

Abstract
Developmental epilepsies are age-dependent seizure disorders for which genetic causes have been increasingly identified. Here we report six unrelated individuals with mutations in salt-inducible kinase 1 (SIK1) in a series of 101 persons with early myoclonic encephalopathy, Ohtahara syndrome, and infantile spasms. Individuals with SIK1 mutations had short survival in cases with neonatal epilepsy onset, and an autism plus developmental syndrome after infantile spasms in others. All six mutations occurred outside the kinase domain of SIK1 and each of the mutants displayed autophosphorylation and kinase activity toward HDAC5. Three mutations generated truncated forms of SIK1 that were resistant to degradation and also showed changes in sub-cellular localization compared to wild-type SIK1. We also report the human neuropathologic examination of SIK1-related developmental epilepsy, with normal neuronal morphology and lamination but abnormal SIK1 protein cellular localization. Therefore, these results expand the genetic etiologies of developmental epilepsies by demonstrating SIK1 mutations as a cause of severe developmental epilepsy.
Main Text
Developmental epilepsies are age-dependent pediatric seizure disorders that include early myoclonic encephalopathy,1,2 Ohtahara syndrome,3,4 malignant migrating partial seizures of infancy,5 infantile spasms,6 and Lennox-Gastaut syndrome.7 These disorders are often devastating, with outcomes ranging from catastrophic neonatal epilepsy with short survival to severe developmental disability, autism, and life-long intractable epilepsy.8,9 Some children present with early myoclonic encephalopathy or Ohtahara syndrome and then progress to infantile spasms and Lennox-Gastaut syndrome as they mature.10,11 Mutations have been identified in several genes in individuals with developmental epilepsies, clustering in several biological pathways, and it is common for persons with similar mutations in genes such as ARX12,13 (MIM 308350), CDKL514,15 (MIM 300672), MEF2C16,17 (MIM 613443), and STXBP118,19 (MIM 612164) to present with a spectrum of developmental epilepsies. However, in many the cause remains unknown.
As part of an ongoing genetic research program on developmental epilepsies, we performed genetic analysis in 101 individuals for whom clinically available molecular genetic tests had failed to detect a mutation. The genetic analysis was performed in accordance with informed consent procedures approved by the Institutional Review Boards at the University of Rochester Medical Center, Washington University, and Seattle Children’s Hospital. Clinical records, electroencephalogram (EEG) tracings and reports, and MRI scans were reviewed retrospectively. In this study, we present six unrelated children with developmental epilepsies in whom mutations in the salt-inducible kinase SIK1 (RefSeq accession number NM_173354.3) were identified.
The first mutation in SIK1 was identified through research-based exome sequencing in subject DB13-001, a boy born at term who presented with seizures consistent with early myoclonic encephalopathy at 20 min of life. An EEG on the second day of life showed a discontinuous background with burst suppression pattern (Figure 1A). He developed intermittent myoclonic jerking movements, and EEG on day 14 of life showed continuous high-amplitude burst activity alternating with periods of voltage suppression. Seizures did not respond to anticonvulsants, and an extensive metabolic and genetic workup was negative. He developed tonic seizures, burst suppression pattern persisted at 8 months of life (Figure 1B), and myoclonic seizures worsened. Brain MRI showed no structural malformations (Figures 1C and 1D). He died after complications of a respiratory illness at 10 months of age. Further clinical details are available in the Supplemental Data.
Figure 1.
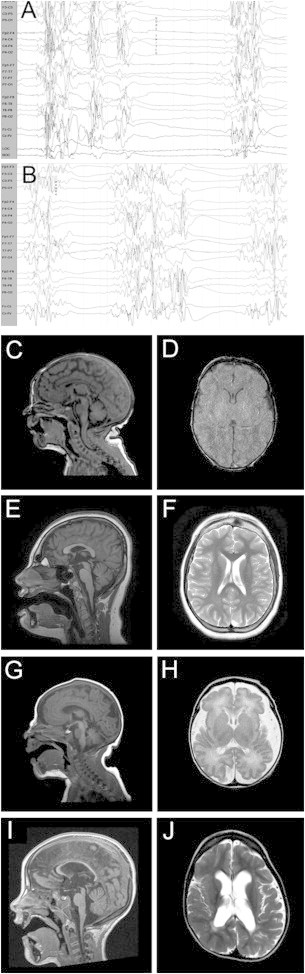
EEG and Brain Imaging Data for Individuals with SIK1 Mutations
(A) Representative EEG tracings from subject DB13-001 at 10 days of age showing burst suppression pattern with myoclonic activity, consistent with a diagnosis of early myoclonic encephalopathy.
(B) At 8 months of age, the EEG had not improved, with continued burst suppression pattern associated with both myoclonic and tonic seizures.
(C–J) MRI scans of subjects with SIK1 mutations showed subtle abnormalities.
(C and D) Subject DB13-001 (p.Pro287Thr) at age 1 week showed normal morphology.
(E and F) Subject IS13-013 (p.Gln614∗) at age 12 years showed low forehead, mild shortening of the corpus callosum, and mild atrophy of the cerebellar vermis.
(G and H) Subject LR05-086 (p.Gly636Ser) at 3 days of age with increased extra-axial spaces and simplified gyral pattern in the frontal lobes.
(I and J) Subject LR05-086 at age 16 months showed thinning of the corpus callosum and asymmetric thinning of the white matter.
