

Abstract
Chromothripsis represents an extreme class of complex chromosome rearrangements (CCRs) with major effects on chromosomal architecture. Although recent studies have associated chromothripsis with congenital abnormalities, the incidence and pathogenic effects of this phenomenon require further investigation. Here, we analyzed the genomes of three families in which chromothripsis rearrangements were transmitted from a mother to her child. The chromothripsis in the mothers resulted in completely balanced rearrangements involving 8–23 breakpoint junctions across three to five chromosomes. Two mothers did not show any phenotypic abnormalities, although 3–13 protein-coding genes were affected by breakpoints. Unbalanced but stable transmission of a subset of the derivative chromosomes caused apparently de novo complex copy-number changes in two children. This resulted in gene-dosage changes, which are probably responsible for the severe congenital phenotypes of these two children. In contrast, the third child, who has a severe congenital disease, harbored all three chromothripsis chromosomes from his healthy mother, but one of the chromosomes acquired de novo rearrangements leading to copy-number changes. These results show that the human genome can tolerate extreme reshuffling of chromosomal architecture, including breakage of multiple protein-coding genes, without noticeable phenotypic effects. The presence of chromothripsis in healthy individuals affects reproduction and is expected to substantially increase the risk of miscarriages, abortions, and severe congenital disease.
Main Text
Complex chromosomal rearrangements (CCRs) underlie congenital abnormalities and are thought to be an important contributor to spontaneous abortions in females and to infertility in males.1–4 Phenotypically normal individuals harboring CCRs have been described, but typically have a copy-number-balanced profile and less-complex rearrangements than phenotypically abnormal individuals. Chromothripsis represents an extreme form of CCRs and has previously been linked to cancer and severe congenital abnormalities. The phenomenon is characterized by local shattering of one or multiple chromosomes and random reassembly of the fragments and typically has a devastating effect on chromosomal architecture and a major impact on human health.5–9
We further examined the genomes of two previously described children referred to our Medical Center for a variety of complex congenital abnormalities, and we also investigated the genome of one additional child (Table S1)8,10,11. We obtained appropriate informed consent from the involved subjects to analyze their genomes and publish the findings. By using Illumina BeadChip arrays or custom Agilent 105k microarrays, we identified from two to five de novo copy-number changes per child; changes ranged in size from 150 kb to 27 Mb and involved 2 or 3 chromosomes per child (Figure S1). Giemsa (G)-banded chromosome analysis revealed the presence of CCRs involving 1–3 chromosomes in each of the three children (Figures S2A–S2D). Chromosome analysis of the parents showed that in all three cases the mother’s karyotype contained all derivative chromosomes identified in her child (Figures S2E–S2H), whereas each of the three fathers displayed a normal karyotype. Furthermore, we identified an additional three derivative chromosomes in the mother of child 1 and one additional derivative chromosome in the mother of child 2 (Figures S2F and S2G). Karyotyping did not reveal any differences between derivative chromosomes identified in child 3 and her mother (Figures S2D and S2H), but previously published FISH (fluorescence in situ hybridization) studies identified de novo rearrangements occurring in the child and resulting in the de novo deletion and duplication.11 Notably, mothers 1 and 3 are healthy, whereas mother 2 displays a much milder phenotype than her affected child; this phenotype consists of delayed psychomotor development and major learning difficulties but no facial dysmorphisms.10 We analyzed chromosome spreads from at least 20 lymphocytes and did not find evidence for mosaicism in the mothers.
