

Abstract
Mutations in genes encoding aminoacyl-tRNA synthetases are known to cause leukodystrophies and genetic leukoencephalopathies—heritable disorders that result in white matter abnormalities in the central nervous system. Here we report three individuals (two siblings and an unrelated individual) with severe infantile epileptic encephalopathy, clubfoot, absent deep tendon reflexes, extrapyramidal symptoms, and persistently deficient myelination on MRI. Analysis by whole exome sequencing identified mutations in the nuclear-encoded alanyl-tRNA synthetase (AARS) in these two unrelated families: the two affected siblings are compound heterozygous for p.Lys81Thr and p.Arg751Gly AARS, and the single affected child is homozygous for p.Arg751Gly AARS. The two identified mutations were found to result in a significant reduction in function. Mutations in AARS were previously associated with an autosomal-dominant inherited form of axonal neuropathy, Charcot-Marie-Tooth disease type 2N (CMT2N). The autosomal-recessive AARS mutations identified in the individuals described here, however, cause a severe infantile epileptic encephalopathy with a central myelin defect and peripheral neuropathy, demonstrating that defects of alanyl-tRNA charging can result in a wide spectrum of disease manifestations.
Main Text
Advances in next-generation sequencing have reduced the number of unsolved cases in genetic leukoencephalopathies and leukodystrophies; however, many still remain a diagnostic mystery. Mutations in genes encoding aminoacyl-transfer RNA (tRNA) synthetases have received increased attention in individuals with white-matter abnormalities on MRI due to the recent identification of a series of related diseases involving these enzymes. Mutations in the majority of tRNA synthetases (22 of 37) and many tRNA synthetase cofactors have been implicated in human disease (Table S1),1–15 and of these, mutations in eight loci (DARS [MIM 615281], RARS [MIM 616140], AIMP1 [MIM 260600], AARS2 [MIM 615889], DARS2 [MIM 611105], EARS2 [MIM 614924], LARS2 [MIM 615300], MARS2 [MIM 611390], FARS2 [MIM 614946]) have been implicated in leukodystrophies or leukoencephalopathies.2,8,9,13,14,16–18 Aminoacyl-tRNA synthetases are ubiquitously expressed, essential enzymes responsible for charging tRNA molecules with cognate amino acids—the first step of protein translation. These enzymes are present in both mitochondria and the cytoplasm and, interestingly, certain tRNA synthetases have non-canonical functions in biological processes such as angiogenesis, regulation of gene transcription, and RNA splicing.19 These non-canonical tRNA synthetase functions are conserved across the complete phylogeny of animals and are now established as playing key roles in a number of pathophysiological processes.19
Here we report three individuals with remarkably similar clinical presentations and biallelic loss-of-function mutations in alanyl-tRNA synthetase (AARS [MIM 601065]). The clinical presentations of these individuals include severe infantile epileptic encephalopathy with myoclonic seizures and extrapyramidal features, peripheral neuropathy based on absent peripheral reflexes, congenital vertical tali, and neuroradiologic features consistent with persistently deficient myelination. Our functional analyses reveal that the identified mutations reduce aminoacylation activity, thus implicating AARS mutations in an autosomal-recessive early infantile epileptic encephalopathy. To date, AARS mutations have been implicated only in autosomal-dominant Charcot-Marie-Tooth type 2N (CMT2N [MIM 613287]), an axonal peripheral neuropathy characterized by muscle weakness, wasting, and impaired sensation in the extremities.20,21 Thus, our findings broaden the genetic and clinical spectrum associated with AARS mutations.
The three affected individuals and their families were collected prospectively as part of the Myelin Disorders Bioregistry Project (MDBP) and the NIH Undiagnosed Diseases Program with approval from the institutional review boards at Children’s National Health System and the National Human Genome Research Institute, respectively. Written informed consent was obtained for each study participant including specific informed consent for whole exome sequencing (WES). Genomic DNA samples were collected from blood samples provided to the biorepository.
The clinical features of each individual are summarized in Table 1 and full clinical descriptions are provided in the Supplemental Data case reports. LD_0115.0A and LD_0115.0B are sibling of mixed-European descent with a history of hypomyelinating leukoencephalopathy, epileptic encephalopathy with congenital vertical tali, microcephaly, failure to thrive, and developmental delay. On examination the children had severe encephalopathy, extrapyramidal symptoms including blepharospasm, spasticity, and absent peripheral reflexes. Both siblings are similarly affected, although LD_0115.0A, the elder of the sibling pair, is more severely affected.