After clinical chromosomal microarray and epilepsy gene panel sequencing was negative, we performed whole exome sequencing (WES) on saliva-derived DNA from subject DB13-001 and both parents. We used the Agilent SureSelect 50 Mb whole exome capture kit, and 100 bp paired-end reads were generated on an Illumina HiSeq2500 machine at the University of Rochester Genomics Research Center. Sequence was aligned to hg19 via BWA v.0.6.2 and analyzed with Picard v.1.84, SAMtools v.0.1.18, and GATK v.2.3-9. Mean read depth across the exome was 72× determined by GATK’s DepthOfCoverage walker, with 89% of bases covered at more than 20×. Annotation of variants was performed with ANNOVAR, and de novo, autosomal-recessive, and X-linked variants were identified and common variants in dbSNP 137 excluded via SOLVE-Brain v.1.0.1. Common population variants were identified with the NHLBI Exome Variant Server. Ensembl’s Variant Effect Predictor provided SIFT and PolyPhen predictions of pathogenicity. GERP scores were obtained from the UCSC Genome Browser. Our analyses revealed four de novo heterozygous and two compound heterozygous variants in this individual (Table S1).
Of particular interest was a de novo heterozygous c.895C>A (p.Pro287Thr) variant identified in SIK1 (RefSeq NM_173354.3) and validated with standard bidirectional Sanger sequencing (primer sequences provided in Table S2) (Figure 2A). Though the p.Pro287Thr amino acid substitution in SIK1 was not predicted to be deleterious by SIFT or PolyPhen, and the amino acid residue was not highly conserved (Table 1), this genomic variant was not present in >6,500 individuals in the NHLBI Exome Variant Server or in the >61,000 individuals in the Exome Aggregation Consortium (ExAC) Browser. The other de novo heterozygous and compound heterozygous variants were determined not to be compelling disease-causing candidates, as detailed in Table S1.
Figure 2.
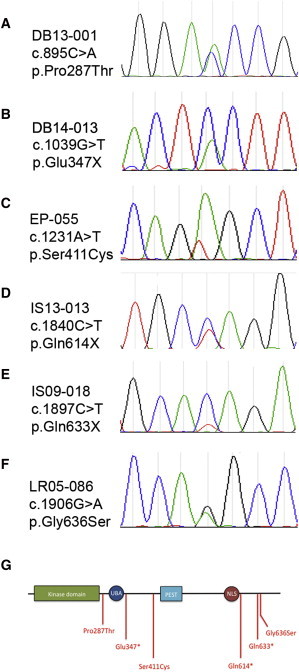
Heterozygous Mutations in SIK1
(A) Whole exome sequencing identified a c.895C>A variation resulting in a p.Pro287Thr amino acid substitution, confirmed de novo by Sanger sequencing of proband and parents.
(B–F) Unidirectional Sanger sequencing of the coding and intron-exon boundaries of SIK1 in 100 subjects with developmental epilepsy identified heterozygous mutations in 5 additional unrelated subjects, including c.1030G>T (p.Glu347∗) (B), c.1231A>T (p.Ser411Cys) (C), c.1840C>T (p.Gln614∗) (D), c.1897C>T (p.Gln633∗) (E), and c.1906G>A (p.Gly636Ser) (F). SIK1 sequences were compared to reference NM_173354.3. Primer sequences used are provided in Table S2. Sequence variations not found in dbSNP 137 were then confirmed with bidirectional sequencing, and all were confirmed de novo except for DB14-013 and IS09-018 where parental DNA was unavailable.
(G) SIK1 contains a protein kinase domain (green), a ubiquitin binding area (UBA), a proline-glutamate-serine-threonine (PEST) domain, and a nuclear localization signal (NLS). Amino acid substitutions (red) resulting from mutations in SIK1 occurred in regions outside of known domains and in a cluster between the nuclear localization signal and the C terminus.
Table 1.
Summary of SIK1 Mutations Found in Six Subjects with Developmental Epilepsy
| Subject | Genomic Coordinates (hg19) | cDNA | Amino Acid Substitution | Inheritance | Present in EVS | Present in ExAC | SIFT/PolyPhen | GERP |
|---|---|---|---|---|---|---|---|---|
| DB13-001 | chr21: g.44840227G>T | c.895C>A | p.Pro287Thr | de novo | no | no | 0.22/0.081 | −1.55 |
| DB14-013 | chr21: g.44839819C>A | c.1039G>T | p.Glu347∗ | ND | no | no | – | 5.16 |
| EP-055 | chr21: g.44839247T>A | c.1231A>T | p.Ser411Cys | de novo | no | no | 0.03/0.751 | −0.88 |
| IS13-013 | chr21: g.44837559G>A | c.1840C>T | p.Gln614∗ | de novo | no | no | – | 4.07 |
| IS09-018 | chr21: g.44837502G>A | c.1897C>T | p.Gln633∗ | ND | no | no | – | 4.07 |
| LR05-086 | chr21: g.44837493C>T | c.1906G>A | p.Gly636Ser | de novo | no | no | 0.46/0.093 | −0.48 |
Abbreviations are as follows: ND, no data because parental samples not available; EVS, NHLBI Exome Variant Server; ExAC, Exome Aggregation Consortium; SIFT, Sorting Intolerant From Tolerant score; PolyPhen, Polymorphism Phenotyping score; GERP, Genomic Evolutionary Rate Profiling score.
