 16 months and had no significant language delay. His neurological examination was normal. Brain MRI performed in the first few months of life revealed a thin and shortened corpus callosum but no other anomaly except microcephaly. He had a horseshoe kidney. He is now 9 years old, and although he is on the lower range of intellectual ability, he attends a normal school. The facial gestalt shows mainly downslanting palpebral fissures, large ears, large first incisors, and mild retrognathism. His younger sister (AII-3) was diagnosed with microcephaly at birth (Figure 1B). She was noted to have bilateral visual impairment and nystagmus, and this prompted fundus examination, which showed bilateral punched-out retinal lesions. She has normal growth. She walked at 18 months and has no significant language delay. Further neurological examination was normal. Her MRI scan was technically poor but showed no major anomaly (Table 1). Morphological examination showed the same features as in her brother.
Figure 1.
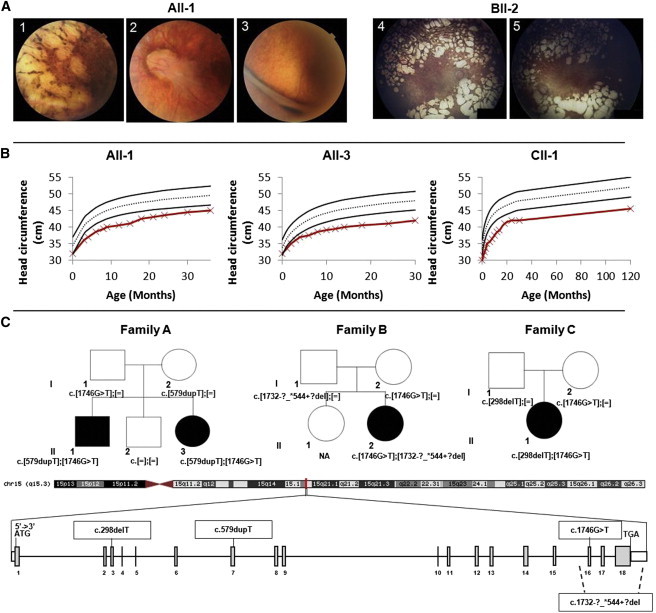
Clinical Presentation and Pedigrees of Families Affected by Microcephaly and Chorioretinal Dysplasia and TUBGCP4 Mutations
(A) Fundus photographs of individual AII-1 show, on the right side, multiple punched-out retinal lesions (1) and widespread pigmentary changes and, on the left side, a large retinal fold emerging from the optic disc (2) and a nasal extension (3) complicated a few months later by total retinal detachment (not shown). Fundus photographs of individual BII-2 show multiple areas of chorioretinal atrophy with punched-out lesions, absence of retinal vessels, and optic nerve remnants (4 and 5).
(B) Head circumference of individuals AII-1, AII-3, and CII-1 shows congenital microcephaly (affected individuals’ curves are in red, and the black curves show the SD).
(C) Schematic representation of TUBGCP4: this gene extends over 38 kb in chromosomal region 15q15.3 and contains 18 exons (vertical gray hatches). The positions of the start codon (ATG) and the stop codon (TGA) are indicated. The mutations detected in this study occur in exons 3, 7, and 16. The deletion includes exons 16–18. The segregation analysis confirmed autosomal-recessive inheritance (affected individuals are blackened). In the family A pedigree, affected individuals (AII-1 and AII-3) harbor compound-heterozygous mutations c.[578_579insT];[1746G>T] in TUBGCP4. In the family B pedigree, the affected girl (BII-2) harbors compound-heterozygous mutations c.[298delT];[1746G>T]. In the family C pedigree, the affected girl (CII-1) harbors compound-heterozygous mutations c.[1746G>T];[1732-?_∗544+?del]. NA indicates not available.
