

Abstract
The endothelin receptor type A (EDNRA) signaling pathway is essential for the establishment of mandibular identity during development of the first pharyngeal arch. We report four unrelated individuals with the syndrome mandibulofacial dysostosis with alopecia (MFDA) who have de novo missense variants in EDNRA. Three of the four individuals have the same substitution, p.Tyr129Phe. Tyr129 is known to determine the selective affinity of EDNRA for endothelin 1 (EDN1), its major physiological ligand, and the p.Tyr129Phe variant increases the affinity of the receptor for EDN3, its non-preferred ligand, by two orders of magnitude. The fourth individual has a somatic mosaic substitution, p.Glu303Lys, and was previously described as having Johnson-McMillin syndrome. The zygomatic arch of individuals with MFDA resembles that of mice in which EDNRA is ectopically activated in the maxillary prominence, resulting in a maxillary to mandibular transformation, suggesting that the p.Tyr129Phe variant causes an EDNRA gain of function in the developing upper jaw. Our in vitro and in vivo assays suggested complex, context-dependent effects of the EDNRA variants on downstream signaling. Our findings highlight the importance of finely tuned regulation of EDNRA signaling during human craniofacial development and suggest that modification of endothelin receptor-ligand specificity was a key step in the evolution of vertebrate jaws.
Introduction
During early embryonic head development, neural crest cells (NCCs) migrate from the dorsal neural tube to populate the pharyngeal arches (PAs), where they give rise to the cartilage and bones of the face. Caudally, NCCs contribute to the conotruncus of the heart, the peripheral nervous system, the enteric nervous system, melanocytes, and para-endocrine cells.1 The endothelin signaling system (i.e., ligands plus receptors) is a vertebrate-specific innovation2 implicated in the development of several NCC-derived tissues. In mammals, the system is comprised of three ligands (encoded by EDN1 [MIM 131240], EDN2 [MIM 131241], and EDN3 [MIM 131242]) and two receptors (encoded by EDNRA [MIM 131243] and EDNRB [MIM 131244]) belonging to the G protein-coupled seven transmembrane domain receptor family.3 All three ligands bind to EDNRB with similar affinity, and EDN1 and EDN2 bind to EDNRA with an affinity two orders of magnitude higher than that of EDN3.4,5 The first PA is comprised of maxillary and mandibular prominences, which give rise to the upper and lower jaws, respectively. EDN1-EDNRA signaling plays a critical role in specification of mandibular identity in post-migratory NCCs in animal models,3 with Edn1 expressed in the mandibular but not the maxillary prominence and Ednra expressed throughout the first PA.6,7 Mutations in EDN1 have recently been reported in individuals with auriculocondylar syndrome (ACS, a disorder affecting derivatives of the first and second PAs [MIM 615706]) or isolated question mark ears (MIM 612798).8 Mutations in PLCB4 (MIM 600810) and GNAI3 (MIM 139370), which encode signaling components downstream of EDNRA, have also been identified in ACS (MIM 602483 and 614669).9
The mandibulofacial dysostoses (MFDs) are characterized by malar and mandibular hypoplasia, typically associated with abnormalities of the ears and eyelids.10 These defects are likely to result from abnormal development of cranial NCCs or their derivatives. MFDs can occur in an isolated form, as in Treacher Collins syndrome (MIM 154500), or can be part of a broader developmental defect, as in Nager (MIM 154400) and Miller (MIM 263750) syndromes. Here we delineate a syndrome that we have named mandibulofacial dysostosis with alopecia (MFDA), shared by four unrelated individuals in whom we identified two de novo missense substitutions in EDNRA, one of which is recurrent. Functional evidence suggested that both substitutions perturb normal endothelin signaling.
Material and Methods
Whole-Exome Sequencing
Genetic analyses were performed in accordance with approved institutional protocols (CPP Ile-de-France II and the Internal Review Board at Cincinnati Children’s), and informed consent for genetic testing was obtained from all participants.
For individual 3 whole-exome sequencing (WES), genomic DNA was extracted from whole blood by standard methods. Library construction was performed on dsDNA, sheared by sonication to an average size of 200 bp, in an automated fashion on an IntegenX Apollo324. After nine cycles of PCR amplification with the Clontech Advantage II kit, 1 μg of genomic library was recovered for exome enrichment by the NimbleGen EZ Exome V2 kit. Libraries were sequenced on an Illumina HiSeq2500, generating approximately 30 million 100 bp paired end reads. Data analysis utilized the Broad Institute’s Genome Analysis Toolkit (GATK).11 Reads were aligned with the Illumina Chastity Filter with the Burrows Wheeler Aligner (BWA).12 Variant sites were called with the GATK Unified Genotyper module. Single-nucelotide variant calls were filtered with the variant quality score recalibration method.11 Golden Helix SNP and Variation Suite v.8.1.5 was used to filter the variants. For individual 4 WES, protocols for library preparation, exome capture, sequencing (performed on an Illumina HiSeq2500 sequencer), read mapping, and variant calling and filtering were similar to that described previously.8
Cell Culture
MC3T3-E1 cells (ATCC) were cultured in MEM alpha (Invitrogen) supplemented with 20% fetal bovine serum (Sigma) at 37°C in a HeraCell humidified incubator (Heraues) containing 5% CO2.
Whole-Mount In Situ Hybridization
Whole-mount in situ hybridization (ISH) was performed essentially as previously described6 with a digoxigenin (DIG)-labeled antisense cRNA riboprobe against Edn3. Embryos were photographed with an Olympus SZX12 microscope fitted with an Olympus DP11 digital camera.