To further explore the complexity of the chromosomal rearrangements, we performed whole-genome mate-pair sequencing for all three children and their mothers (Table S2).7,8 We selected breakpoints by filtering data for these three mother-child pairs against a set of 150 control mate-pair datasets and against data from the Genome of the Netherlands Project.12 In addition, we performed validation assays with PCR and Sanger sequencing. We identified 13 (mother 1), 23 (mother 2), and 8 (mother 3) unique breakpoint junctions in the mothers; their sequences are consistent with non-homologous repair mechanisms (Figure 1A, Table S2). Furthermore, plotting of the breakpoint junctions onto the reference genome revealed signatures of double-stranded DNA breaks (Figure 1B, Figure S3). We used the orientations and positions of the breakpoint junctions to reconstruct digital karyotypes, resulting in four derivative chromosomes for mother 1 and five for mother 2 (Figure 2).6–8,13 We were unable to completely reconstruct the derivative chromosomes for mother 3; probably, we missed some breakpoint junctions because the repetitive character of the affected genomic regions (e.g., 3q29) hampered the unique mapping of sequence reads (Figure S3B). The reconstructed chromosomes match the G-banded chromosome analysis for mother 1 (Figure 2A). However, mate-pair sequencing revealed that five (chr6, chr7, chr9, chr10, and chr12) rather than two chromosomes were involved in the rearrangements in mother 2, emphasizing the importance of next-generation sequencing for revealing the full complexity of the rearrangements (Figure 2B). Taken together, the presence of large numbers of clustered double-stranded breaks affecting a single haplotype, the randomness of breakpoint-junction orientations and DNA-segment order, and the ability to walk the derivative chromosomes provide strong evidence that the rearrangements in the three mothers and their children resulted from germline chromothripsis (Figure 1B, Figure S3A, Table S3).13 Regularly oscillating copy-number states—typical for cancer chromothripsis—were not observed, consistent with the more balanced state of previously described germline-chromothripsis–affected individuals. Seven out of 13 published cases are completely balanced, whereas the other six cases show only 2–4 copy-number changes.6–8,14,15 The more balanced state of germline chromothripsis is generally thought to be a consequence of selective pressure during embryogenesis.6,16
Figure 1.
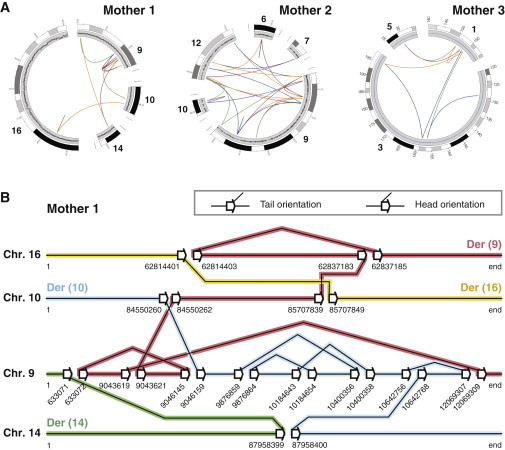
Chromothripsis Involving Multiple Chromosomes Can Be Stably Present in Healthy Individuals
(A) Circos plots of breakpoint junctions (solid lines) in the three mothers. Lines are colored according to the orientation of the breakpoint junction, from low to high chromosomal coordinate: tail-head (blue), head-tail (green), head-head (red), and tail-tail (orange). 13, 23, and 8 rearrangements were detected in mothers 1, 2, and 3, respectively.
(B) Schematic diagram showing the exact genomic positions and orientations of breakpoint junctions detected in mother 1. Sets of adjacent white arrows indicate a double-strand break (DSB), and connecting lines between two arrows indicate breakpoint junctions. This panel shows our rationale for resolving chromosomal structure on the basis of the breakpoint junctions. We produced digital karyotypes by following the breakpoint junctions. By doing so, we predicted that the breakpoint junctions in mother 1 gave rise to four derivative chromosomes, indicated by the colored lines: der(9) (red), der(10) (blue), der(14) (green), and der(16) (yellow). This configuration is fully in line with the karyotypes derived from G-banded chromosome analysis (Figure S2).
Figure 2.