Table 1.
Summary of Key Clinical Features of Affected Individuals
| LD_0115.0A | LD_0115.0B | LD_0857.0 | |
|---|---|---|---|
| Nucleotide variants | c.242A>C, c.2251A>G | c.242A>C, c.2251A>G | c.2251A>G |
| Protein variants | p.Lys81Thr, p.Arg751Gly | p.Lys81Thr, p.Arg751Gly | p.Arg751Gly |
| Status | compound heterozygous | compound heterozygous | homozygous |
| Intrauterine growth retardation | yes | no | yes |
| Age at presentation | 36 weeks GA | 3 months | birth |
| Congenital vertical tali | yes | yes | yes |
| Failure to thrive | yes | yes | yes |
| Microcephaly | << 4 SD below the norm | << 4 SD below the norm | << 4 SD below the norm |
| Refractory myoclonic epilepsy | onset at 2 months | onset at 3 months | onset at 6 months |
| Spasticity | yes | yes | yes |
| Chorea/adventitious movement | blepharospasm, orobuccal dyskinesia, dystonia of limbs, chorea | blepharospasm, orobuccal dyskinesia, dystonia of limbs, chorea | blepharospasm, orobuccal dyskinesia, dystonia of limbs, chorea |
| Loss of peripheral deep tendon reflexes | yes | yes | yes |
Abbreviation is as follows: GA, gestational age.
LD_0857.0 is of mixed European descent and is unrelated to the siblings described above. He presented after a full-term birth with intrauterine growth retardation, abnormal movements characterized as dystonia and tremulousness, microcephaly, and congenital vertical tali. In the first months of life, he developed progressive microcephaly, failure to thrive, refractory myoclonic epilepsy, feeding difficulties, and nystagmus. On examination he also had severe encephalopathy, extrapyramidal symptoms including blepharospam, spasticity, and absent peripheral reflexes. LD_0857.0 is the most severely affected of the three children and has been critically ill numerous times, requiring mechanical ventilation. He is currently dependent on nasal BiPAP support for home pulmonary care.
EEGs were obtained in the newborn period for LD_0115.0A and LD_0857.0 and were indicative of mild encephalopathy with excessive sharp waves seen in the central-temporal areas. Later recordings in all three subjects demonstrated moderate diffuse background slowing (or suppression in LD_0857.0) with multifocal posterior-predominant interictal epileptiform discharges, typically polyspike, and slow-wave discharges or high-amplitude occipital paroxysmal fast (beta) activity (Figure S1). In one individual (LD_0115.0B), routine EEG performed at age 5 months showed multiple tonic extensor spasms associated with a generalized electrodecrement, but no hypsarrhythmia.
Several brain MRI findings were common among all three individuals (Figure 1). A relentlessly progressive diffuse cerebral atrophy affects both gray and white matter, evolving from mild to severe with onset in the first few weeks of life. Early scans from LD_0115.0A and LD_0857.0 performed on days 0 and 27, respectively, showed normal brain volume. Slow progression of mild to moderate generalized brainstem and cerebellar volume loss was present in all individuals. Posterior fossa volume loss commenced after the initiation of cerebral volume loss, no earlier than 2 to 8 months of age. A progressive micro-encephaly was consequent.
Figure 1.