Wild-Type and Mutant EDNRA Overexpression in MC3T3-E1 Cells
For in vitro analysis of wild-type and mutant EDNRA activity, an expression vector (pCMV6-XL5) containing the full cDNA sequence of human EDNRA transcript variant 1 (RefSeq accession number NM_001957.1) (Origene) was used. To introduce the p.Tyr129Phe substitution (which corresponds to a c.386A>T mutation, codon TAT to TTT), we used the QuickChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s instructions for annealing and extension times and cycle numbers. In brief, the mutant strand synthesis was accomplished by PCR via mismatched primers containing the c.386A>T mutation. Primer sequences were 5′-tcttgcccttggagaccttatctttgtggtcattga-3′; 5′-tcaatgaccacaaagataaggtctccaagggcaaga-3′. The PCR product was DpnI treated to digest the template vector and transformed into XL10-Gold ultracompetent cells (Agilent Technologies). The sequence of both the wild-type and mutant vectors was verified by DNA sequencing. For transfection, X-tremeGENE 9 DNA transfection reagent (Roche) was used. 0.8 μg of either the EDNRA wild-type or EDNRA p.Tyr129Phe-encoding expression vector was transfected into MC3T3-E1 cells. Transfection complexes were made according to the manufacturer’s protocol, added to culture media, and removed 6 hr after transfection. Cells were then starved overnight with MEM alpha supplemented with 0.1% fetal bovine serum (Sigma-Aldrich). At 1 hr prior to cell harvest, 10 μM BQ788 (Sigma-Aldrich), an EDNRB-specific antagonist, was added to the media followed by 5 μl of ddH20 or 5 nM of either EDN1 (Sigma-Aldrich) or EDN3 (Sigma-Aldrich).
Quantitative Real-Time PCR
Transfected MC3T3-E1 cells were harvested 24 hr after transfection and RNA was extracted with the RNeasy Micro Kit (QIAGEN). cDNA was prepared from total RNA with the Quantitect cDNA Synthesis Kit (QIAGEN). qRT-PCR was performed on 5 ng of cDNA and the Quantitect SYBR Green PCR Kit (QIAGEN), including Quantitect Assay primers for Dlx5 and Hand2 (QIAGEN). RT-PCR and data analysis was performed with a MyiQ2 thermocycler (BioRad). Statistical analysis was conducted with the statistical package in Excel.
Zebrafish Morpholino and mRNA Injections
Zebrafish (Danio rerio) were raised and mated as described.13 Embryos were injected at the 1- to 2-cell stage into the yolk sac with 1 nl of solution containing MO, mRNA, or both via a microinjector. Phenotyping was carried out at 5 days post-fertilization (dpf). Alcian blue staining was performed as described.14 Photographs were taken on a Nikon SMZ6 microscope. Measurements were performed with ImageJ. Splice-blocking MO antisense oligonucleotides against the two orthologs of EDNRA, ednraa (previously known as ednra2) and ednrab (previously known as ednra1), were previously designed15 and obtained from GeneTools. MO sequences are as follows: ednraa 5′-ATCAGACTTTTCTTTACCTGCTTAA-3′ and ednrab 5′-AGTGGTGTGTTCACCTGTTTGAGGT-3′. Knockdown efficiency was assessed by RT-PCR across the exon-intron boundary targeted by each MO (see Figure S8).
Zebrafish Expression Constructs
Wild-type human EDNRA was amplified and cloned into the pCS2+ expression vector as previously described.16 Site-directed mutagenesis was performed to introduce the p.Tyr129Phe, p.Ser167Ala, and p.Glu303Lys variants. Each mutant construct was fully sequenced after mutagenesis to confirm presence of the point mutation. Full-length wild-type human EDN1 and EDN3 cDNA clones were obtained from the human ORF clone collection (clone ID: IOH80822 and IOH57032, Life Technologies). ORF clones were fully sequenced and full-length cDNAs were then transferred from pENTR to pCS2+ utilizing the Gateway Cloning system (Life Technologies). Capped mRNA was generated for all constructs utilizing the mMessage mMachine SP6 transcription kit (Life Technologies).
Ednralacz/+ Expression Analysis
Ednralacz/+ mouse strain generation and X-gal staining procedure have been previously described.17 Experiments in this report were performed in accordance with institutional ethical guidelines.
Results
Phenotype of Individuals with MFDA
Individuals 1–3 presented with a highly similar facial dysmorphism, and individual 4, previously described as having Johnson-McMillin syndrome18 (JMS [MIM 147770]), displayed a milder phenotype (Figure 1 and Table 1). A phenotypic description of individual 2 up to the age of 4 years has been previously published.19 Individuals 1–3 all had micrognathia and cleft palate. Individuals 1 and 3 had limited jaw mobility and individual 3 had glossoptosis. Upper airway obstruction necessitated tracheostomy for individuals 1 and 3. Individual 3 has remained tracheostomy dependent despite three mandibular distraction osteogenesis surgeries (performed at 3 months, 2 years, and 5.5 years of age). Individual 1 presented with irregular placement of primary teeth in both upper and lower jaws. Delayed eruption of primary teeth was previously noted in individual 2,19 and at age 7, primary teeth were still present with only 16 permanent tooth buds visible on radiographs, indicating agenesis of multiple permanent teeth. Individual 3 did not have anomalies of tooth eruption or morphology but had poor dental hygiene related to mandibular ankylosis and dental crowding. Individuals 1–3 had an everted lower lip (see Figure S1 for pre-distraction pictures of individual 3), and all four individuals had a similar short nose with a squared nasal tip. In individuals 1–3 the ears were small, cupped, and dysplastic with ectopic tissue at the attachment of the helix to the scalp (see Figure S1 for ectopic post-auricular tissue in individual 1); in individuals 1 and 3 this protuberance contained a posteriorly facing pit. The ears of individual 4 were also severely abnormal, with pre-auricular pits and tags.18 Conductive hearing loss has been treated with hearing aids in individuals 1, 3, and 4. Individuals 1–3 had sparse eyelashes and severe hypoplasia or aplasia of the eyelids requiring reconstructive surgery, and individual 4 had a lower eyelid coloboma. All four individuals had near-complete alopecia, with less hair on the vertex relative to the occipital region in individuals 1, 3, and 4. White hair was present in individual 2.19 Although hyposmia was suspected at a younger age in individual 2,19 it was not present upon evaluation at 7 years of age. Growth and intellectual development have been normal in all. Echocardiograms revealed no defects in individuals 1, 2, and 4,18 although a bicuspid aortic valve was detected in individual 3.