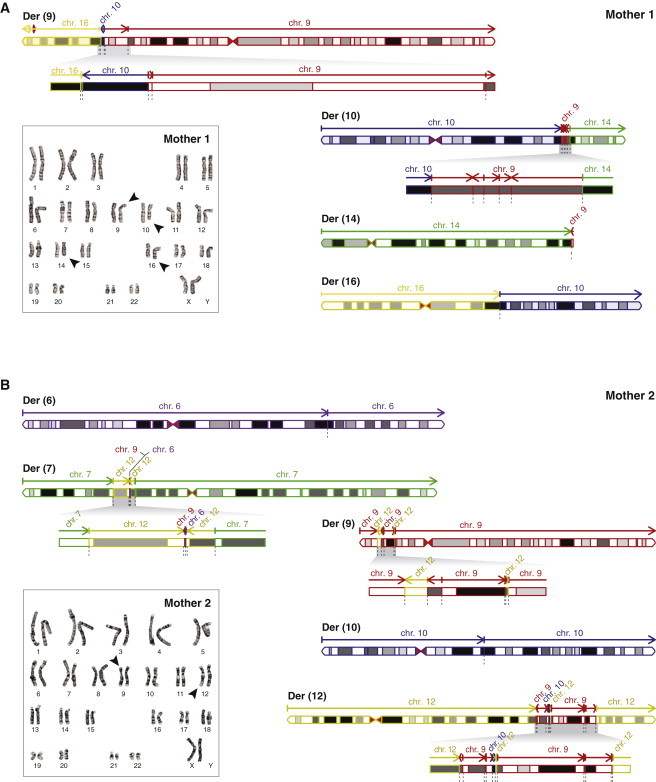
Reconstruction of Digital Karyotypes Based on the Breakpoint Junctions in Mothers 1 and 2
(A) Karyogram and digital karyotype as derived from the sequencing data of mother 1. Chromosome segments are colored according to their origin: chr9 (red), chr10 (blue), chr14 (green), and chr16 (yellow). Der(9) and der(10) harbor a region with a large number of small rearranged fragments (zoom panels). Arrows indicate the orientation of the chromosomal fragments; dotted gray lines indicate breakpoint junctions. The predicted structure of the rearranged chromosomes matches the G-banded karyotype of the mother.
(B) Karyogram and digital karyotype as derived from sequencing data of mother 2. Chromosome segments are colored according to their origin: chr6 (purple), chr7 (green), chr9 (red), chr10 (blue), and chr12 (yellow). Der(7), der(9), and der(12) each harbor one or multiple regions with a large number of small rearrangements (zoom panels). In contrast to that of mother 1, karyotyping of mother 2 revealed only two [der(9) and der(12)] out of five derivative chromosomes detected by mate-pair sequencing (Figure S2G).
The large numbers of breakpoints in each of the three mothers, and the lack of a phenotype in two of them, raised the question of whether breakpoints affected their gene function. By examining the overlap between breakpoints and protein-coding genes, we found that 3, 9, and 13 genes have breakpoints in, or in close proximity (<20 kb distance) to, such a gene in mothers 1, 2, and 3, respectively (Figure 3, Figure S4A, Table S4). The affected genes contain breakpoints in introns (14 genes), exons (one gene), the promoter region (five genes), or the 3′ region of the gene (five genes). Five of the affected genes are annotated as disease-associated genes in OMIM; three of these are found in mother 2, who suffers from delayed psychomotor development and major learning difficulties (Table S1). Seven genes are affected by intronic or exonic breakpoints in the two healthy mothers (mothers 1 and 3; Figures 3B and 3C). Because all gene disruptions are heterozygous, next we tested for the probability of haploinsufficiency of the affected genes by applying a metric previously described by Huang et al.17 This did not categorize any of the genes as very likely to be haploinsufficient, consistent with the absence of a phenotype in two out of three mothers (Figure S4B). In addition, we examined exome sequencing data from the Exome Aggregation Consortium (ExAC) and found that multiple loss-of-function mutations are reported for 22/25 affected genes (Figure S4C). Previous studies have shown that every human genome contains around 100 loss-of-function variants.18 Our data further emphasize the permissiveness of the genome to gene-disrupting changes and to massive relocations of chromosomal segments within a single, healthy individual.
Figure 3.
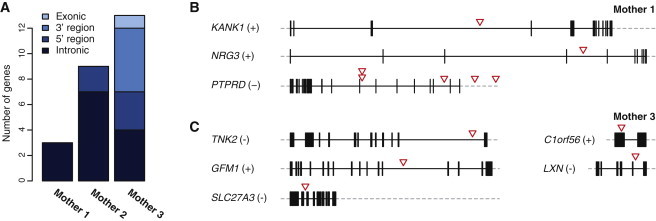
Chromothripsis Breakage Affects Protein-Coding Genes
(A) Number of genes affected by chromothripsis breakpoints in mothers 1, 2, and 3. Genes are considered affected if a break occurred in, or in close proximity (within <20 kb distance) to, the gene.
(B) Three genes are disrupted by breakpoints in mother 1; red arrowheads indicate the location of the break.
(C) Five out of 13 genes affected by breakpoints in mother 3.