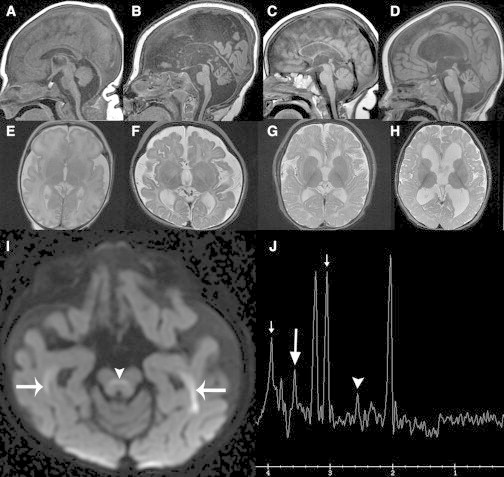
MRI Findings in Individuals with AARS Mutations
(A–D) Sagittal T1-weighted images show varying degrees of parenchymal volume loss (B–D) manifesting different degrees of thinning of the corpus callosum, enlarged sulci/fissures, and ventriculomegaly along with relative hypointensity of the corpus callosum reflecting hypomyelination. Cerebral volume was normal in LD_0857.0 at day 0 (A) with progressive cerebral atrophy, severe at 14 months (B). The vermis evolved from being hypoplastic (A) to atrophic (B) and the brainstem showed no interval growth (B). LD_0115.0A (C) had moderate cerebral atrophy and mild cerebellar volume loss by 14 months, and LD_0115.0B (D) had mild cerebral and cerebellar volume loss at 17 months. Disproportionate lateral ventriculomegaly reflects superimposed hydrocephalus from prior intraventricular hemorrhage (D).
(E–H) Axial T2-weighted images demonstrate variable degrees of cerebral volume loss/atrophy (F–H) and hypomyelination (E–H). Generalized, excessive-for-age white matter hyperintensity reflects hypomyelination. At birth, the normal hypointense PLIC signal is not visible in LD_0857.0 (E). By 14 months, only mild myelination progress has occurred with myelination similar to that of a normal 3 month old; the optic radiations and PLIC are now hypointense. Similar findings are present at 14 months in LD_0115.0A (G) and 17 months in LD_0115.0B (H).
(I) Axial DWI image from LD_0857.0 at 14 months shows hyperintense signal consistent with restricted diffusion and myelin edema in the optic radiations (arrows) and superior cerebellar peduncle decussations (arrowhead). Similar myelin edema was present in the occipitofrontal fascicles, central tegmental tracts, and globi pallidi (not shown).
(J) SV MRS (144 ms echo time) over the left basal ganglia reveals elevation of citrate at 2.6 ppm (arrowhead), glycine at 3.5 ppm (long arrow), and creatine at 3 ppm and 3.9 ppm (short arrows).
Hypomyelination was universal, present in all individuals, and clinically suspected on all exams from day 0 to 30 months, but was not formally diagnosed until follow-up MRI was performed at 12 months. A slow myelin progression occurred in all individuals without normalization up to at least 30 months. Myelination progressed to no more than the degree expected for a normal 6-month-old subject. Cerebellar and brainstem myelination ranged from normal to mildly decreased. All individuals had evidence of prior choroid plexus hemorrhage in the lateral ventricular glomus. One individual (LD_0115.0B) had a chronic non-communicating hydrocephalus probably secondary to intraventricular hemorrhage causing cerebral aqueduct adhesions.
WES was performed on all affected individuals and their parents as part of a cohort of 70 families diagnosed with unresolved leukodystrophies recruited to the MDBP. However, prior to exome sequencing, the three individuals were subgrouped within the broader cohort on the basis of their phenotypic homogeneity. Exome capture and sequencing was performed at the Queensland Centre for Medical Genomics with either Illumina Nextera Rapid Capture or the SeqCap EZ Human Exome Library v.3.0 kits with captured libraries sequenced with the Illumina platform. Reads were aligned to the reference human genome (GRCh37) and pedigree-informed variant calling was performed with the Real Time Genomics (RTG) integrated analysis tool rtgFamily v.3.2.22 All variants were annotated with SnpEff v.3.423 and filtered to include SNPs and indels predicted to result in altered mRNA splicing or protein sequence. Rare variants were identified on the basis of a MAF < 0.01 in both the NHLBI Exome Sequencing Project (ESP) Exome Variant Server and 1000 Genomes database (October 2013). Subsequent identification and prioritization of candidate variants was performed with an in-house workflow incorporating the annotated variant data and segregation within each pedigree.