Figure 1.

Phenotype of Mandibulofacial Dysostosis with Alopecia
Front and side views of facial features of individual 1 at age 1 year 8 months (A and B), of individual 2 at age 3 years 4 months (C and D), of individual 3 at age 10 years (E and F), and of individual 4 at age 13 years (G and H). Although individual 4 has a shaved head, patches devoid of follicles can be observed.
Table 1.
Clinical Features of Individuals Studied in This Report
| Individual 1 | Individual 2 | Individual 3 | Individual 4 | |
|---|---|---|---|---|
| Previous publication | not previously reported | Zechi-Ceide et al.19 | not previously reported | Cushman et al.18 |
| Diagnosis | MFDA | MFDA | MFDA | MFDA/JMS |
| Gender | M | M | M | F |
| MFD | + | + | + | NA |
| Zygomatic arch (by CT scan) | dysplastic | dysplastic | dysplastic | NA |
| Mandible (by CT scan) | dysplastic | dysplastic | dysplastic | NA |
| Cleft palate | + | + | + | − |
| Alopecia | + | + | + | + |
| Eyelid anomalies | + | + | + | + |
| Auricular dysmorphism | + | + | + | + |
| Hearing loss | + | + | + | + |
| Dental anomalies | + | + | − | − |
| Other phenotypes | megaureter | white hairs | bicuspid aortic valve | − |
| EDNRA mutationa | c.386A>T | c.386A>T | c.386A>T | c.907G>A |
| Protein change | p.Tyr129Phe | p.Tyr129Phe | p.Tyr129Phe | p.Glu303Lys |
| Inheritance | de novo (somatic mosaic) | de novo | de novo | de novo (somatic mosaic) |
Abbreviations are as follows: MFDA, mandibulofacial dysostosis with alopecia; JMS, Johnson-McMillin syndrome; F, female; M, male; +, finding present; −, finding not present; NA, information not available.
Co-ordinates refer to RefSeq transcript NM_001957.3.
Computed tomography scans of individuals 1–3 confirmed a severe MFD (Figure 2). In all three, the zygomatic process of each temporal bone was absent, the maxillae were dysmorphic, and the malar bones were thickened with grossly dysplastic ventral orbital margins. The lateral margin of each orbit was essentially absent in individual 3 and was hypoplastic in individuals 1 and 2. The mandible was hypoplastic with reduced gonial angle in individuals 1 and 2 and retro-positioned dental arcade in individual 1. Note that the images of individual 3 in Figure 2 are 6 months after his second mandibular distraction surgery. In individuals 1–3, the temporomandibular joints were absent and the condyles were broad, flat, and articulated with a proximal surface of the dysmorphic malar bones. In individuals 1–3 the coronal suture was displaced anteriorly; the wings of the sphenoid were hypoplastic in individual 1. Individuals 1 and 2 displayed deviation of the nasal cavities, and in individuals 1 and 3 the external auditory canals were narrow, the mastoids were underpneumatized, the middle ear cavities were small, and the ossicular chains were hypoplastic. Individual 3 had absent maxillary sinuses and hypoplastic pterygoid processes. Skeletal imaging was not available for individual 4, but of note, Cushman et al.18 described individual 4 as having depressions at the lateral aspects of the eyes, consistent with underlying lateral orbital defects as seen for individuals 1–3.
Figure 2.
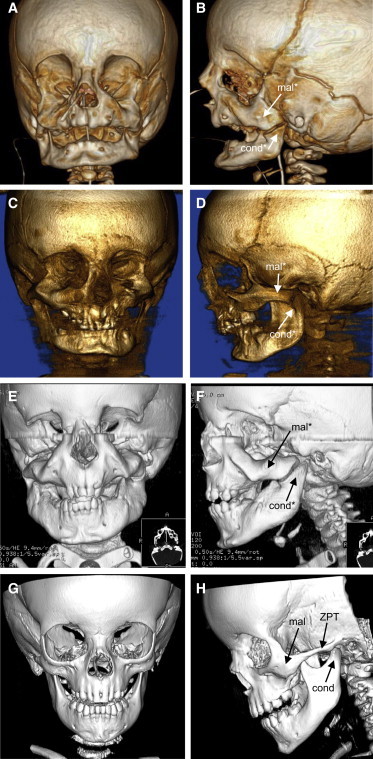
Craniofacial Skeletal Anomalies in Mandibulofacial Dysostosis with Alopecia
Front and side views of 3D computed tomography (CT) scans of individual 1 at age 6 months (A and B), of individual 2 at age 7 years (C and D), of individual 3 at age 2.5 years (E and F), and of a normal individual at age 2.5 years (G and H).