We performed whole-exome sequencing for the two affected siblings (AII-1 and AII-3), the healthy brother (AII-2), and the healthy mother (AI-2). Exons of DNA samples were captured with in-solution enrichment methodology (Agilent SureSelect All Exon XT2 50 Mb Kit) and sequenced with an Illumina HiSeq 2500 instrument, resulting in a coverage of at least ten reads for more than 93% of the targeted exomes. SNPs and indels were called with the Genome Analysis Toolkit v.2.5.2, and 60,041–62,197 genetic variants per proband were identified (Tables S1 and S4). Identification of the disease-causing variants was performed with the VaRank program for the annotation, ranking, and filtering of genetic variants. Very stringent criteria were used for excluding the following non-pathogenic variants in particular: (1) variants represented with an allele frequency of more than 1% in dbSNP 138 or the NHLBI Exome Sequencing Project Exome Variant Server (EVS), (2) variants found at the homozygous state or more than once at the heterozygous state in 48 control exomes, (3) variants in the 5′ or 3′ UTR, (4) variants with intronic locations and no prediction of local splice effect, and (5) synonymous variants without prediction of local splice effect. Finally, we identified three candidates genes (TRPM2 [transient receptor potential cation channel, subfamily M, member 2 (MIM 603749)], ERICH6B [glutamate-rich 6B], and TUBGCP4) with compound-heterozygous variants present only in the affected offspring and with one heterozygous variant found in the mother (filtering by segregation). TRPM2 was excluded because the two variants (c.3653C>A [p.Ala1218Glu] [RefSeq accession number NM_003307.3] and c.1762C>T [p.Leu588Phe] [RefSeq NM_003307.3]) were not predicted to be pathogenic according to SIFT7 and PolyPhen-2.8 Moreover, this gene was implicated in inflammation rather than developmental disease according to the animal model.9 In the second gene, ERICH6B, one variant was at the end of six reads and was probably an artifact, and the second variant was in a GGGG repeat and was probably not pathogenic. We then focused the subsequent study on compound-heterozygous variants in TUBGCP4: a heterozygous frameshift (c.579dupT [p.Gly194Trpfs∗8] [RefSeq NM_014444.2]) and a heterozygous synonymous variant with a strongly activated cryptic acceptor (c.1746G>T [p.=] [RefSeq NM_014444.2]). Both mutations are rare—their frequencies in the NHLBI EVS and ExAC are 1/11,597 and 14/120,608 (MAF = 0.0001161), respectively, for c.579dupT and 6/12,096 and 24/66,538 (MAF = 0.00036), respectively, for c.1746G>T (Figure 1C).
Family B consists of one affected child (BII-2) and one healthy sister born to non-consanguineous parents from the UK. The girl was born at 36 weeks of gestation after a normal delivery. Intrauterine growth retardation was noticed. At 6 weeks of age, she was noticed to have microcephaly with nystagmus and poor vision. MRI scans performed when she was 3 months and 5 years of age were within normal limits. Her general health was good, but she was developmentally delayed and had learning difficulties. Pattern visual evoked potentials indicated rudimentary to poor vision levels for both eyes. Full-field electroretinography (ERG) was markedly attenuated with a small residual response, indicating severe rod and cone dysfunction. Fundus examination showed multiple round areas of chorioretinal atrophy (Figure 1A), and fluorescein angiogram showed absent retinal circulation but the presence of choroidal circulation. Vision remained very poor on follow-up (Table 1). For exome sequencing purposes, the DNA was analyzed by high-throughput sequencing (exon capture: Illumina TruSeq or Nextera Kit; sequencing: Illumina HiSeq 2000) at AROS Applied Biotechnology. Reads were aligned to the hg19 human reference sequence (UCSC Genome Browser) with Novoalign (Novocraft) v.2.05. The ANNOVAR tool10 was used to annotate SNPs and small indels. Analysis of copy-number variation was performed with ExomeDepth11 on the basis of read-depth data. We identified the two compound-heterozygous variants in TUBGCP4: c.1746G>T and c.1732-?_∗544+?del (RefSeq NM_014444.2; corresponding to a deletion of exons 16–18, including at least 544 nucleotides of the 3′ UTR) (Figure 1C).
To explore additional cases of autosomal-recessive MCCRP, we performed Sanger sequencing of TUBGCP4 (primers used are detailed in Table S2) on a cohort of 12 additional French microcephaly-affected families reported to also have eye anomalies (these families were included in the National Programme Hospitalier de Recherche Clinique study carried out at the Robert Debré Hospital in Paris). In one French family (family C) with one affected girl (CII-1), we identified two mutations in TUBGCP4: c.298delT (p.Tyr100Ilefs∗27) (RefSeq NM_014444.2) and again the c.1746G>T synonymous mutation (Figure 1C). The individual had been referred by the University Hospital of Tours and presented with congenital microcephaly (Figure 1B). Her height was unremarkable, but she had been progressively overweight (+3 SDs) since the age of 2 years. She walked at 16 months, and her speech was delayed. The presence of nystagmus prompted an ocular examination, which revealed numerous punched-out chorioretinal lesions throughout the retina, optic nerve hypoplasia, and absence of retinal vessels (Table 1; Figure 1A). ERG showed absent responses when she was 6 months and 8 months of age.
Thus, we report herein on three families who are affected by autosomal-recessive MCCRP and carry compound-heterozygous TUBGCP4 mutations. They all share the same synonymous variant (c.1746G>T), and each has an additional severe mutation (one of two frameshifts [c.579dupT (p.Gly194Trpfs∗8) and c.298delT (p.Tyr100Ilefs∗27)] or one three-exon deletion).