Analysis of the exomes revealed that all three affected individuals carry rare, biallelic missense variants in AARS with no other compelling pathogenic variants common to the two families (Figure 2). The affected siblings LD_0115.0A and LD_0115.0B are compound heterozygous carriers of two AARS missense mutations. The maternally inherited mutation (RefSeq accession number NM_001605.2; c.242A>C [p.Lys81Thr]) is not present in the dbSNP v.141, 1000 Genomes, NHLBI ESP6500, or Exome Aggregation Consortium (ExAC) databases and is predicted to be damaging by both SIFT (score: 0.000) and PolyPhen2 (score: 1.00). The paternally inherited mutation (RefSeq NM_001605.2; c.2251A>G [p.Arg751Gly]) is present in dbSNP (rs143370729)—however, from a cohort of more than 60,000 individuals, the ExAC project identified only six heterozygous carriers of this allele, giving an allele frequency of less than 0.00005 in this predominantly European population. This mutation is also predicted to be damaging by both SIFT (score: 0.000) and PolyPhen2 (score: 1.00). The third affected individual LD_0857.0 is homozygous for the same p.Arg751Gly mutation identified in the LD_0115 siblings. Neither the p.Lys81Thr nor p.Arg751Gly missense mutations were detected in any other individuals in an in-house control dataset of more than 350 exomes.
Figure 2.
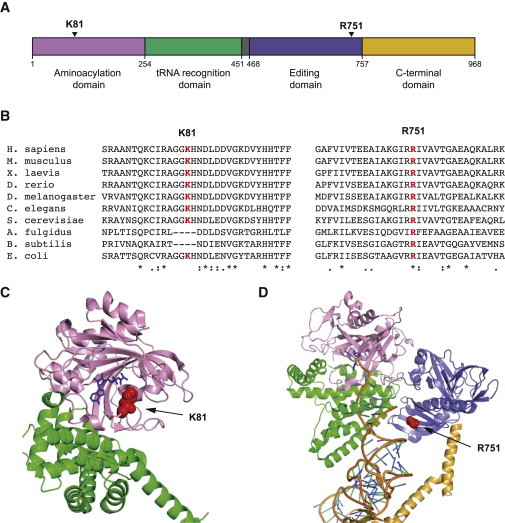
Pathogenic Mutations in Human AARS
(A) The p.Lys81Thr mutation and p.Arg751Gly mutations mapped to the known functional domains of human AARS, showing the aminoacylation domain, the tRNA recognition domain, the editing domain, and the C-terminal domain.
(B) Multiple-species sequence alignment of AARS showing the p.Lys81Thr mutation and p.Arg751Gly mutation along with the flanking protein sequences in multiple and evolutionarily diverse species.
(C) Structure of an E. coli alanyl-tRNA synthetase in complex with alanine analog.26 Arrow indicates the conserved residue homologous to human p.Lys81.
(D) Structure of archaea A. fulgidus alanyl-tRNA synthetase in complex with tRNA and an alanine analog.24 Arrow indicates the conserved residue homologous to human p.Arg751.
In (C) and (D), the AARS domains are colored as shown in (A).
AARS consists of four highly conserved functional domains: the aminoacyl-tRNA synthetase domain (residues 1–254), the tRNA recognition domain (residues 255–451), the editing domain (residues 468–757), and the C-terminal domain (residues 758–968) (Figure 2).24 The p.Lys81Thr variant affects a lysine residue that resides in the aminoacyl-tRNA synthetase domain, whereas the p.Arg751Gly variant affects an arginine residue localized in the AARS editing domain (Figure 2A). Both residues are highly evolutionarily conserved: p.Lys81 is conserved in animals, yeast, and bacteria, and p.Arg751 is also strictly conserved in archaea (Figure 2B).
AARS catalyzes the aminoacylation of tRNAAla with alanine in a reaction dependent on ATP. The majority of disease-associated mutations in aminoacyl-tRNA synthetases, including those in AARS associated with CMT2N, have been shown to impair aminoacylation activity.20,21 We tested the ability of p.Lys81Thr and p.Arg751Gly mutant AARS to catalyze aminoacylation in vitro compared to wild-type AARS by monitoring incorporation of tritium-labeled alanine onto in vitro transcribed tRNAAla. Steady-state assays for aminoacylation showed that the wild-type enzyme displayed Km (tRNA) and kcat values similar to those reported previously, showing a kcat/Km value of 0.13 ± 0.04 μM−1s−1 (Table 2).21 The p.Lys81Thr mutant was mildly defective, due to a 2-fold increase in Km (tRNA) with no change in kcat, leading to an overall 2-fold decrease in kcat/Km. In contrast, the p.Arg751Gly mutant was more severely defective, showing both a 2-fold increase in Km (tRNA) and a 5-fold decrease in kcat, leading to an overall 10-fold decrease in kcat/Km. Thus, both mutations impaired the aminoacylation activity of AARS in vitro. The p.Arg751Gly mutation is localized within the AARS editing domain, so we also investigated the impact of this variant on editing activity; however, there was no detectable effect of the mutation on editing function (Figure 3).