Abbreviations are as follows: mal, malar bone; cond, condyle of the mandible; ZPT, zygomatic process of the temporal bone. An asterisk indicates structures that are dysmorphic relative to normal.
Genetic Analyses
Initial genetic analyses of the four MFDA-affected individuals included chromosomal microarray analysis by Agilent 60k array-CGH for individuals 1 and 4, GeneChip SNP array for individual 2,19 and SignatureSelect V2 BAC array and Infinium Assay (Illumina HD Human610-quad BeadChip platform) for individual 3. No likely pathogenic abnormalities were detected. Individuals 1, 2,19 and 3 also had normal karyotypes.
Given the strong phenotypic similarities of individuals 1–3, we hypothesized that they could have mutations in the same gene. Trio WES was therefore performed for individual 3 and his parents (II:1, I:1, and I:2, respectively, in Figure 3). WES was also done for individual 4 and her parents (II:1, I:1, and I:2, respectively, in Figure 3), because her milder phenotype suggested the possibility of another locus. The absence of other affected family members for all individuals suggested an autosomal-dominant mode of inheritance with de novo mutations in the probands as the most likely etiology.
Figure 3.
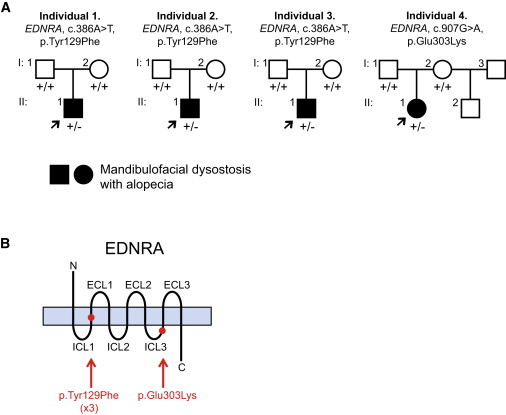
Pedigrees and EDNRA Mutations Identified for Individuals 1–4
(A) Pedigrees of individuals 1–4. Arrows indicate probands. Presence of a mutant allele is indicated by a minus sign.
(B) Schematic of the EDNRA protein, with red dots indicating the positions of the MFDA substitutions (listed below the schema). Protein domains not drawn to scale. Abbreviations are as follows: ECL, extra-cellular loop; ICL, intra-cellular loop.
WES of individual 3 and his parents was performed with a mean depth of coverage for each sample of at least 89-fold and with at least 97% of the exome covered 15-fold or greater. We identified de novo rare variants predicted to have a deleterious effect on protein function in ITGAE (MIM 604682), WNT4 (MIM 603490), and EDNRA (Table S1). The frameshift variant in ITGAE (RefSeq NM_002208.4, c.2986delG [p.Glu996fs∗24]) was considered unlikely to be pathogenic because a recurrent frameshift variant in this gene is listed in the Exome Variant Server (EVS), and mice deficient in Itgae do not have morphogenesis defects.20 Heterozygous missense WNT4 variants are associated with a human phenotype in females (Mullerian aplasia and hyperandrogenism [MIM 158330]). We confirmed the WNT4 variant (RefSeq NM_030761.4, c.114dupA [p.Glu39fs∗38]) in individual 3 by Sanger sequencing but we found no coding variants in WNT4 in the other three MFDA-positive individuals; the WNT4 variant was therefore considered unlikely to contribute to the phenotype of individual 3. Finally, the variant in EDNRA (RefSeq NM_001957.3, c.386A>T [p.Tyr129Phe]; Figure S2) was confirmed de novo by Sanger sequencing (Figures 3 and S3). Variants affecting Tyr129 of EDNRA have not been reported in public SNP databases, and a tyrosine is conserved at this position in all sequenced vertebrates, including lamprey.
Trio WES of individual 4 was performed with a mean depth of coverage for each sample of at least 130-fold, with 98% of the exome covered 15-fold or greater. Analysis of the exome data under a recessive model yielded only compound heterozygous missense variants in PAXIP1 (Table S2). One of the affected PAXIP1 residues was not absolutely conserved in sequenced mammals, and neither affected residue fell within the known Pfam domains of the protein; although we cannot exclude the influence of these variants, PAXIP1 was not considered a strong candidate. Under a de novo model, the only variant predicted to affect protein function and not previously reported in public SNP databases (dbSNP138, EVS) or in-house exomes was in EDNRA: c.907G>A (p.Glu303Lys) (RefSeq NM_001957.3). Analysis of exome sequencing reads indicated a 75%:25% bias in reference- versus variant-containing reads for individual 4, with 100% reference-containing reads for each parent (Figure S2). Sanger sequencing from two independent blood samples from individual 4 (Figure S4) and an alternate pair of primers (data not shown) indicated a similar bias in wild-type versus mutant chromatogram peak height, and cloning of PCR products yielded a 12:3 ratio of bacterial colonies harboring wild-type:mutant sequences, suggesting that individual 4 was somatic mosaic for the p.Glu303Lys EDNRA variant. The residue Glu303 of EDNRA, as well as the equivalent amino acid of EDNRB (Glu320), is highly conserved in vertebrates and maps to the C-terminal portion of the third intra-cellular loop2 (Figure 3). Deletion analysis has shown that the equivalent region of the third intra-cellular loop of EDNRB is required for EDN1-induced signaling.21
We subsequently performed Sanger sequencing of EDNRA in individuals 1 and 2, and in both we identified the same mutation found in individual 3 (RefSeq NM_001957.3, c.386A>T [p.Tyr129Phe], Figure S3). The mutations were confirmed as de novo in both individuals 1 and 2 (Figures 3 and S3). In individual 1, the mutation appeared to be mosaic in DNA extracted from blood (the same result was obtained from PCR products amplified with two alternate pairs of primers) and from a buccal swab (Figure S3 and data not shown). Mosaicism was confirmed by cloning three PCR products from individual 1: two products contained the major allele of the SNP rs6841473 (433 bp from the mutation) plus either the mutant or wild-type allele at position 386 (cDNA numbering), and the third product harbored the rs6841473 minor allele and wild-type at position 386. Given that a similar degree of mosaicism (judged by chromatogram peak intensity) was observed in two tissue sources, the mutation is likely to have arisen within the first few divisions after conception. The absence of visible skin pigmentation anomalies in individual 1, which might occur with somatic mutations that arise later in development, is consistent with this (assuming that an EDNRA mutation could indeed affect pigmentation). Paternity was confirmed in individuals 1 and 2 by analysis of more than ten unlinked and informative microsatellite markers.