After the reconstruction of the digital karyotypes, we set out to determine the link between the chromothripsis in the mothers and the severe congenital phenotypes in their children. We used mate-pair sequencing and did not identify any de novo breakpoint junctions in the children, despite the presence of multiple unique copy-number changes. For family 1, 5 out of the 13 rearrangements were detected in both the mother and her child. These represent all junctions on der(9), whereas none of the breakpoint junctions on the other three derivative chromosomes present in mother 1 were detected in her child (Figure 1B, Figure 4A); these results are consistent with the G-banded chromosome analysis. Out of the 23 breakpoint junctions detected in mother 2, her son harbored 16, representing the complete der(7) and der(12) chromosomes, whereas none of the junctions on der(6), der(9), or der(10) were detected in him (Figure S5A, Figure 2B). These findings indicate that the children inherited a subset of derivative chromosomes from their mothers and did not acquire any additional breakpoints upon germline transfer. In support of this, partial inheritance of the chromothripsis chromosomes explains all de novo copy-number changes in children 1 and 2 (Figure S1, Figure 4A, Figure S5A). Remarkably, the interstitial deletion on chr9 of child 1 is a result of five distinct, sequential chromosomal fragments that have been inserted in der(10), which was not transmitted to this child (Figure 4B). Similarly, the three copy gains in 9p21–24 in child 2 are a direct consequence of the presence of eight, rather than three, distinct segments of chromosome 9 inserted into der(12) (Figure 4B). Finally, we examined the transmission of breakpoint junctions in family 3. Seven out of eight junctions identified in the mother were also detected in the child (Table S2). The breakpoint junction missing in the child flanks the de novo deletion on 1q21.3 and matches the loss of this segment from der(3) in the child (Figure S5B).11 Unfortunately, we did not identify a de novo breakpoint junction that explains the terminal 3q29 duplication in child 3.
Figure 4.
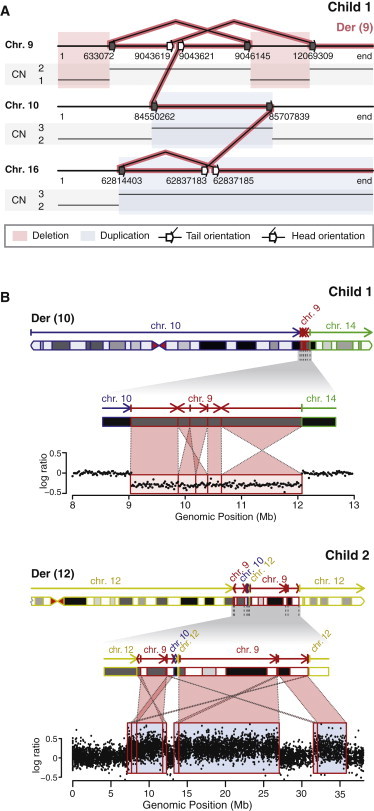
Stable but Partial Inheritance of Chromothripsis Chromosomes Can Lead to Highly Complex Copy-Number Changes
(A) Schematic representation of the breakpoint junctions detected in child 1. The CNVs detected in this child are explained by the structure of the inherited der(9). Connected lines between two arrows indicate breakpoint junctions. Adjacent white arrows indicate DSBs, and gray arrows indicate a single end of a break that was found to be a DSB in the mother. The other single end of these DSBs is located on der(10), der(14), or der(16), which were not inherited by the child, explaining the CNVs.
(B) Apparently simple CNVs can consist of multiple, highly rearranged, sequential chromosomal fragments rather than one solid fragment. Top: a deletion on chr9 in child 1 is a consequence of a highly complex rearrangement of five sequential chromosome fragments that were translocated to der(10) in the mother. Bottom: each of the three chr9 duplications detected in child 2 resulted from the translocation of multiple distinct chr9 segments into der(12) in the mother.