Table 2.
Aminoacylation Kinetics of AARS Variants
| Km(μM) | kcat(s−1) | kcat/Km(μM−1s−1) | Ratio to WT | |
|---|---|---|---|---|
| WT | 3.1 ± 0.7 | 0.4 ± 0.1 | 0.13 ± 0.04 | 1 |
| p.Lys81Thr | 6.0 ± 1.8 | 0.4 ± 0.1 | 0.07 ± 0.03 | 1/2 |
| p.Arg751Gly | 6.3 ± 1.4 | 0.07 ± 0.03 | 0.010 ± 0.005 | 1/10 |
Steady-state assays of aminoacylation of tRNAAla with 3H-alanine (20 μM) were performed at 37°C with each enzyme at 20 nM, tRNAAla in varying concentrations (0.6–16 μM) in a buffer containing 20 mM KCl, 50 mM HEPES (pH 7.5), 4.0 mM DTT, 0.2 mg/ml BSA, 10 mM MgCl2, and 2 mM ATP. Error bars show standard deviations derived from averaging at least four independent sets of experiments. Wild-type and mutants of human AARS were constructed in pET-DEST42 gateway vector (Life Technologies) for expression in E. coli with a 6× His C-terminal fusion tag. Protein was purified with the Co2+-affinity resin was achieved according to the manufacturer’s protocol (Novagen). Human tRNAAla was prepared and purified and annealed as described.1,2 Steady-state aminoacylation assays were performed as described, monitoring incorporation of 3H-Ala (specific activity of 16,500 dpm/pmole) to the synthesis of 3H-Ala-tRNAAla as acid precipitable counts on filter pads.1 Each reaction was initiated by addition of an AARS enzyme (20 nM) to a mixture of increasing concentrations of tRNAAla (0.6–16 mM). Acid precipitable counts were corrected for the filter quench factor and were calculated as pmoles of 3H-Ala-tRNAAla. Data were fit to the Michaelis-Menten equation.
Figure 3.
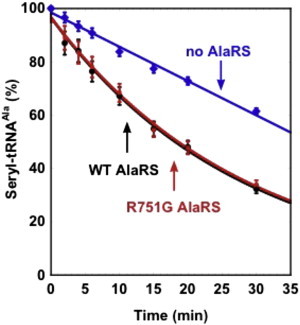
Analysis of the Hydrolytic Editing Activity of AARS
Deacylation of the incorrectly charged Ser-tRNAAla was monitored over time by the wild-type enzyme (black) and the p.Arg751Gly mutant (red). The uncatalyzed deacylation reaction (the no-enzyme reaction, indicated in blue) was run in parallel as a control for background hydrolysis. Values represent the average of two independent experiments and error bars show the standard deviation. The substrate Ser-tRNAAla for analysis of post-transfer editing was prepared by 32P-labeling of the QA A76 nucleotide in the transcript of human tRNAAla with the CCA enzyme,3 followed by aminoacylation with chemically synthesized Ser-DBE using the dFx flexizyme.4,5 Deacylation was monitored in 50 mM HEPES (pH 7.5), 20 mM KCl, 4 mM DTT, and 10 mM MgCl2 at 37°C with 5 nM of WT or p.Arg751Gly human AARS and a mixture of 2.8 μM Ser-tRNAAla and 17.2 μM tRNAAla. At the indicated time points, aliquots of a deacylation reaction were quenched with a buffer containing 200 mM NaOAc (pH 5.0) and treated with S1 nuclease for 20 min at 37°C to digest the tRNA to 5′-phosphate mononucleotides. The aliquots were then subjected to thin layer chromatography through a PEI cellulose matrix with 0.1 M NH4Cl and 5% acetic acid to separate [32P]seryl-AMP from [32P]AMP. Quantification of the two products by a phosphorimager and analysis of the ratio of (ser-AMP) over (AMP) determined the extent of Ser-tRNAAla remaining after the editing reaction.