In Vitro Functional Analysis of the EDNRA p.Tyr129Phe Variant
Tyr129, located within the second transmembrane domain of EDNRA (Figure 3), has been shown to be critical for the selective affinity of endothelin ligand subtype: mutagenesis of tyrosine to several different amino acids, including phenylalanine (the recurrent MFDA substitution), results in an ∼100-fold increase in affinity for EDN3, without change in affinity for EDN1.4,5,22 EDN3 is expressed in the first PA in humans,23 chick,24 and mice (Figure S5 and microarray data from the FaceBase Consortium,25 as displayed in the UCSC Genome Browser), raising the possibility that in MFDA, EDN3 present in the first PA binds to and alters signaling downstream of mutant EDNRA. To better understand the effect of the p.Tyr129Phe variant on EDNRA function, we generated an EDNRA expression construct encoding the p.Tyr129Phe variant and addressed whether the mutated EDNRA could still activate downstream effectors of endothelin signaling, in vitro, in response to EDN1 and EDN3. Cells from the mouse pre-osteoblastic cell line MC3T3-E1 were transfected with either the EDNRA wild-type or EDNRA p.Tyr129Phe-encoding expression constructs and then starved overnight. After 24 hr, cells were stimulated with either EDN1 or EDN3. To limit our analysis to the effects resulting from EDNRA signaling, we also included BQ788, an EDNRB-specific antagonist. One hour later, cells were collected and RNA was isolated. To assess EDNRA signaling activity, we measured the expression levels of two downstream markers of the EDN1-EDNRA pathway, Dlx5 and Hand2, by qRT-PCR. Both EDN1 and EDN3 resulted in a significant increase in Dlx5 and Hand2 mRNA levels in cells in which the wild-type EDNRA expression construct had been introduced (Figure 4). In contrast, neither EDN1 nor EDN3 resulted in significant increases in Dlx5 and Hand2 mRNA levels in cells expressing EDNRA p.Tyr129Phe (Figure 4). When the above experiments were repeated in the absence of BQ788, upregulation of both Dlx5 and Hand2 was less robust or no longer significantly changed, suggesting a negative influence of EDNRB signaling on expression of these genes in this system (Figure S6). These data indicate that, in vitro, EDNRA is able to respond to both EDN1 and EDN3 and that the p.Tyr129Phe substitution disrupts this response.
Figure 4.
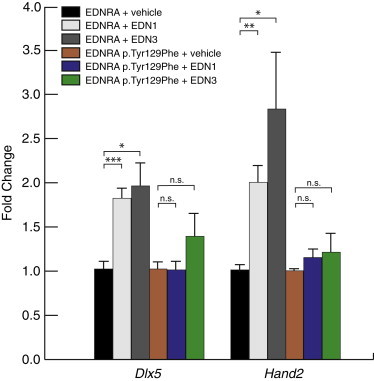
Response of EDNRA and EDNRA p.Tyr129Phe to EDN1 and EDN3 In Vitro
After transfection of MC3T3-E1 cells with an expression vector encoding either wild-type EDNRA or EDNRA p.Tyr129Phe, cells were treated with vehicle, EDN1, or EDN3 and then RNA collected for qRT-PCR analysis of Dlx5 and Hand2 expression. All treatments were performed in the presence of the EDNRB antagonist BQ788 to exclude expression changes due to EDNRB signaling. Each condition represents an average of results from three separate transfection experiments. Error bars represent standard error of the mean. Results are normalized to vehicle within each expression assay (Dlx5 or Hand2). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; n.s., not statistically significant (p > 0.05). Statistical test was a two-tailed t test (assuming equal variance).
Functional Analysis of the EDNRA Variants in Zebrafish
We utilized zebrafish to provide further evidence for the pathogenicity of the EDNRA variants identified in MFDA-affected individuals. First, we injected increasing doses (100, 150, and 300 pg) of mRNAs encoding human wild-type or mutant (p.Tyr129Phe, p.Glu303Lys) EDNRA into 1- to 2-cell stage embryos. At 5 days post-fertilization (dpf), cartilage structures of zebrafish embryos were visualized by whole-mount alcian blue staining. Misexpressing embryos (EDNRA wild-type, n = 163; p.Tyr129Phe, n = 145; p.Glu303Lys, n = 161) were indistinguishable from uninjected embryos (n = 157) from the same clutch, and no quantifiable craniofacial defects were detected in embryos injected with either wild-type or mutant EDNRA mRNAs (Figures 5C and S7 and data not shown for p.Glu303Lys). To exclude the possibility that the absence of phenotype was due to the degradation of the injected human RNAs, total RNA from injected embryos was extracted at 50 hr post-fertilization (hpf), a time point well past the patterning of NCC mesenchyme in the PAs. We confirmed the presence of the p.Tyr129Phe-encoding human message by RT-PCR by utilizing primers to specifically amplify human EDNRA (Figure S7). The absence of phenotype upon misexpression of mutant EDNRAs argues against a dominant-negative effect.