The synonymous c.1746G>T mutation is predicted to have a local splice effect as a strong cryptic acceptor site according to Alamut (Interactive Biosoftware). We performed reverse transcription of mRNA obtained from skin fibroblasts of individual AII-1 and from a healthy unrelated control individual, and we then sequenced the full-length cDNA from both individuals. We obtained three PCR products after amplification of the cDNA of AII-1 and only one PCR product for the control individual (Figure S1). From the cDNA of AII-1, the shortest product (found only in the affected individual) was sequenced and showed exon 16 skipping. The affected individual’s two remaining PCR products, found in the same position as in the control individual (a 1-bp difference), showed on sequencing the maternal allele carrying c.579dupT and the paternal allele carrying c.1746G>T. Together, the presence of three cDNA products—maternal cDNA, paternal cDNA without exon 16, and paternal cDNA with functional exon 16—suggests that the splicing defect results in alternative exon 16 skipping and thus generates a population of unstable mRNA (without exon 16) and residual production of non-spliced cDNA containing exon 16. Moreover, qPCR on cDNA extracted from skin fibroblasts revealed that the amount of TUBGCP4 was drastically lower in the affected individual than in the control individual (Figure S1).
We then examined the cellular consequences of the mutations in fibroblasts derived from a skin biopsy in the index individual, AII-1. Fibroblasts were grown at 37°C in Dulbecco’s modified Eagle’s medium containing 15% fetal calf serum. For immunofluorescence, cells grown on coverslips were fixed in methanol at −20°C and processed according to conventional protocols. Fluorescence microscopy was performed on a microscope (Axiovert, Carl Zeiss) with a 63× objective (1.4 numerical aperture). Images were acquired with a camera (Axiocam MRm, Carl Zeiss) and processed with Adobe Photoshop. For immunoblotting, 40 μg of cell extracts was submitted to SDS-PAGE and immunoblotted with the Odyssey imaging system (Li-Cor Biosciences). For visualization of γ-tubulin ring complexes (γ-TuRCs), extracts were submitted to sucrose gradient sedimentation as described in Haren et al.12 Quantification of protein intensities was performed with Odyssey software. Cells grown on fibronectin-coated coverslips were transferred on ice for 45 min and then into prewarmed medium at 25°C. At the indicated time points, coverslips were transferred into permeabilization buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2, 0.1% saponin, pH 6.9) for 40 s before methanol fixation.
Immunoblotting with two different antibodies revealed a reduction of GCP4 (gamma-tubulin complex protein 4, encoded by TUBGCP4) levels to approximately 40% of that in control fibroblasts (Figure 2A). No smaller GCP4 variants were detected, indicating that any potential translation products without the full complement of exons are unstable. Reduced levels of GCP4 were confirmed by immunofluorescence of the centrosomes, both in interphase and in mitosis (Figures 2B and 2C). Further examination by immunoblot revealed that levels of all other components (γ-tubulin and GCP2, GCP5, and GCP6) of the γ-TuRC were also reduced. Moreover, we observed a loss of more than 60% of γ-TuRCs from sucrose gradients (Figure 2F, fraction 7) in favor of smaller complexes. This was expected, given that the integrity of the γ-TuRC depends on proteins GCP4, GCP5, and GCP6, and lowering the levels of any of these proteins affects the assembly of γ-TuRCs or destabilizes them.14
Figure 2.
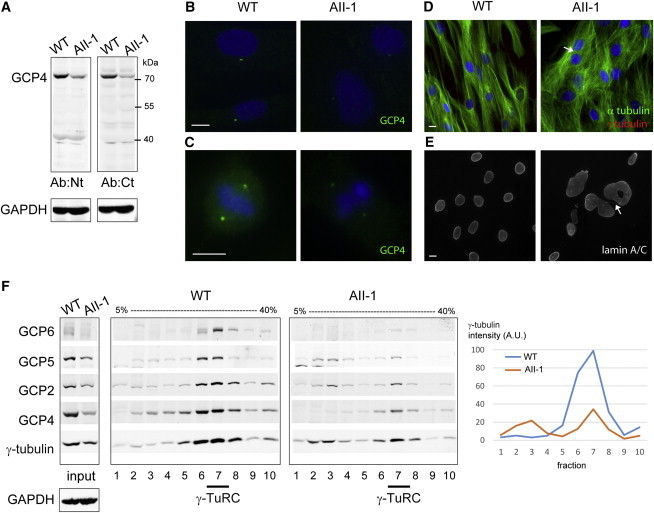
Fibroblasts from Individual AII-1 Have Reduced Amounts of GCP4 and γ-TuRCs and Show Aneuploidy
(A) Immunoblot shows lower GCP4 levels in the fibroblasts from individual AII-1 than in control fibroblasts. Antibodies against the N-terminal domain (Ab:Nt) or a C-terminal peptide (Ab:Ct) of GCP4 were used. GAPDH was used as a loading control.
(B and C) Immunofluorescence of GCP4 (green) shows lower levels in AII-1 fibroblasts than in control fibroblasts in interphase (B) and mitosis (C).