SIK1 is a member of the AMP kinase subfamily with several roles in the central nervous system including regulation of the circadian clock20 and transcription of corticotropin-releasing hormone in the hypothalamus.21 SIK1 abundance and activity are also increased by stimulation with adrenocorticotrophic hormone (ACTH),22 which is interestingly a first-line treatment for infantile spasms.23 ACTH triggers phosphorylation of SIK1 in a Protein Kinase A (PKA)-dependent manner,24 resulting in movement of activated SIK1 from the cytoplasm into the nucleus, where it in turn phosphorylates HDAC5.25 Phosphorylation of HDAC5 allows MEF2C transcriptional activity to proceed.26 MEF2C is a transcription factor important in both dorsal and ventral neuronal developmental pathways17 and its expression is decreased with deficiency of the early forebrain transcription factor ARX.27 Loss-of-function mutations in both ARX and MEF2C cause developmental epilepsies including infantile spasms.12,16
SIK1 also regulates CREB1 transcriptional activity via phosphorylation of TORC1 in an LKB1-dependent manner,28 resulting in downstream effects on dendrite growth during neuronal development.29 Recent transcriptome studies of epilepsy have identified CREB-target genes as involved in seizure pathogenesis.30 SIK1 is therefore positioned to interact with the MEF2C pathway known to be important in developmental epilepsies and the CREB1 pathway implicated in epilepsy pathogenesis.
Because some authors have found mutation tolerance scores helpful in determining a gene’s likelihood to cause disease,31 we performed this evaluation for SIK1. With the NHLBI EVS, we confirmed that SIK1 has 13 common (mean allele frequency > 0.1%) variants out of 105 coding variants, with a residual variance intolerance score (RVIS) of −0.2626, which falls at the 35th percentile.32 Other authors have arbitrarily chosen RVIS scores below the 25th percentile as associated with pathogenicity in epileptic encephalopathies.31 However, given that several known epileptic encephalopathy genes have RVIS scores between the 35th percentile (for FOXG1 [MIM 613454]) and the 58th percentile (for MEF2C),32 we question the usefulness of this statistical approach to identifying causal mutations in genes, particularly in genes associated with rare but very specific phenotypes.
To determine whether mutations in SIK1 are present in other subjects with developmental epilepsies, we performed Sanger sequencing of the coding regions of SIK1 in 100 subjects with developmental epilepsies and identified five additional mutations (Figures 2B–2F), including three truncations and two missense mutations. The mutations were de novo in all subjects where parental DNA was available (four out of six children, including our initial proband). None of these mutations were present in >6,500 individuals in the NHLBI EVS or in >61,000 individuals in the ExAC Browser. The three nonsense mutations were all predicted to result in a truncated protein. None of the mutations identified in SIK1 in our subjects with developmental epilepsies localized to the functional kinase domain of the protein (Figure 2G) or other well-characterized regulatory regions.
A truncating mutation (c.1039G>T [p.Glu347∗]; RefSeq NM_173354.3) was identified in postmortem brain tissue from subject DB14-013, a male infant who presented with early myoclonic encephalopathy shortly after birth with death at 3 months of age attributed to intractable seizures. Three subjects presenting with infantile spasms (EP-055, IS13-013, and IS09-018) subsequently developed intractable epilepsy and an autism plus developmental disorder with absent speech, impaired socialization, and repetitive behaviors. Brain MRI in our subjects was either normal (Figures 1C and 1D), showed mild hypoplasia of the frontal lobes (Figures 1E and 1F), or in one subject exhibited simplification of the gyral pattern and asymmetric thinning of the white matter (Figures 1G–1J). These are nonspecific findings commonly identified in individuals with severe developmental disability. Genomic features of the SIK1 mutations are summarized in Table 1 and further clinical details are in the Supplemental Data.
Because SIK1 is known to exhibit a high rate of turnover in C2C12 cells and primary myoblasts,33 we first tested whether the mutations identified affected protein stability. Wild-type and variant forms of SIK1 were expressed in HEK293T cells by transfection, and the half-life of each protein determined by cycloheximide treatment and quantitative immunoblotting. Our analysis revealed that the c.1039G>T (p.Glu347∗), c.1840C>T (p.Gln614∗), and c.1897C>T (p.Gln633∗) proteins exhibited an increase in stability compared to wild-type SIK1 (Figures 3A and 3B). Phosphorylation of Thr473 by PKA increases the half-life of SIK1, as does deleting an internal segment (residues 451–472) that resembles a PEST sequence.33 Thus, the absence of these residues in the p.Glu347∗ protein probably explains the increase in protein stability. Even more striking was the stability of the p.Gln614∗ and p.Gln633∗ mutants, which might indicate that interdomain interactions are critical for kinase stability and occur after the Ser575, but might interact with the stability domains. The variant SIK1 proteins p.Pro287Thr, p.Ser411Cys, and p.Gly363Ser had turnover rates similar to that of wild-type SIK1.
Figure 3.