Figure 5.
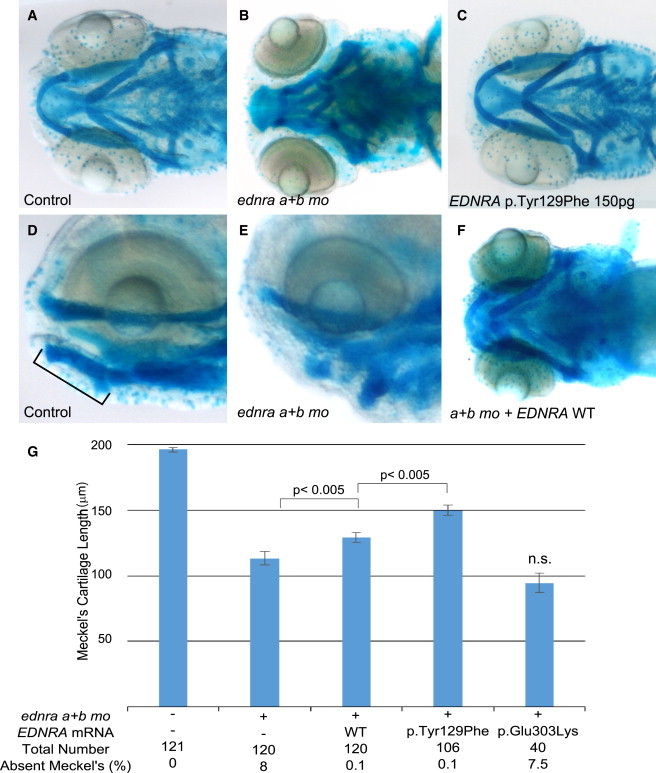
EDNRA p.Tyr129Phe but Not p.Glu303Lys Rescues Ventral Pharyngeal Arch Defects Caused by ednra Knockdown in Zebrafish
(A, B, D, and E) Ventral and lateral views of uninjected controls (A and D) and embryos injected with ednraa and ednrab morpholinos (MOs) (B and E) and stained with alcian blue. Meckel’s cartilage is indicated by a bar in (D).
(C) Injection of mRNA encoding human EDNRA p.Tyr129Phe (150 pg) did not result in a discernable phenotype.
(F) Injection of mRNA encoding wild-type (WT) human EDNRA partially rescues the ventral cartilage defect induced by MO knock-down of the zebrafish ednra orthologs.
(G) Bar graphs showing measurements of the length of Meckel’s cartilage. The first column represents uninjected embryos. For each co-injection, 100 pg of mRNA was injected. Meckel’s cartilage reduction is rescued significantly by both EDNRA WT and EDNRA p.Tyr129Phe-encoding mRNAs but not by EDNRA p.Glu303Lys-encoding mRNA. Statistical significance (Student’s t test) is denoted by p values or n.s. (not significant) on the bar graphs. Error bars represent standard error of the mean. Data are from three independent experiments.
We next tested the possibility that the variants act as loss-of-function alleles. EDNRA has two known orthologs in zebrafish: Ednraa (73% identity to human EDNRA protein) and Ednrab (72% identity). 1- to 2-cell stage zebrafish embryos were injected with MOs previously shown to target ednraa and ednrab.15 To quantify the phenotype, we measured the length of Meckel’s cartilage in controls and MO-injected embryos. MO knockdown of both EDNRA orthologs in zebrafish recapitulated the reported loss of function of edn1 (sucker mutant)26 with either complete absence or reduction of Meckel’s cartilage length (Figures 5B, 5E, and 5G); besides the jaw phenotype, no appreciable gross abnormalities were observed (Figures S8A and 8B). We observed a significant rescue of the morphant phenotype by co-injecting both MOs with mRNAs encoding wild-type human EDNRA (p < 0.005, Figure 5G) or the p.Tyr129Phe variant; the latter rescued significantly better than wild-type EDNRA (p < 0.005 between wild-type and mutant rescue, Figure 5G), suggesting that p.Tyr129Phe is not a loss-of-function variant in this assay. By contrast, the p.Glu303Lys variant did not rescue the phenotype (non-significant compared to MOs alone, Figure 5G).