Previously detected CCRs in healthy individuals harbor relatively few breaks.4 In contrast, the three mothers presented here show a 1.1- to 2.6-fold increase in the number of rearrangements compared to those of their severely affected children, indicating that a larger number of breakpoints does not necessarily lead to more severe disease. Thus, the massive genome breakage and reassembly that occurred in the mothers is not the primary determinant of the phenotypic consequences in their children. Instead, the congenital abnormalities in the three children are caused by the CNVs that resulted from the partial or unstable transmission of the chromothripsis chromosomes. In support of this, trisomy 16qter, which is found in child 1, has previously been found to cause severe psychomotor retardation, facial dysmorphisms, and multiple other congenital defects, including heart, skeletal, kidney, gall bladder, and genital abnormalities.19,20 Most, if not all, abnormalities observed in this child can probably be attributed to the 27 Mb trisomy of 16qter. The phenotypes of children 2 and 3 can also largely be explained by their CNVs, as described previously.10,11 For child 2, this is further supported by the findings in two of his siblings, who only share the mother’s much milder phenotype and who did not harbor the chr9 duplications and chr12 deletions found in child 2 (Figure S6A). Interestingly, PCR and Sanger sequencing revealed that both siblings have also only partially inherited the chromothripsis chromosomes from their mother, albeit a different subset of them (Figure S6B). Like child 2, the siblings inherited der(7) and der(12) but not der(6) or der(10) from their mother; however, unlike child 2, they additionally inherited der(9), leading to copy-number balanced chr9 and chr12.
In conclusion, our results give important insights into the permissiveness of the human genome to extreme CCRs by demonstrating that it can tolerate massive chromothripsis rearrangements, disrupting multiple protein-coding genes, without phenotypic consequences. This suggests that chromothripsis, although rare, might be more common in the general population than previously expected. To date, ∼100 apparently balanced CCRs have been described in phenotypically normal individuals experiencing a broad range of reproductive problems.4 Some of these individuals might be affected by chromothripsis, given that the relatively low-resolution techniques used to identify these individuals are unable to uncover the full complexity of their CCRs.
In line with previous findings, including the fact that the presence of a CCR often leads to infertility in males, the chromothripsis chromosomes in this study are transferred from mother to child.3 We demonstrate that chromothripsis in healthy females can severely impact reproduction by causing miscarriages, abortions, and the birth of children with multiple congenital abnormalities and developmental delay (Table S1). The copy-number-neutral character of the chromothripsis rearrangements found in the mothers in this study shows the necessity of the use of a combination of detection methods rather than the use of CNV analysis alone for couples experiencing a broad range of reproductive problems.
Acknowledgments
We are grateful to the families for participating in this study. This research was supported by the Child Health priority program at the University Medical Center Utrecht. We thank M.E.M. Swinkels and E.F. Ippel for counseling family 2 and family 3, respectively. We would like to thank M. Poot for providing access to SNP array data for family 2.
Accession Numbers
The accession number for the microarray data reported in this paper is GSE65454 (NCBI Gene Expression Omnibus). The accession number for the mate-pair sequencing data reported in this paper is PRJEB8343 (European Nucleotide Archive).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl Genome Browser, http://www.ensembl.org/index.html
European Nucleotide Archive (ENA), http://www.ebi.ac.uk/ena
ExAC Browser, http://exac.broadinstitute.org/
Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo
OMIM, http://www.omim.org/
References
- 1.Batista D.A., Pai G.S., Stetten G. Molecular analysis of a complex chromosomal rearrangement and a review of familial cases. Am. J. Med. Genet. 1994;53:255–263. doi: 10.1002/ajmg.1320530311. [DOI] [PubMed] [Google Scholar]
- 2.Gorski J.L., Kistenmacher M.L., Punnett H.H., Zackai E.H., Emanuel B.S. Reproductive risks for carriers of complex chromosome rearrangements: analysis of 25 families. Am. J. Med. Genet. 1988;29:247–261. doi: 10.1002/ajmg.1320290202. [DOI] [PubMed] [Google Scholar]