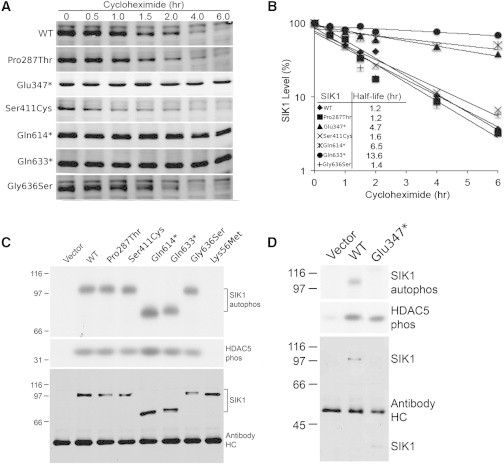
SIK1 Biochemical Studies
(A and B) To study the effects of SIK1 mutations in vitro, myc-SIK1 expression constructs were created by using human SIK1 amplified by PCR (Phusion Polymerase, NEB) from a cDNA construct (Thermo Scientific, clone ID, 4831049; accession, BC038504). Primer sequences are available in Table S3. Both myc and hSIK1 fragments were cloned with T4 ligase (NEB) into the XhoI and NotI site of a lentiviral bi-cistronic farneslyated GFP construct derived from a combination of pGIPZ non-silencing (Thermo Scientific), pLVX-IRES-tdTomato (Clontech), pDsRed-Monomer-F (Clontech), and pIRES2-acGFP (Clontech) vectors. Individual mutations were introduced into SIK1 through PCR amplification with Phusion polymerase (NEB) and mutagenic primers, followed by recombination with an In-Fusion HD cloning system (Clontech). HEK293 cells (Systems Biosciences) were cultured in growth medium with DMEM/F12 ([+] L-glut and [+] sodium bicarbonate from Life Technologies), 5% FBS (Atlanta Biologicals), 1% MEM NEAA (Life Technologies), and 1% sodium pyruvate (Life Technologies) in a 5% CO2 humidified incubator (Forma Scientific). SIK1 constructs were transfected into cells with Fugene-6 (Promega) according to the manufacturer’s procedure. Six hours after transfection, cells were split into smaller plates, grown overnight, and given drug treatments (cycloheximide 100 μg/ml [Sigma]) for various durations. Cells were washed once with PBS, lysed in SDS-loading buffer, heated at ∼97°C for 5 min, and then sonicated. Lysates were separated by SDS-PAGE. Proteins were transferred onto nitrocellulose membrane for immunoblots. Membranes were blocked with 5% NFDM (non-fat dry milk) in PBS-T solution and probed with an anti-myc primary antibody (9E10, DSHB), followed by Alexa Fluor 680-goat anti-mouse-IgG (H+L) (Invitrogen). The signal was visualized and quantified with Odyssey (LICOR). The truncating p.Glu347∗, p.Gln614∗, and p.Gln633∗ mutants showed increased protein stability (A) and prolonged half-life (B) compared to wild-type SIK1. The missense mutations were comparable to wild-type in their protein stability and half-life. SIK1 kinase activity (C and D) was measured by IP kinase assay with two GST-tagged fragments of human HDAC5 (243–276, 423–538; expressed in E. coli) that contain published sites for SIK1 (Ser259, Ser498). Myc-tagged WT and SIK1 mutants were expressed in HEK293 cells, immunoprecipitated with anti-myc antibody preloaded onto protein G beads, and incubated with 1 μg substrate and 32P-ATP (100 μM ATP total) in kinase buffer (50 mM HEPES [pH 7], containing 5 mM MgCl2, 5 mM B-glycerophosphate, 150 μM Na3CO4, 1 mM DTT, and 1 mg/ml BSA) for 30 min at 30°C. SIK1 mutants p.Glu347∗, p.Gln614∗, and p.Gln633∗ express at higher levels, and therefore bead volume inputs were adjusted accordingly. Kinase assay samples were subjected to SDS-PAGE and phosphorimaging. SIK1 (WT and mutants) does not phosphorylate GST under these assay conditions (data not shown). Auto-phosphorylation and HDAC5 phosphorylation was observed for all mutants, compared to wild-type SIK1 and the p.Lys56Met kinase-dead control (C and D).
We were next interested in whether the observed SIK1 mutations altered kinase activity. Using immunoprecipitation-kinase assays, we evaluated both autophosphorylation of SIK1 and phosphorylation of HDAC5, a well-characterized substrate of SIK1.26,34 We used recombinant fragments of HDAC5 expressed as GST fusion proteins that contained SIK1 phosphorylation sites Ser359 and Ser498. A kinase-dead form of SIK1 (p.Lys56Met) was used as a negative control in our activity assays.33 All SIK1 variant proteins identified in individuals with developmental epilepsy demonstrated both autophosphorylation activity as well as the ability to phosphorylate HDAC5 fragments in vitro (Figures 3C and 3D). These data suggest that both variant and truncated SIK1 proteins retain kinase activity.
We then sought to determine whether variant and truncated SIK1 proteins exhibited normal protein localization patterns. In control conditions, wild-type and kinase-dead p.Lys56Met SIK1 localize to a punctate pattern in the nuclei of HEK293TN cells (Figures 4A and 4C). Whereas the p.Pro287Thr, p.Ser411Cys, and p.Gly363Ser proteins showed a similar pattern of punctate nuclear localization, the p.Glu347∗, p.Gln614∗, and p.Gln633∗ truncated proteins exhibited a broader pattern of localization within the nucleus and in the cytoplasm. Because PKA is known to regulate the expression and distribution of SIK1,22 we treated HEK293TN cells with Forskolin (Fsk) and 3-isobutyl-1-methylxanthine (IBMX) to activate PKA signaling and determine its effects on the SIK1 mutant proteins. As expected, wild-type SIK1 protein levels increased with prolonged Fsk/IBMX treatment and its localization shifted to both the cytoplasm and the nucleus (Figure 4B). A similar response to Fsk/IBMX was seen with the kinase-dead protein, although additional foci of protein product were observed in the cytoplasm of these cells (Figures 4C and 4D). A similar observation was made in cells expressing the p.Glu347∗ variant (Figures 4G and 4H). Additionally, all three truncated proteins (p.Glu347∗, p.Gln614∗, and p.Gln633∗) showed a broader nuclear localization and increased cytoplasmic localization compared to wild-type SIK1 (Figures 4G–4L). The protein localization pattern for the missense variant proteins after Fsk/IBMX treatment resembled that of wild-type protein (Figures 4M–4R). Furthermore, unlike the wild-type SIK1 protein, the expression level of the p.Gln614∗ and p.Gln633∗ proteins did not increase as much under Fsk/IBMX treatment, although their overall expression levels were higher than that of the wild-type protein or the missense mutants (Figure S1). These data suggest that the missense SIK1 proteins localized to the nucleus and shuttled to the cytoplasm with PKA stimulation, similar to wild-type protein. The truncated proteins appear to have improper protein localization and regulation, perhaps due to lack of a regulatory function normally contained in the C terminus that promotes nuclear localization. Our biochemical and cellular observations on the effects of these mutations are summarized in Table 2.