Next, we sought to determine whether p.Tyr129Phe and p.Glu303Lys are capable of altering the response of the receptor to the human ligands EDN1 and EDN3 in zebrafish embryos by co-injecting wild-type or mutant EDNRA mRNAs with either human EDN1 or EDN3 transcripts. Co-injections of mRNAs encoding ligand and receptor were toxic at high doses; we therefore injected suboptimal doses (5 pg of each mRNA) to decrease the penetrance of the phenotype and to give the dynamic range necessary to detect possible interaction between ligands and wild-type or mutant receptors. Co-injection of various ligand-receptor combinations resulted in a range of rostral hypoplasia phenotypes of variable penetrance, which included a reduction of the volume of the telencephalon bringing the eyes closer together, frequent cyclopia, eye anteversion (rotation inward), and complex cartilage defects probably secondary to the forebrain and eye defects, including absence of the ethmoid plate and ventral displacement of the trabecular, Meckel’s, and ceratohyal (Figures 6D–6I). These phenotypes were found in approximately 5% of the wild-type EDNRA and EDN1 mRNA co-injected embryos, whereas no appreciable phenotype was observed in embryos injected with wild-type EDNRA and EDN3 mRNAs (Figures 6J and 6K). Notably, the percentage of affected embryos increased to 20% upon co-injection of EDN1 mRNA with either EDNRA p.Tyr129Phe- or p.Glu303Lys-encoding mRNAs (p < 0.001 compared to EDN1 and wild-type EDNRA; Figure 6K), whereas 10% or 15% of the embryos co-injected with EDN3 mRNA and EDNRA p.Tyr129Phe- or p.Glu303Lys-encoding mRNAs, respectively, displayed rostral hypoplasia phenotypes (p < 0.001 compared to EDN3 and wild-type EDNRA; Figure 6J). Given that Glu303 maps to an intracellular loop of EDNRA, the molecular mechanism underlying the EDN3-mediated effect on p.Glu303Lys remains unclear, but is probably different to that of p.Tyr129Phe.
Figure 6.

Co-expression of Endothelins and EDNRA Variants in Zebrafish
(A–C) Embryos injected with 5 pg of EDN1 mRNA.
(D–F) Embryos co-injected with 5 pg each of EDN1 and wild-type (WT) EDNRA mRNAs.
(G–I) Co-injection of 5 pg each of mRNAs encoding EDNRA p.Glu303Lys and EDN1.
All embryos are shown at 5 dpf. Abbreviations are as follows: mc, Meckel’s; pq, palatoquadrate; ch, ceratohyal; tr, trabecula.
(A, D, and G) Brightfield images.
(B, E, and H) Lateral views of alcian blue-stained embryos.
(C, F, and I) Ventral views of alcian blue-stained embryos.
Co-injected embryos in (D)–(I) exhibit a pronounced rostral hypoplasia phenotype including severe eye separation defects and loss of the ethmoid plate (ep; asterisk when reduced or absent) with relative preservation and downward displacement of the facial cartilages.
(J and K) Percentage of embryos with rostral hypoplasia is shown for co-injections with EDN3 (J) and EDN1 (K) mRNAs. The first column of each graph represents uninjected embryos. Error bars represent the 95% confidence interval of the mean. Fisher’s exact test was performed and p values are as follows: #p < 0.001 versus EDN3 and WT, %p < 0.001 versus EDN1 and WT, $p < 0.001 versus EDN3 and WT; n.s. is compared to respective EDN-alone injection.
The rostral hypoplasia phenotypes were unlikely to be driven by overall developmental delay; misexpressing embryos had no apparent pathology in other internal organs, such as the heart or the swim bladder, and their body length was indistinguishable from that of control embryos from the same clutch (Figure S8F; EDN1 and wild-type EDNRA co-injected embryo shown as representative). We note that the forebrain and eye phenotypes reported above are not representative of the phenotypes in individuals with MFDA, nor of other human or animal models of perturbed endothelin signaling. These phenotypes are probably caused by misexpression of endothelin ligand-receptor combinations in the developing neuroepithelium. Despite being the product of ectopic expression, these phenotypes do nevertheless provide a readout for wild-type versus mutant EDNRA protein activity in response to ligand. In the above assays, it appears that the p.Tyr129Phe and p.Glu303Lys variants have the capacity to perturb EDNRA activity in an endothelin ligand-dependent manner.
In addition to p.Tyr129Phe, Krystek et al. tested the selectivity of endothelin ligand binding to another variant, p.Ser167Ala, because as for Tyr129, Ser167 is predicted to fall at the ligand binding cavity of EDNRA, and differs between EDNRA and EDNRB. However, unlike p.Tyr129Phe, p.Ser167Ala appeared to display decreased binding affinity for EDN3.4 We found that mRNA encoding the p.Ser167Ala variant significantly rescued the ednra morphant phenotype in zebrafish (p = 9.7 × 10−10; Figure S8D), suggesting that this variant does not perturb normal endothelin signaling. To test further the possibility that direct binding of EDN3 to EDNRA p.Tyr129Phe can lead to dysregulation of downstream signaling, we generated mRNA encoding the double-mutant EDNRA p.Tyr129Phe/p.Ser167Ala and co-injected with EDN3 mRNA. We hypothesized that binding of EDN3 to p.Tyr129Phe might be inhibited by the presence of p.Ser167Ala on the same receptor molecule, and the double mutant might therefore reduce the penetrance of the phenotype observed upon co-injection of EDN3 mRNA and mRNA encoding the single-mutant EDNRA p.Tyr129Phe. Indeed, we observed that co-injection of mRNAs encoding EDN3 and EDNRA p.Tyr129Phe/p.Ser167Ala led to a significant reduction in the percentage of affected embryos compared to co-injection of mRNAs encoding EDN3 and EDNRA p.Tyr129Phe (p < 0.001, Figure 6J). Upon co-injection of mRNAs encoding EDN1 and the double-mutant EDNRA p.Tyr129Phe/p.Ser167Ala, the percentage of affected embryos was not significantly different from that observed upon co-injection of mRNAs encoding EDN1 and the single p.Tyr129Phe mutant (Figure 6K), suggesting that although p.Ser167Ala abrogates the effects of EDN3 on p.Tyr129Phe, p.Ser167Ala has no effect on the consequences of EDN1-mediated signaling via p.Tyr129Phe.