To further assess the functional consequences of p.Lys81Thr and p.Arg751Gly in AARS in vivo, we modeled each mutation in the yeast ortholog ALA1 and compared the ability of mutant and wild-type ALA1 to support yeast cell growth in a haploid yeast strain with the endogenous ALA1 deleted.21 The wild-type ALA1 vector rescued yeast viability, whereas the empty vector was unable to complement loss of the endogenous ALA1 allele (Figure 4). Yeast expressing p.Arg751Gly ALA1 showed survival comparable to those expressing wild-type ALA1. Yeast expressing p.Lys81Thr ALA1, however, demonstrated dramatically reduced, but not ablated, growth (Figure 4), indicating that p.Lys81Thr AARS represents a hypomorphic AARS allele in vivo.
Figure 4.

Yeast Complementation Studies
Yeast complementation assays were performed as previously described.1 Haploid Δala1 yeast strains carrying a wild-type ALA1 on a URA3-bearing vector were transformed with a LEU2-bearing vector containing wild-type ALA1, p.Lys85Thr ALA1 (orthologous to human p.Lys81Thr AARS), p.Arg747Gly ALA1 (orthologous to human p.Arg751Gly AARS), or no insert (pRS315 Empty). Single colonies from two independent transformations were grown to saturation in SD -leu-ura liquid media (Teknova) then spotted (undiluted or diluted 1:10 and 1:100 in H2O) on plates containing 0.1% 5-fluoroorotic acid (5-FOA) or SD -leu-ura growth media (Teknova) and incubated at 30°C for 48 hr. Survival was determined by visual inspection of growth. Culture dilutions are indicated on the left and media conditions are indicated on the right.
This study has provided insight into the pathogenic mechanism of the p.Lys81Thr and p.Arg751Gly AARS mutations identified in three individuals with a phenotypically identical recessive, early-onset epileptic encephalopathy. Modeling of the E. coli crystal structure of AARS24 indicates that the p.Lys81Thr mutation affects a highly conserved lysine residue (Figure 2B) in a loop adjacent to the AARS synthetic active site (Figure 2C). The p.Lys81Thr variant reduced yeast growth in vivo and decreased aminoacylation activity by 50% in vitro. The p.Arg751Gly mutation affects a highly conserved arginine residue (Figure 2B) at the interface with the backbone of the 5′ nucleotide in the acceptor stem of tRNAAla when modeled on the A. fulgidus AARS, possibly playing a role in orienting the acceptor end for aminoacylation (Figure 2D).24 The pronounced aminoacylation activity impairment seen for the p.Arg751Gly mutation, decreasing the kcat/Km value 10-fold, is consistent with the more severe phenotype of LD_0857.0, who is homozygous for this allele.
The data presented here implicate loss-of-function AARS mutations in a previously unreported recessive phenotype of severe epileptic encephalopathy with central myelin deficits and peripheral neuropathy based on absent peripheral reflexes. The severe delay in myelination seen in these cases is not consistent with classic hypomyelinating leukodystrophies, in which there is typically no progression of myelination over time. At this point, it is unclear whether AARS mutations result in a primary defect of myelin development or whether the delay in myelination can be attributed to primary neuronal dysfunction.25
It is intriguing to note that two previously reported AARS missense mutations20,21 cause Charcot-Marie-Tooth type 2N (CMT2N), a dominant axonal peripheral neuropathy characterized by muscle weakness, wasting, and impaired sensation in the extremities. These mutations have catastrophic effects on AARS activity (50-fold and 4,130-fold decreases in kcat/Km). This is in stark contrast to the relatively mild 10-fold and 2-fold reduction in aminoacylation activity associated with the mutations described here. This suggests a model by which heterozygosity for a variant that reduces AARS aminoacylation activity below a certain threshold presents as a dominant peripheral neuropathy, probably through the compensatory function of the remaining wild-type allele. However, carrying two hypomorphic AARS alleles results in a more severe overall reduction in AARS function and causes not only a peripheral neuropathy, but also a severe infantile epileptic encephalopathy. In summary, our findings implicate an autosomal-recessive phenotype caused by AARS mutations and reveal a broader-than-previously-appreciated spectrum of disorders.