Figure 4.
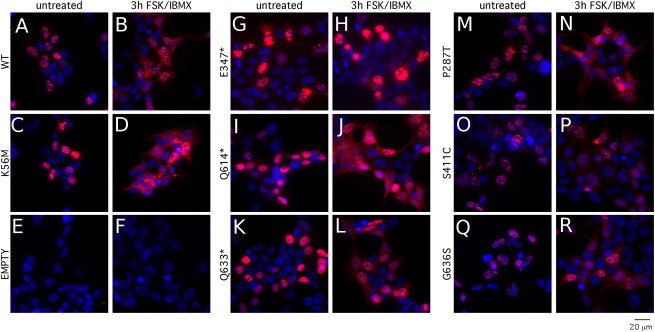
SIK1 Protein Localization Studies
To study protein localization, HEK293TN cells were plated on coverslips in a plasma cleaner and coated with poly-L-lysine (Sigma). The day after plating, cells were transfected with CMV::myc-SIK1-IRES2-acGFP-F expression constructs via Lipofectamine 2000 according to the manufacturer’s procedure. The day after transfection, cells received drug treatments (Forskolin 10 μM [Cayman Chemicals], IBMX 18 μM [Calbiochem]) for 3 hr. Cells were fixed with a 4% paraformaldehyde PBS solution and blocked with PBS containing 1% serum and 0.1% Triton X-100. Cells were incubated with a mouse anti-myc (9E10, DSHB) primary antibody diluted in blocking solution, followed by a second incubation with goat anti-mouse Alexa594 conjugated secondary antibody (Invitrogen Molecular Probes) and Hoechst 33342 nuclear stain (Sigma) also diluted in blocking solution. Coverslips were mounted on slides with Mowiol and imaged with an Olympus BX61 fluorescent microscope and Metamorph software. Wild-type SIK1 (A and B), the kinase-dead p.Lys56Met (C and D), and empty vector (E and F) were used as controls. The mutant p.Glu347∗ (G and H), p.Gln614∗ (I and J), and p.Gln633∗ (K and L) SIK1 proteins showed abnormal localization in both control (A, C, E, G, I, K, M, O, and Q) and Fsk/IBMX-treated conditions (B, D, F, H, J, L, N, P, R). The missense mutants p.Pro287Thr (M and N), p.Ser411Cys (O and P), and p.Gly636Ser (Q and R) had localization patterns resembling wild-type protein.
Table 2.
Summary of Effects of SIK1 Mutations on Protein Stability, Cellular Localization, and HDAC5 Phosphorylation
| Subject | Amino Acid Substitution | Increased Protein Stability | Abnormal Cellular Localization | HDAC5 Phosphorylation |
|---|---|---|---|---|
| DB13-001 | p.Pro287Thr | − | − | + |
| DB14-013 | p.Glu347∗ | + | + | + |
| EP-055 | p.Ser411Cys | − | − | + |
| IS13-013 | p.Gln614∗ | + | + | + |
| IS09-018 | p.Gln633∗ | + | + | + |
| LR05-086 | p.Gly636Ser | − | − | + |
Because of the availability of a brain specimen from subject DB14-013 with the p.Glu347∗ SIK1 mutation, we were able to compare SIK1 distribution in the presence of this mutation as compared with control and mutation-negative epileptic infant brain. Brain weight for subject DB14-013 was 584 g (average 516 g). On microscopic examination, no malformations were identified and neuronal morphology and lamination were normal. SIK1 immunofluorescence (IF) was detected in neurons and proximal dendrites within the hippocampus and cortex (Figures 5A–5C). SIK1 IF was also seen in GFAP+ astrocytes in the cortical white matter (Figures 5D–5F). However, the intracellular distribution of SIK1 IF differed between control infant brain, subject DB14-013 (SIK1 p.Glu347∗), and that of an infant with epileptic encephalopathy but normal SIK1 sequencing. In subject DB14-013, neuronal SIK1 IF was predominant in the cytoplasm only, whereas in the other specimens it was equivalent in both the nucleus and cytoplasm (Figures 5G–5I). These findings indicated that SIK1 protein expression levels were not substantially altered by the presence of the heterozygous p.Glu347∗ mutation and that this SIK1 mutation did not result in gross abnormalities of brain formation or neuronal morphology. However, alteration of SIK1 intracellular distribution was detected, suggesting that nuclear localization of the p.Glu347∗ mutant might be impaired, consistent with our in vitro studies.
Figure 5.