(D) Immunofluorescence of microtubules (green) and γ-tubulin (red) shows abnormal microtubule organization and altered cell morphology in AII-1 fibroblasts. The arrow points to a binucleated cell.
(E) Immunofluorescence of lamin A/C shows nuclear defects in AII-1 fibroblasts: large nuclei, abnormal shapes, and multiple nuclei, connected by chromatin bridges (arrow), per cell.
(F) Immunoblots of extracts from control and AII-1 fibroblasts before (input) or after fractionation in gradients of 5%–40% sucrose. Antibodies against γ-tubulin and GCP2, GCP4, GCP5, and GCP6 were used for visualizing γ-TuRC components. The peak position of the γ-TuRC (fraction 7) is indicated below the figure. On the right, the quantification of γ-tubulin signal intensity shows a 63% reduction of the γ-TuRC in fraction 7 from AII-1 fibroblasts.
DNA is blue in merged images. Scale bars represent 10 μm. Antibodies are anti-GCP4 Ab:Nt (R7629/30), Ab:Ct (R801),13 anti-GCP2,12 anti-γ-tubulin TU-30 (Abcam), anti-GCP5 E-1 (Santa Cruz biotech.), anti-GCP6 (Abcam), anti-α-tubulin (Sigma), and anti-lamin A/C (Santa Cruz Biotech).
Consequently, cells from individual AII-1 showed reduced levels of microtubule nucleation after depolymerization in the cold (Figure S2B). We also observed that microtubule organization was abnormal, such that microtubule arrays radiating from the centrosomes were less pronounced and there were fewer microtubules orienting along the long axis of the cells (Figure 2D). This was accompanied by changes in cellular shape, such that affected cells were less elongated than control cells (Figure 2D). In mitosis, microtubule organization of the spindle apparatus seemed normal at first glance, although γ-tubulin staining was reduced in mutant fibroblasts (Figure S2A). However, in interphase, we observed a 9-fold increase in nuclear anomalies, including binucleated cells, increased nuclear size, and abnormal nuclear shape, such as doughnut or dumbbell shapes (2.5% in controls and 22% in mutants; Figure 2E). These anomalies mark the presence of aneuploid cells in the population of mutant fibroblasts. They can be explained by defects occurring late in mitosis, such as during cytokinesis (e.g., see chromatin bridge in Figure 2E, arrow).
Next, we used the zebrafish model to elucidate the roles of GCP4 during embryonic development. The GCP4 zebrafish ortholog (ENSDART00000009649) exhibits 88% identity with the human protein. In situ hybridization revealed ubiquitous expression of tubgcp4 mRNA at 48 hr post-fertilization (hpf) (data not shown). A morpholino was designed against the start codon of its mRNA (tubgcp4-mo, 0.9 mM) and injected into zebrafish embryos (morpholino sequences are detailed in Table S3). Injection of tubgcp4-mo caused 70% (n = 240) of embryos to have a smaller head and eyes, as well as a slightly shorter body axis (“reduced tail,” Figures 3A–3D). Eye diameter was reduced by 28% (n = 20, ±6%) at 48 hpf and by 24% (n = 20; ±5%) at 72 hpf (Figures 3E–3G). In addition, 20% of morphants (n = 240) had a severe reduction of the body axis (dwarf phenotype, Figure 3C). Sections through the head of the less strongly affected embryos (Figures 3B and 3F) confirmed the reduction of the size of the head and eyes (Figures 3I and 3J). In contrast, only 3% of uninjected embryos (n = 96) had a smaller head and eyes, and only 6% of embryos injected with three different control morpholinos (cont-mo) of unrelated genes showed these phenotypes (n = 366, data not shown). Partial rescue was obtained by co-injection of 50 ng/μl tubgcp4-mo-resistant mRNA. At 3 days post-fertilization (dpf), the proportion of wild-type embryos was increased by a factor of two, and the proportion of embryos with a “reduced tail” or “dwarf” phenotype was reduced accordingly (Figure 3H, n > 200). The retina of morphants showed all the expected layers. However, the retina was punctuated with rounded cells, probably representing photoreceptors that did not mature (Figures 3K and 3L, red arrow head). Retinal differentiation in control embryos is basically completed at 72 hpf.15 Specific immunostaining of the double-cone photoreceptor subtype by Zpr-1 antibody15,16 revealed a continuous, uniform layer of elongated photoreceptor cells in uninjected and cont-mo-injected embryos (Figures 3M and 3O), whereas the morphants displayed only a single patch of immunofluorescent cones (Figure 3N). This lack of cones was confirmed with a second morpholino directed against the exon-intron boundary (0.9 mM; Figures S3A–S3D). Similar to the tubgcp4-mo, the “spliced morpholino” caused a reduced size of the head and eyes (75% embryos, n = 200). In addition, cones as revealed by immunostaining with Zpr-1 antibody developed only in a restricted area, whereas cones covered the whole retina in control embryos (Figure S3E; data not shown). Section through embryos exhibiting discontinuous Zpr-1 staining revealed the presence of a distinct layer corresponding to the photoreceptor layer (Figure S3F). The same restricted Zpr-1 immunostaining was obtained at 4 and 5 dpf, excluding the possibility that the morphants were simply developmentally delayed (Figure S3G; data not shown). The corresponding control embryos revealed a thick and well-organized photoreceptor layer (Figures 3P, 3S, and 3U). Next, we stained rod photoreceptors with the 1D1 antibody directed against rhodopsin.17 In the uninjected and cont-mo-injected embryos, 1D1-positive cells were scattered in the photoreceptor layer (Figure 3V; data not shown), whereas no specific staining was detected in the morphants (Figure 3W). Taken together, our results indicate that the retina of morphants is composed of photoreceptors that have not fully differentiated. Although we regard it as less likely, we cannot totally exclude, however, that the rounded cells are the result of degeneration of differentiated photoreceptors.