Acknowledgments
We thank the individuals described in this manuscript and their families. The participation of G.H. and A.V. was supported by the Myelin Disorders Bioregistry Project. G.H. is also supported by the Delman Fund for Pediatric Neurology Education. This publication was supported by Award Number UL1TR000075 from the NIH National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Advancing Translational Sciences or the NIH. This work was also supported by the National Institute of General Medical Sciences (GM110184 to A.A.), the NIH Cellular and Molecular Biology Training Grant (T32 GM007315 to L.B.G.), the Tolz Foundation for Research and the Keystone Innovation Network award (to Y.M.H.), the NIH Medical Scientist Training Grant (T32 GM007863 to L.B.G.), and other NIH grants (F30 NS092238 to L.B.G.). The NIH Undiagnosed Diseases Program evaluated LD_0115.0A and LD_0115.0B. We thank the QCMG and IMB sequencing facility teams for their assistance with this project. Computational support was provided by the NeCTAR Genomics Virtual Laboratory and QRIScloud programs. R.J.T. was supported by an Australian Research Council Discovery Early Career Research Award. This work was supported by National Health and Medical Research Council, Australia Grant (APP1068278), and a University of Queensland Foundation Research Excellence Award. We also thank Marjo van der Knaap for her manuscript revision.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
ExAC Browser, http://exac.broadinstitute.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
References
- 1.Almalki A., Alston C.L., Parker A., Simonic I., Mehta S.G., He L., Reza M., Oliveira J.M.A., Lightowlers R.N., McFarland R. Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency. Biochim. Biophys. Acta. 2014;1842:56–64. doi: 10.1016/j.bbadis.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dallabona C., Diodato D., Kevelam S.H., Haack T.B., Wong L.J., Salomons G.S., Baruffini E., Melchionda L., Mariotti C., Strom T.M. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology. 2014;82:2063–2071. doi: 10.1212/WNL.0000000000000497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMillan H.J., Schwartzentruber J., Smith A., Lee S., Chakraborty P., Bulman D.E., Beaulieu C.L., Majewski J., Boycott K.M., Geraghty M.T. Compound heterozygous mutations in glycyl-tRNA synthetase are a proposed cause of systemic mitochondrial disease. BMC Med. Genet. 2014;15:36. doi: 10.1186/1471-2350-15-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steenweg M.E., Ghezzi D., Haack T., Abbink T.E., Martinelli D., van Berkel C.G., Bley A., Diogo L., Grillo E., Te Water Naudé J. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain. 2012;135:1387–1394. doi: 10.1093/brain/aws070. [DOI] [PubMed] [Google Scholar]
- 5.Yamashita S., Miyake N., Matsumoto N., Osaka H., Iai M., Aida N., Tanaka Y. Neuropathology of leukoencephalopathy with brainstem and spinal cord involvement and high lactate caused by a homozygous mutation of DARS2. Brain Dev. 2013;35:312–316. doi: 10.1016/j.braindev.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Riley L.G., Menezes M.J., Rudinger-Thirion J., Duff R., de Lonlay P., Rotig A., Tchan M.C., Davis M., Cooper S.T., Christodoulou J. Phenotypic variability and identification of novel YARS2 mutations in YARS2 mitochondrial myopathy, lactic acidosis and sideroblastic anaemia. Orphanet J. Rare Dis. 2013;8:193. doi: 10.1186/1750-1172-8-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pierce S.B., Chisholm K.M., Lynch E.D., Lee M.K., Walsh T., Opitz J.M., Li W., Klevit R.E., King M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA. 2011;108:6543–6548. doi: 10.1073/pnas.1103471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pierce S.B., Gersak K., Michaelson-Cohen R., Walsh T., Lee M.K., Malach D., Klevit R.E., King M.C., Levy-Lahad E. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am. J. Hum. Genet. 2013;92:614–620. doi: 10.1016/j.ajhg.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayat V., Thiffault I., Jaiswal M., Tétreault M., Donti T., Sasarman F., Bernard G., Demers-Lamarche J., Dicaire M.J., Mathieu J. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012;10:e1001288. doi: 10.1371/journal.pbio.1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belostotsky R., Ben-Shalom E., Rinat C., Becker-Cohen R., Feinstein S., Zeligson S., Segel R., Elpeleg O., Nassar S., Frishberg Y. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet. 2011;88:193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diodato D., Melchionda L., Haack T.B., Dallabona C., Baruffini E., Donnini C., Granata T., Ragona F., Balestri P., Margollicci M. VARS2 and TARS2 mutations in patients with mitochondrial encephalomyopathies. Hum. Mutat. 2014;35:983–989. doi: 10.1002/humu.22590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolf N.I., Salomons G.S., Rodenburg R.J., Pouwels P.J., Schieving J.H., Derks T.G., Fock J.M., Rump P., van Beek D.M., van der Knaap M.S., Waisfisz Q. Mutations in RARS cause hypomyelination. Ann. Neurol. 2014;76:134–139. doi: 10.1002/ana.24167. [DOI] [PubMed] [Google Scholar]
- 14.Taft R.J., Vanderver A., Leventer R.J., Damiani S.A., Simons C., Grimmond S.M., Miller D., Schmidt J., Lockhart P.J., Pope K. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet. 2013;92:774–780. doi: 10.1016/j.ajhg.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X., Ling J., Barcia G., Jing L., Wu J., Barry B.J., Mochida G.H., Hill R.S., Weimer J.M., Stein Q. Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am. J. Hum. Genet. 2014;94:547–558. doi: 10.1016/j.ajhg.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheper G.C., van der Klok T., van Andel R.J., van Berkel C.G., Sissler M., Smet J., Muravina T.I., Serkov S.V., Uziel G., Bugiani M. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 2007;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 17.Almalki A., Alston C.L., Parker A., Simonic I., Mehta S.G., He L., Reza M., Oliveira J.M., Lightowlers R.N., McFarland R. Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency. Biochim. Biophys. Acta. 2014;1842:56–64. doi: 10.1016/j.bbadis.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feinstein M., Markus B., Noyman I., Shalev H., Flusser H., Shelef I., Liani-Leibson K., Shorer Z., Cohen I., Khateeb S. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am. J. Hum. Genet. 2010;87:820–828. doi: 10.1016/j.ajhg.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao P., Poruri K., Martinis S., Fox P. Non-catalytic Regulation of Gene Expression by Aminoacyl-tRNA Synthetases. In: Kim S., editor. Aminoacyl-tRNA Synthetases in Biology and Medicine. Springer; 2014. pp. 167–187. [DOI] [PubMed] [Google Scholar]
- 20.Latour P., Thauvin-Robinet C., Baudelet-Méry C., Soichot P., Cusin V., Faivre L., Locatelli M.C., Mayençon M., Sarcey A., Broussolle E. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2010;86:77–82. doi: 10.1016/j.ajhg.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLaughlin H.M., Sakaguchi R., Giblin W., Wilson T.E., Biesecker L., Lupski J.R., Talbot K., Vance J.M., Züchner S., Lee Y.C., NISC Comparative Sequencing Program A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N) Hum. Mutat. 2012;33:244–253. doi: 10.1002/humu.21635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cleary J.G., Braithwaite R., Gaastra K., Hilbush B.S., Inglis S., Irvine S.A., Jackson A., Littin R., Nohzadeh-Malakshah S., Rathod M. Joint variant and de novo mutation identification on pedigrees from high-throughput sequencing data. J. Comput. Biol. 2014;21:405–419. doi: 10.1089/cmb.2014.0029. [DOI] [PubMed] [Google Scholar]
- 23.Cingolani P., Platts A., Wang L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naganuma M., Sekine S., Chong Y.E., Guo M., Yang X.-L., Gamper H., Hou Y.-M., Schimmel P., Yokoyama S. The selective tRNA aminoacylation mechanism based on a single G•U pair. Nature. 2014;510:507–511. doi: 10.1038/nature13440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanderver A., Prust M., Tonduti D., Mochel F., Hussey H., Helman G., Garbern J., Eichler F., Labauge P., Aubourg P. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol. Genet. Metab. 2015 doi: 10.1016/j.ymgme.2015.01.006. Published online January 29, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo M., Chong Y.E., Shapiro R., Beebe K., Yang X.L., Schimmel P. Paradox of mistranslation of serine for alanine caused by AlaRS recognition dilemma. Nature. 2009;462:808–812. doi: 10.1038/nature08612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.