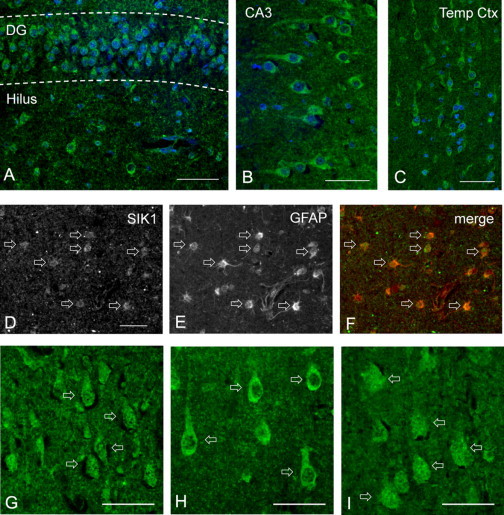
SIK1 Brain Immunohistochemistry
Formalin-fixed human brain sections from subject DB14-013 (p.Glu347∗ mutation) and two other infants were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, embedded in paraffin, and sectioned at a thickness of 4 μm. Specimens were deparaffinized by incubation in xylene, followed by rehydration through graded ethanol/water solutions and equilibration in PBS. Antigen retrieval was performed by heating in 0.01 M sodium citrate (pH 6.0) in a 90°C water bath for 45 min. Specimens were then blocked in PBS with 10% donkey serum, 0.1% Triton X-100, and 2% BSA for 30 min and incubated overnight at 4°C in primary antibodies diluted in blocking solution. The SIK1 primary antibody (M01, clone 2C12, Abnova) is a mouse monoclonal IgG2a directed against an epitope mapping to the N-terminal 100 amino acids of human SIK1. Other primary antibodies included goat polyclonal MAP2 and GFAP (Santa Cruz). Slides were then incubated in fluorescent secondary antibodies at room temperature for 2 hr. After incubation in DAPI nuclear counterstain and Sudan Black to block endogenous fluorescence, sections were examined and photographed with a Nikon Eclipse Ni-U epifluorescence microscope.
(A–C) SIK1 immunofluorescence (IF) in subject DB14-013 was detected in neuronal somas and proximal dendrites within the dentate gyrus and hilus (A) and CA3 of the hippocampus (B), as well as in the temporal cortex (C).
(D–F) SIK1 expression in GFAP+ astrocytes in white matter of the temporal cortex in subject DB14-013, illustrated by staining for SIK1 (D), GFAP (E), or both (F). Scale bar represents 100 μm.
(G–I) SIK1 IF in temporal cortex of a control infant (G), subject DB14-013 (H), and an epileptic infant with normal SIK1 sequencing (I). Pyramidal neurons exhibited both nuclear and cytoplasmic SIK IF in the control and epileptic subjects, but localization predominantly in the cytoplasm in subject DB14-013. Scale bars represent 50 μm.
Our individuals with SIK1 mutations had universally severe outcomes. Subjects DB13-001 and DB14-013 both presented with early myoclonic encephalopathy and died in infancy from intractable epilepsy with respiratory failure. The other subjects had severe developmental impairment with autistic features and continued refractory seizures, and subject LR05-086 had respiratory failure requiring tracheostomy placement and long-term ventilation. None of the subjects with infantile spasms responded to ACTH. Nearly all of the subjects required the placement of gastrostomy tubes for nutrition, and two of the four persons who survived past infancy remained nonambulatory. Scoliosis was present in two of the subjects. The clinical features of SIK1-related developmental epilepsy are summarized in Table 3 as well as in the Supplemental Data.
Table 3.
Genotype-Phenotype Correlations in Six Subjects Presenting with Developmental Epilepsy and SIK1 Mutations
| Subject | Amino Acid Substitution | Presenting Epilepsy | Death in Infancy | ID | Absent Language | Impaired Socialization | Repetitive Behaviors | Non- ambulatory | Gastrostomy Tube | Respiratory Failure | Other Findings |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DB13-001 | p.Pro287Thr | EME | + | NA | NA | NA | NA | NA | + | + | − |
| DB14-013 | p.Glu347∗ | EME | + | NA | NA | NA | NA | NA | + | + | − |
| EP-055 | p.Ser411Cys | ISS | − | + | + | + | + | − | − | − | anomalous pulmonary arterial circulation |
| IS13-013 | p.Gln614∗ | ISS | − | + | + | + | + | − | + | − | disordered sleep, breath-holding spells, scoliosis |
| IS09-018 | p.Gln633∗ | ISS | − | + | + | + | + | + | + | − | − |
| LR05-086 | p.Gly636Ser | OS | − | + | + | + | + | + | + | + | diabetes insipidus, scoliosis |
Abbreviations are as follows: EME, early myoclonic encephalopathy; OS, Ohtahara syndrome; ISS, infantile spasms; ID, intellectual disability; NA, not applicable.
In conclusion, we identified de novo heterozygous mutations in SIK1 in children presenting with developmental epilepsies including early myoclonic encephalopathy, Ohtahara syndrome, and infantile spasms. Overall, our data indicated that truncating mutations resulted in increased protein stability and abnormal cellular localization and that all mutant proteins were capable of phosphorylating HDAC5. We also present human neuropathology of SIK1-related developmental epilepsy and confirm our in vitro observations of abnormal SIK1 protein cellular localization in that specimen. Our study identified 6 SIK1 mutations in 101 individuals sequenced, suggesting that this might be a relatively common cause of developmental epilepsy and that kinases involved in the nuclear control of gene transcription can contribute to neurodevelopmental disease and severe epilepsy.