Figure 3.
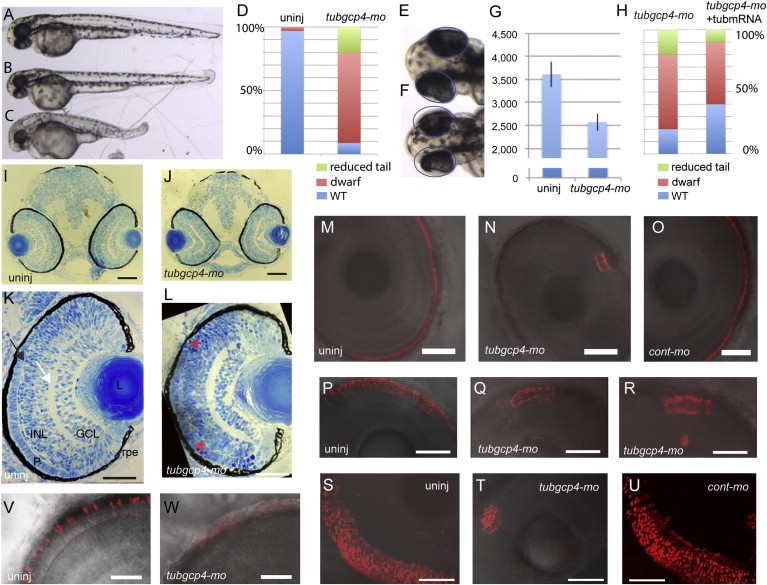
tubgcp4 Morphants Exhibit Head and Photoreceptor Developmental Anomalies in Zebrafish
(A–C) Injection of tubgcp4-mo into zebrafish embryos gave rise to dwarf embryos. (A) Uninjected embryo. (B and C) Morphants with a mild (dwarf, B) or severe (reduced tail, C) phenotype.
(D) Charts represent the percentage of each category of phenotypes in uninjected and morphant embryos.
(E and F) Uninjected embryo (E) and tubgcp4-mo morphant (F). Same-sized circles indicate how eyes were smaller in morphant embryos than in uninjected embryos.
(G) The chart shows the size of eyes (in pixels) of uninjected embryos and tubgcp4-mo-injected morphants.
(H) The chart shows that morphants injected with 0.9 mM of morpholino could be partially rescued with 50 ng/μl of tubgcp4-mo-resistant mRNA (tubmRNA).
(I and J) Transverse sections through the head of uninjected embryos (I) and morphant embryos (J) revealed no obvious abnormality in the overall morphology except that the eyes and head of the morphants were smaller.
(K and L) Higher magnification of transverse sections through the eye of a morphant reveals rounded retinal cells (K, arrowhead), which were absent from the uninjected embryos (L).
(M–U) Zpr1 antibody staining shows a continuous, uniform layer of elongated retinal cone cells in uninjected embryos (M, P, and S) or cont-mo-injected embryos (O and U), whereas only a restricted small patch was observed in the morphants (N, Q, R, and T). Images in (P)–(U) are superpositions of 15 confocal images representing 24-μm thickness.
(V and W) 1D1 antibody staining shows the rhodopsin-positive rods uniformly distributed in the retina of uninjected embryos (V). This staining was not detectable in the morphants (W).
Embryos were imaged at 48 hpf (A–G), 72 hpf (H–O, V, and W), 4 dpf (P–R), or 5 dpf (S–U). Abbreviations are as follows: L, lens; INL, inner nuclear layer; GCL, ganglion cell layer; P, photoreceptor layer; and RPE, retinal pigment epithelium. The white arrow indicates the inner plexiform layer, and the black arrow shows the outer plexiform layer. Scale bars represent 100 μm (I and J), 80 μm (K and L), 47 μm (M–R), 56 μm (S–U), and 23 μm (V and W).