- 3.Pellestor F., Anahory T., Lefort G., Puechberty J., Liehr T., Hédon B., Sarda P. Complex chromosomal rearrangements: origin and meiotic behavior. Hum. Reprod. Update. 2011;17:476–494. doi: 10.1093/humupd/dmr010. [DOI] [PubMed] [Google Scholar]
- 4.Madan K. Balanced complex chromosome rearrangements: reproductive aspects. A review. Am. J. Med. Genet. A. 2012;158A:947–963. doi: 10.1002/ajmg.a.35220. [DOI] [PubMed] [Google Scholar]
- 5.Stephens P.J., Greenman C.D., Fu B., Yang F., Bignell G.R., Mudie L.J., Pleasance E.D., Lau K.W., Beare D., Stebbings L.A. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiang C., Jacobsen J.C., Ernst C., Hanscom C., Heilbut A., Blumenthal I., Mills R.E., Kirby A., Lindgren A.M., Rudiger S.R. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat. Genet. 2012;44:390–397, S1. doi: 10.1038/ng.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kloosterman W.P., Guryev V., van Roosmalen M., Duran K.J., de Bruijn E., Bakker S.C., Letteboer T., van Nesselrooij B., Hochstenbach R., Poot M., Cuppen E. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum. Mol. Genet. 2011;20:1916–1924. doi: 10.1093/hmg/ddr073. [DOI] [PubMed] [Google Scholar]
- 8.Kloosterman W.P., Tavakoli-Yaraki M., van Roosmalen M.J., van Binsbergen E., Renkens I., Duran K., Ballarati L., Vergult S., Giardino D., Hansson K. Constitutional chromothripsis rearrangements involve clustered double-stranded DNA breaks and nonhomologous repair mechanisms. Cell Rep. 2012;1:648–655. doi: 10.1016/j.celrep.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 9.Zhang C.Z., Leibowitz M.L., Pellman D. Chromothripsis and beyond: rapid genome evolution from complex chromosomal rearrangements. Genes Dev. 2013;27:2513–2530. doi: 10.1101/gad.229559.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Pater J.M., Ippel P.F., van Dam W.M., Loneus W.H., Engelen J.J. Characterization of partial trisomy 9p due to insertional translocation by chromosomal (micro)FISH. Clin. Genet. 2002;62:482–487. doi: 10.1034/j.1399-0004.2002.620611.x. [DOI] [PubMed] [Google Scholar]
- 11.van Binsbergen E., Hochstenbach R., Giltay J., Swinkels M. Unstable transmission of a familial complex chromosome rearrangement. Am. J. Med. Genet. A. 2012;158A:2888–2893. doi: 10.1002/ajmg.a.35580. [DOI] [PubMed] [Google Scholar]
- 12.Francioli L.C., Menelaou A., Pulit S.L., Van Dijk F., Palamara P.F., Elbers C.C., Neerincx P.B.T., Ye K., Guryev V., Kloosterman W.P., Genome of the Netherlands Consortium Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 2014;46:818–825. doi: 10.1038/ng.3021. [DOI] [PubMed] [Google Scholar]
- 13.Korbel J.O., Campbell P.J. Criteria for inference of chromothripsis in cancer genomes. Cell. 2013;152:1226–1236. doi: 10.1016/j.cell.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 14.Macera M.J., Sobrino A., Levy B., Jobanputra V., Aggarwal V., Mills A., Esteves C., Hanscom C., Pereira S., Pillalamarri V. Prenatal diagnosis of chromothripsis, with nine breaks characterized by karyotyping, FISH, microarray and whole-genome sequencing. Prenat. Diagn. 2014 doi: 10.1002/pd.4456. Published online July 9, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nazaryan L., Stefanou E.G., Hansen C., Kosyakova N., Bak M., Sharkey F.H., Mantziou T., Papanastasiou A.D., Velissariou V., Liehr T. The strength of combined cytogenetic and mate-pair sequencing techniques illustrated by a germline chromothripsis rearrangement involving FOXP2. Eur. J. Hum. Genet. 2014;22:338–343. doi: 10.1038/ejhg.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kloosterman W.P., Cuppen E. Chromothripsis in congenital disorders and cancer: similarities and differences. Curr. Opin. Cell Biol. 2013;25:341–348. doi: 10.1016/j.ceb.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Huang N., Lee I., Marcotte E.M., Hurles M.E. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., 1000 Genomes Project Consortium A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laus A.C., Baratela W.A., Laureano L.A., Santos S.A., Huber J., Ramos E.S., Rebelo C.C., Squire J.A., Martelli L. Karyotype/phenotype correlation in partial trisomies of the long arm of chromosome 16: case report and review of literature. Am. J. Med. Genet. A. 2009;158A:821–827. doi: 10.1002/ajmg.a.32988. [DOI] [PubMed] [Google Scholar]
- 20.Brisset S., Joly G., Ozilou C., Lapierre J.M., Gosset P., LeLorc’h M., Raoul O., Turleau C., Vekemans M., Romana S.P. Molecular characterization of partial trisomy 16q24.1-qter: clinical report and review of the literature. Am. J. Med. Genet. 2002;113:339–345. doi: 10.1002/ajmg.b.10740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.