Acknowledgments
The authors would like to thank the families of our research subjects. Research reported in this work was supported by the NIH, National Institute of Neurologic Disorders and Stroke under award numbers K02NS072162 (to L.A.J.), R01NS046616 (to W.B.D.), R01NS076617 (to M.W.H.), and K08NS078054 (to A.R.P.), and the National Institute on Aging R01AG040162 (to B.M.P.). We would like to acknowledge the University of Rochester Genomics Research Center for sequencing support, and the University of Rochester Center for Integrated Research Computing for providing high-performance computing resources. We wish to thank Dr. Timothy McKinsey (University of Colorado, Denver) for HDAC5 constructs.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
ExAC Browser, http://exac.broadinstitute.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Picard, http://picard.sourceforge.net/
SAMtools, http://samtools.sourceforge.net/
SOLVE-Brain, https://paciorkowski-lab.urmc.rochester.edu/solve_brain
Variant Effect Predictor, http://useast.ensembl.org/info/docs/tools/vep/index.html
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Ohtahara S., Yamatogi Y. Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res. 2006;70(1):S58–S67. doi: 10.1016/j.eplepsyres.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 2.Djukic A., Lado F.A., Shinnar S., Moshé S.L. Are early myoclonic encephalopathy (EME) and the Ohtahara syndrome (EIEE) independent of each other? Epilepsy Res. 2006;70(1):S68–S76. doi: 10.1016/j.eplepsyres.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 3.Ohtahara S., Ohtsuka Y., Yamatogi Y., Oka E. The early-infantile epileptic encephalopathy with suppression-burst: developmental aspects. Brain Dev. 1987;9:371–376. doi: 10.1016/s0387-7604(87)80110-9. [DOI] [PubMed] [Google Scholar]
- 4.Yamatogi Y., Ohtahara S. Early-infantile epileptic encephalopathy with suppression-bursts, Ohtahara syndrome; its overview referring to our 16 cases. Brain Dev. 2002;24:13–23. doi: 10.1016/s0387-7604(01)00392-8. [DOI] [PubMed] [Google Scholar]
- 5.Coppola G., Plouin P., Chiron C., Robain O., Dulac O. Migrating partial seizures in infancy: a malignant disorder with developmental arrest. Epilepsia. 1995;36:1017–1024. doi: 10.1111/j.1528-1157.1995.tb00961.x. [DOI] [PubMed] [Google Scholar]
- 6.Lux A.L., Osborne J.P. A proposal for case definitions and outcome measures in studies of infantile spasms and West syndrome: consensus statement of the West Delphi group. Epilepsia. 2004;45:1416–1428. doi: 10.1111/j.0013-9580.2004.02404.x. [DOI] [PubMed] [Google Scholar]
- 7.Dulac O., N’Guyen T. The Lennox-Gastaut syndrome. Epilepsia. 1993;34(7):S7–S17. doi: 10.1111/j.1528-1157.1993.tb04593.x. [DOI] [PubMed] [Google Scholar]
- 8.Saemundsen E., Ludvigsson P., Rafnsson V. Risk of autism spectrum disorders after infantile spasms: a population-based study nested in a cohort with seizures in the first year of life. Epilepsia. 2008;49:1865–1870. doi: 10.1111/j.1528-1167.2008.01688.x. [DOI] [PubMed] [Google Scholar]
- 9.Partikian A., Mitchell W.G. Neurodevelopmental and epilepsy outcomes in a North American cohort of patients with infantile spasms. J. Child Neurol. 2010;25:423–428. doi: 10.1177/0883073809341664. [DOI] [PubMed] [Google Scholar]
- 10.Dulac O. Epileptic encephalopathy. Epilepsia. 2001;42(3):23–26. doi: 10.1046/j.1528-1157.2001.042suppl.3023.x. [DOI] [PubMed] [Google Scholar]
- 11.You S.J., Kim H.D., Kang H.-C. Factors influencing the evolution of West syndrome to Lennox-Gastaut syndrome. Pediatr. Neurol. 2009;41:111–113. doi: 10.1016/j.pediatrneurol.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Strømme P., Mangelsdorf M.E., Scheffer I.E., Gécz J. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev. 2002;24:266–268. doi: 10.1016/s0387-7604(02)00079-7. [DOI] [PubMed] [Google Scholar]
- 13.Guerrini R., Moro F., Kato M., Barkovich A.J., Shiihara T., McShane M.A., Hurst J., Loi M., Tohyama J., Norci V. Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology. 2007;69:427–433. doi: 10.1212/01.wnl.0000266594.16202.c1. [DOI] [PubMed] [Google Scholar]
- 14.Weaving L.S., Christodoulou J., Williamson S.L., Friend K.L., McKenzie O.L., Archer H., Evans J., Clarke A., Pelka G.J., Tam P.P. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004;75:1079–1093. doi: 10.1086/426462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bahi-Buisson N., Kaminska A., Boddaert N., Rio M., Afenjar A., Gérard M., Giuliano F., Motte J., Héron D., Morel M.A. The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia. 2008;49:1027–1037. doi: 10.1111/j.1528-1167.2007.01520.x. [DOI] [PubMed] [Google Scholar]