Inherited congenital microcephaly is a clinically and genetically heterogeneous disorder. The biallelic TUBGCP4 mutations described herein, as well as the very recent TUBGCP6 mutations, confirm the existence of autosomal-recessive cases described as MCCRP. Thus, the common hypothesis of germline mosaicism in affected siblings with healthy parents has to be questioned. In addition, simplex cases previously considered to involve de novo dominant mutations could very well be linked to recessive inheritance.1,18,19
The recent identifications of the causative genetic variants in some individuals with primary or syndromic microcephaly have greatly improved our understanding of the pathogenesis of these conditions. The disease phenotype is associated with defects in a number of interconnected cellular functions, including alterations of mitotic spindle structure, of the centrosome, of ciliary function, of DNA repair, of DNA-damage-response signaling, and of DNA replication.20 In eukaryotic cells, the microtubule cytoskeleton is essential for intracellular transport, organelle position, motility, signaling, and cell division. Assembly of new microtubules relies on efficient microtubule nucleation induced by the γ-TuRC. The γ-TuRC is a complex resulting from the assembly of γ-tubulin with γ-tubulin complex proteins GCP2, GCP3, GCP4, GCP5, and GCP6 (encoded by TUBGCP2, TUBGCP3, TUBGCP4, TUBGCP5 [MIM 608147], and TUBGCP6, respectively). The five GCPs share similar core structures, although with low sequence homology.21
The TUBGCP4 mutations described herein point to the γ-TuRC, involved in microtubule organization, as a major pathway related to syndromic microcephaly associated with chorioretinal development abnormalities (MCCRP). Moreover, TUBGCP6 has recently been found to be mutated in the original MCCRP-affected family in whom a homozygous read-through mutation was found in affected members. However, functional investigations are still needed to ascertain the role of the γ-TuRC in the pathogenesis of the phenotype in this family.5 Very recently, a study on families affected by growth failure, microcephaly, and retinopathy has identified mutations in PLK4 (MIM 605031), a master regulator of centriole duplication, as well as mutations in its substrate TUBGCP6, in three families,6 confirming the role of TUBGCP6 in the pathogenesis of this phenotype. CDK5RAP2 (MIM 608201) and CENPJ (MIM 609279) have been found to be mutated in individuals with autosomal-recessive primary non-syndromic microcephaly and to encode centrosomal proteins that interact with the γ-TuRC.22,23 Interestingly, severe microcephaly with malformation of cortical development has been reported in individuals with mutations in TUBG1 (MIM 191135), one of two human genes encoding γ-tubulin.24 Defects in α- and β-tubulin and in microtubule-interacting proteins (i.e., encoded by KIF2A [MIM 602591] and KIF5C [MIM 604593]) have been reported in microcephaly associated with marked cortical developmental anomalies—such as pachygyria, polymicrogyria, or lissencephaly—manifesting with epilepsy and profound cognitive impairment.24 In contrast, the microcephaly-affected individuals reported to have chorioretinal dysplasia, related to microtubule-interacting proteins, such as those encoded by KIF11, TUBGCP6, and TUBGCP4 (reported here), have not been reported to have any major cerebral malformation. However, epilepsy was reported in 8% of MCLMR with KIF11 mutations, and learning difficulties of varying degrees are very common (73% of affected individuals).25 It would be conceivable that such GCP4-dependent defects might lead to developmental abnormalities in the neocortex and therefore result in reduced brain growth. The compound-heterozygous mutations identified in family A do not lead to total absence of GCP4. It is likely that such total absence would be lethal in the embryo. Our hypothesis is that the synonymous mutation, identified in three families, impairs splicing and thus leads to reduced amounts of functional GCP4.
We have demonstrated the impact of the TUBGCP4 mutations on cell morphology in cells from an affected individual (AII-1). Our findings are consistent with previous reports documenting that reduced amounts of GCP4 led to downregulation of other GCPs and to reduced amounts of the γ-TuRC.14,26 Consequently, microtubule nucleation was reduced in fibroblasts from affected individuals. Although it has been shown in organisms such as Drosophila that loss of GCP4 orthologs does not prevent the formation of mitotic spindles, subtle defects in mitosis, including altered spindle morphology and defects in spindle orientation, have nevertheless been observed.14,27 Moreover, neuroblasts from mutant Drosophila larvae exhibited delayed mitosis and low frequencies of aneuploidy.14 We also detected aneuploidy in the affected individual’s fibroblasts, marked by the presence of binucleated cells or nuclei of increased size.