- 16.Le Meur N., Holder-Espinasse M., Jaillard S., Goldenberg A., Joriot S., Amati-Bonneau P., Guichet A., Barth M., Charollais A., Journel H. MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J. Med. Genet. 2010;47:22–29. doi: 10.1136/jmg.2009.069732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paciorkowski A.R., Traylor R.N., Rosenfeld J.A., Hoover J.M., Harris C.J., Winter S., Lacassie Y., Bialer M., Lamb A.N., Schultz R.A. MEF2C haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways. Neurogenetics. 2013;14:99–111. doi: 10.1007/s10048-013-0356-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saitsu H., Kato M., Mizuguchi T., Hamada K., Osaka H., Tohyama J., Uruno K., Kumada S., Nishiyama K., Nishimura A. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat. Genet. 2008;40:782–788. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- 19.Otsuka M., Oguni H., Liang J.S., Ikeda H., Imai K., Hirasawa K., Imai K., Tachikawa E., Shimojima K., Osawa M., Yamamoto T. STXBP1 mutations cause not only Ohtahara syndrome but also West syndrome—result of Japanese cohort study. Epilepsia. 2010;51:2449–2452. doi: 10.1111/j.1528-1167.2010.02767.x. [DOI] [PubMed] [Google Scholar]
- 20.Jagannath A., Butler R., Godinho S.I., Couch Y., Brown L.A., Vasudevan S.R., Flanagan K.C., Anthony D., Churchill G.C., Wood M.J. The CRTC1-SIK1 pathway regulates entrainment of the circadian clock. Cell. 2013;154:1100–1111. doi: 10.1016/j.cell.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y., Poon V., Sanchez-Watts G., Watts A.G., Takemori H., Aguilera G. Salt-inducible kinase is involved in the regulation of corticotropin-releasing hormone transcription in hypothalamic neurons in rats. Endocrinology. 2012;153:223–233. doi: 10.1210/en.2011-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takemori H., Doi J., Horike N., Katoh Y., Min L., Lin X.Z., Wang Z.N., Muraoka M., Okamoto M. Salt-inducible kinase-mediated regulation of steroidogenesis at the early stage of ACTH-stimulation. J. Steroid Biochem. Mol. Biol. 2003;85:397–400. doi: 10.1016/s0960-0760(03)00199-7. [DOI] [PubMed] [Google Scholar]
- 23.Pellock J.M., Hrachovy R., Shinnar S., Baram T.Z., Bettis D., Dlugos D.J., Gaillard W.D., Gibson P.A., Holmes G.L., Nordl D.R. Infantile spasms: a U.S. consensus report. Epilepsia. 2010;51:2175–2189. doi: 10.1111/j.1528-1167.2010.02657.x. [DOI] [PubMed] [Google Scholar]
- 24.Takemori H., Katoh Y., Horike N., Doi J., Okamoto M. ACTH-induced nucleocytoplasmic translocation of salt-inducible kinase. Implication in the protein kinase A-activated gene transcription in mouse adrenocortical tumor cells. J. Biol. Chem. 2002;277:42334–42343. doi: 10.1074/jbc.M204602200. [DOI] [PubMed] [Google Scholar]
- 25.Takemori H., Katoh Hashimoto Y., Nakae J., Olson E.N., Okamoto M. Inactivation of HDAC5 by SIK1 in AICAR-treated C2C12 myoblasts. Endocr. J. 2009;56:121–130. doi: 10.1507/endocrj.k08e-173. [DOI] [PubMed] [Google Scholar]
- 26.Finsterwald C., Carrard A., Martin J.-L. Role of salt-inducible kinase 1 in the activation of MEF2-dependent transcription by BDNF. PLoS ONE. 2013;8:e54545. doi: 10.1371/journal.pone.0054545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fulp C.T., Cho G., Marsh E.D., Nasrallah I.M., Labosky P.A., Golden J.A. Identification of Arx transcriptional targets in the developing basal forebrain. Hum. Mol. Genet. 2008;17:3740–3760. doi: 10.1093/hmg/ddn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katoh Y., Takemori H., Lin X.Z., Tamura M., Muraoka M., Satoh T., Tsuchiya Y., Min L., Doi J., Miyauchi A. Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J. 2006;273:2730–2748. doi: 10.1111/j.1742-4658.2006.05291.x. [DOI] [PubMed] [Google Scholar]
- 29.Li S., Zhang C., Takemori H., Zhou Y., Xiong Z.-Q. TORC1 regulates activity-dependent CREB-target gene transcription and dendritic growth of developing cortical neurons. J. Neurosci. 2009;29:2334–2343. doi: 10.1523/JNEUROSCI.2296-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beaumont T.L., Yao B., Shah A., Kapatos G., Loeb J.A. Layer-specific CREB target gene induction in human neocortical epilepsy. J. Neurosci. 2012;32:14389–14401. doi: 10.1523/JNEUROSCI.3408-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allen A.S., Berkovic S.F., Cossette P., Delanty N., Dlugos D., Eichler E.E., Epstein M.P., Glauser T., Goldstein D.B., Han Y., Epi4K Consortium. Epilepsy Phenome/Genome Project De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrovski S., Wang Q., Heinzen E.L., Allen A.S., Goldstein D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stewart R., Akhmedov D., Robb C., Leiter C., Berdeaux R. Regulation of SIK1 abundance and stability is critical for myogenesis. Proc. Natl. Acad. Sci. USA. 2013;110:117–122. doi: 10.1073/pnas.1212676110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dietrich J.-B., Takemori H., Grosch-Dirrig S., Bertorello A., Zwiller J. Cocaine induces the expression of MEF2C transcription factor in rat striatum through activation of SIK1 and phosphorylation of the histone deacetylase HDAC5. Synapse. 2012;66:61–70. doi: 10.1002/syn.20988. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.