The zebrafish model has been proven to be an efficient model for investigating the role of previously reported genes (namely STIL, ASPM, WDR62, and ODF2) in human microcephaly, given that the phenotype in zebrafish results in a marked reduction in head and eye size.28 Moreover, microcephaly models in the developing zebrafish have pointed to an underlying defect in metaphase progression in the retinal neuroepithelium.28 The tubgcp4 zebrafish model described herein reproduces head anomalies and interestingly demonstrates clustered abnormalities in photoreceptor development, pointing to a common developmental denominator for rods and cones. The human-specific ocular phenotype shows major chorioretinal developmental abnormalities, ranging from atrophic punched-out retinal lesions surrounded by zones with absent retinal vasculature to retinal folds (a typical stigmata of early-onset abnormality of retinal vascular development). One hypothesis would be that the zones in which the clusters of abnormal photoreceptor cells exist have altered development of other retinal layers, explaining this punched-out appearance.
In summary, because KIF11, TUBGCP6, and TUBGCP4 are all implicated in both dominant and recessive forms of microcephaly with chorioretinal dysplasia, this common pathway in the critical stages of brain and retinal development warrants further investigations and has implications for the genetic counselling of MCCRP-affected individuals.
Acknowledgments
We wish to warmly thank the individuals and their families for their participation. We would like to thank Jean-Louis Mandel and the financial support of the Centre Régional de Génétique Médicale de Strasbourg, Caisse d’Assurance Retraite et de la Santé au Travail Alsace-Moselle, and RETINA France. Sequencing was performed by the IGBMC Microarray and Sequencing platform, a member of the “France Génomique” consortium (ANR-10-INBS-0009). This work was funded in part by the National Institute for Health Research and Biomedical Research Centre at Moorfields Eye Hospital and the UCL Institute of Ophthalmology (London), Fight For Sight (London), Rosetrees Trust (Edgware), and the Agence Nationale de la Recherche (L.H. and A.M.). We are grateful for support from the European Commission through IP ZF-Health (FP7-HEALTH-2007-B2) and the Interreg Network for Synthetic Biology in the Upper Rhine valley (NSB-Upper Rhine, C.E. and U.S.). This work was approved by local ethics committees at Moorfields Eye Hospital, Hôpitaux Universitaires de Strasbourg (Strasbourg University Hospital), and Robert Debré Hospital.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ANNOVAR, http://www.openbioinformatics.org/
ExAC Browser, http://exac.broadinstitute.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
Orphanet, http://www.orpha.net/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
VaRank, http://www.lbgi.fr/VaRank
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Alzial C., Dufier J.L., Brasnu C., Aicardi J., de Grouchy J. [“True” microcephaly with dominant-inheritance chorioretinal dysplasia] Ann. Genet. 1980;23:91–94. [PubMed] [Google Scholar]
- 2.Ostergaard P., Simpson M.A., Mendola A., Vasudevan P., Connell F.C., van Impel A., Moore A.T., Loeys B.L., Ghalamkarpour A., Onoufriadis A. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am. J. Hum. Genet. 2012;90:356–362. doi: 10.1016/j.ajhg.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKusick V.A., Stauffer M., Knox D.L., Clark D.B. Chorioretinopathy with hereditary microcephaly. Arch. Ophthalmol. 1966;75:597–600. doi: 10.1001/archopht.1966.00970050599003. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen T.N., Der Kaloustian V.M., Barsoum-Homsy M., Dembinska O., Koenekoop R.K. Congenital microcephaly, juvenile retinal dystrophy and normal mentation in a mildly dysmorphic child. Can. J. Ophthalmol. 2005;40:195–199. doi: 10.1016/S0008-4182(05)80034-1. [DOI] [PubMed] [Google Scholar]
- 5.Puffenberger E.G., Jinks R.N., Sougnez C., Cibulskis K., Willert R.A., Achilly N.P., Cassidy R.P., Fiorentini C.J., Heiken K.F., Lawrence J.J. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE. 2012;7:e28936. doi: 10.1371/journal.pone.0028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin C.A., Ahmad I., Klingseisen A., Hussain M.S., Bicknell L.S., Leitch A., Nürnberg G., Toliat M.R., Murray J.E., Hunt D. Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat. Genet. 2014;46:1283–1292. doi: 10.1038/ng.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 8.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto S., Shimizu S., Kiyonaka S., Takahashi N., Wajima T., Hara Y., Negoro T., Hiroi T., Kiuchi Y., Okada T. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008;14:738–747. doi: 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plagnol V., Curtis J., Epstein M., Mok K.Y., Stebbings E., Grigoriadou S., Wood N.W., Hambleton S., Burns S.O., Thrasher A.J. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–2754. doi: 10.1093/bioinformatics/bts526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haren L., Remy M.H., Bazin I., Callebaut I., Wright M., Merdes A. NEDD1-dependent recruitment of the gamma-tubulin ring complex to the centrosome is necessary for centriole duplication and spindle assembly. J. Cell Biol. 2006;172:505–515. doi: 10.1083/jcb.200510028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fava F., Raynaud-Messina B., Leung-Tack J., Mazzolini L., Li M., Guillemot J.C., Cachot D., Tollon Y., Ferrara P., Wright M. Human 76p: A new member of the gamma-tubulin-associated protein family. J. Cell Biol. 1999;147:857–868. doi: 10.1083/jcb.147.4.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vérollet C., Colombié N., Daubon T., Bourbon H.M., Wright M., Raynaud-Messina B. Drosophila melanogaster gamma-TuRC is dispensable for targeting gamma-tubulin to the centrosome and microtubule nucleation. J. Cell Biol. 2006;172:517–528. doi: 10.1083/jcb.200511071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raymond P.A., Barthel L.K., Curran G.A. Developmental patterning of rod and cone photoreceptors in embryonic zebrafish. J. Comp. Neurol. 1995;359:537–550. doi: 10.1002/cne.903590403. [DOI] [PubMed] [Google Scholar]
- 16.Biehlmaier O., Lampert J.M., von Lintig J., Kohler K. Photoreceptor morphology is severely affected in the beta,beta-carotene-15,15′-oxygenase (bcox) zebrafish morphant. Eur. J. Neurosci. 2005;21:59–68. doi: 10.1111/j.1460-9568.2004.03830.x. [DOI] [PubMed] [Google Scholar]
- 17.Morris A.C., Fadool J.M. Studying rod photoreceptor development in zebrafish. Physiol. Behav. 2005;86:306–313. doi: 10.1016/j.physbeh.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jarmas A.L., Weaver D.D., Ellis F.D., Davis A. Microcephaly, microphthalmia, falciform retinal folds, and blindness. A new syndrome. Am. J. Dis. Child. 1981;135:930–933. doi: 10.1001/archpedi.1981.02130340036013. [DOI] [PubMed] [Google Scholar]
- 19.Trzupek K.M., Falk R.E., Demer J.L., Weleber R.G. Microcephaly with chorioretinopathy in a brother-sister pair: evidence for germ line mosaicism and further delineation of the ocular phenotype. Am. J. Med. Genet. A. 2007;143A:1218–1222. doi: 10.1002/ajmg.a.31717. [DOI] [PubMed] [Google Scholar]
- 20.Alcantara D., O’Driscoll M. Congenital microcephaly. Am. J. Med. Genet. C. Semin. Med. Genet. 2014;166C:124–139. doi: 10.1002/ajmg.c.31397. [DOI] [PubMed] [Google Scholar]
- 21.Guillet V., Knibiehler M., Gregory-Pauron L., Remy M.H., Chemin C., Raynaud-Messina B., Bon C., Kollman J.M., Agard D.A., Merdes A., Mourey L. Crystal structure of γ-tubulin complex protein GCP4 provides insight into microtubule nucleation. Nat. Struct. Mol. Biol. 2011;18:915–919. doi: 10.1038/nsmb.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bond J., Roberts E., Springell K., Lizarraga S.B., Scott S., Higgins J., Hampshire D.J., Morrison E.E., Leal G.F., Silva E.O. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet. 2005;37:353–355. doi: 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- 23.Issa L., Mueller K., Seufert K., Kraemer N., Rosenkotter H., Ninnemann O., Buob M., Kaindl A.M., Morris-Rosendahl D.J. Clinical and cellular features in patients with primary autosomal recessive microcephaly and a novel CDK5RAP2 mutation. Orphanet J. Rare Dis. 2013;8:59. doi: 10.1186/1750-1172-8-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poirier K., Lebrun N., Broix L., Tian G., Saillour Y., Boscheron C., Parrini E., Valence S., Pierre B.S., Oger M. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013;45:639–647. doi: 10.1038/ng.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones G.E., Ostergaard P., Moore A.T., Connell F.C., Williams D., Quarrell O., Brady A.F., Spier I., Hazan F., Moldovan O. Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): review of phenotype associated with KIF11 mutations. Eur. J. Hum. Genet. 2014;22:881–887. doi: 10.1038/ejhg.2013.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouissou A., Vérollet C., Sousa A., Sampaio P., Wright M., Sunkel C.E., Merdes A., Raynaud-Messina B. gamma-Tubulin ring complexes regulate microtubule plus end dynamics. J. Cell Biol. 2009;187:327–334. doi: 10.1083/jcb.200905060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouissou A., Vérollet C., de Forges H., Haren L., Bellaïche Y., Perez F., Merdes A., Raynaud-Messina B. γ-Tubulin Ring Complexes and EB1 play antagonistic roles in microtubule dynamics and spindle positioning. EMBO J. 2014;33:114–128. doi: 10.1002/embj.201385967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Novorol C., Burkhardt J., Wood K.J., Iqbal A., Roque C., Coutts N., Almeida A.D., He J., Wilkinson C.J., Harris W.A. Microcephaly models in the developing zebrafish retinal neuroepithelium point to an underlying defect in metaphase progression. Open Biol. 2013;3:130065. doi: 10.1098/rsob.130065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.