Ednralacz Expression
All MFDA-affected individuals have alopecia, yet very little is known about the normal role of EDNRA in hair follicle development. By utilizing a mouse line in which lacz is knocked-in to the Ednra locus,17 we observed strong expression of Ednralacz in hair follicles at multiple stages of follicular development (Figure 7). Ednralacz expression was also observed in other tissues that are strongly affected in MFDA: in the developing eyelids and in the pinnae, particularly the dorsal and ventral regions of the pinnae that fuse with the scalp (Figure 7).
Figure 7.

Ednralacz/+ Expression in Mouse Embryos
(A and B) Ednra+/+ (A) and Ednralacz/+ (B) whole-mount E15.5 embryos stained with X-gal. White arrows indicate eyelid and pinna expression of Ednralacz. The inset in (B) is a magnified view of an Ednralacz/+ hair follicle.
(C) Cryosection of whole-mount X-gal-stained E13.5 Ednralacz/+ embryo.
(D and E) Ednra+/+ (D) and Ednralacz/+ (E) postnatal day (P) 0 X-gal-stained cryosections of vibrissal follicles. Abbreviations are as follows: dp, dermal papilla; m, matrix; vf, vibrissal follicle.
Discussion
Here we have described a disorder characterized by MFD and alopecia that is caused by de novo missense substitutions in EDNRA. We identified mutations in four unrelated simplex cases. In three of the four individuals, Tyr129 is substituted; this amino acid is the key determinant of preferential affinity of EDNRA for endothelin ligand subtype.4,5 The recurrent MFDA substitution p.Tyr129Phe had previously been shown to increase affinity of EDNRA for EDN3, its non-preferred ligand. Furthermore, the importance of Tyr129 in ligand selectivity has been used as the basis for rational design of EDNRA-specific antagonists.27
The consequences of the MFDA variants on signaling (and by extension, on phenotype) might differ depending on the regional composition of endothelin ligands and downstream signaling components within the PAs. Ednra is expressed by all cranial NCCs soon after they emerge from the neural tube,6,7 whereas Edn1 expression is restricted to the mandibular portion of the first PA and more caudal arches.6,7,28,29 A homeotic transformation of the mandible into a maxilla has been reported in Edn1, Ednra, or Dlx5 and Dlx6 knock-out mice.3 The mandibular alterations in ACS are thought to represent a similar transformation.9,30 The inverse transformation, consisting of the modification of the upper jaw into a mandible-like structure, has been observed upon injection of human EDN1 into the PAs of zebrafish31 and knock-in of the Edn1 cDNA into the Ednra locus in mice (EdnraEdn1/+).7 In the maxillary prominence of EdnraEdn1/+ embryos, ectopic Edn1 expression resulted in upregulation of target genes normally restricted to the mandibular prominence. MFDA-affected individuals 1–3 displayed increased symmetry across the jaw joint, as had been observed in EdnraEdn1/+ mouse embryos, along with absence of the zygomatic process of the temporal bone and enlargement of the malar bone, suggesting a partial homeotic transformation of the upper jaw into a mandible-like structure (Figure 2). MFDA-affected individuals and EdnraEdn1/+ embryos also share eyelid hypoplasia. These phenotypic comparisons support the hypothesis that an ectopic and/or constitutive activation of EDNRA in the developing first PA (particularly the regions giving rise to the upper jaw) underlies the MFDA phenotype. Given the increased affinity of EDN3 for the p.Tyr129Phe variant, this hypothesis would imply that EDN3 present in the maxillary prominence might positively affect signaling via the mutant EDNRA, leading to ectopic activation. In zebrafish, co-misexpression of EDN3 and mutant EDNRA resulted in a higher frequency of rostral neural tube phenotypes than co-misexpression of EDN3 and wild-type EDNRA. This phenotypic readout, although not biologically related to the MFDA phenotype, supports the possibility that EDN3 can elicit aberrant signaling consequences via mutant EDNRA in some contexts. Another argument against the MFDA variants being entirely loss of function is the phenotype of ACS and isolated question mark ears, both of which result from loss-of-function or dominant-negative variants in EDN1, PLCB4, and GNAI3, encoding components of the EDNRA pathway.8,32,33 In ACS the lower jaw is typically extremely hypoplastic and the upper jaw is essentially normal. Similarly, the lower jaw of Ednra-null mice is more severely affected than the upper jaw.6,34 However, we cannot exclude the possibility that a loss of normal EDNRA signaling function contributes in part to the MFDA phenotype. Our analysis of gene expression downstream of wild-type EDNRA or EDNRA p.Tyr129Phe in cultured cells indicated that although the wild-type receptor was able to respond to EDN1 and EDN3, neither EDN1 nor EDN3 appeared to induce Dlx5 or Hand2 expression via mutant EDNRA. Given the above hypothesis that the MFDA phenotype might be due to an EDN3-mediated gain of function of EDNRA in the upper jaw, this in vitro result was surprising. However, published in vitro studies have shown that a p.Tyr129Ala EDNRA variant had reduced function (decreased levels of phosphoinositide turnover and extracellular acidification) when stimulated with EDN1, compared to the wild-type receptor.35 On the other hand, EDNRA p.Tyr129Phe was able to rescue the phenotype of zebrafish in which the endogenous ednra genes were knocked down. This finding argues that at least some wild-type activity is retained by the mutant receptor. It is plausible that although this activity is sufficient to rescue the effect of the MOs, in MC3T3-E1 cells the composition of G proteins and other downstream components necessary for Dlx5/Hand2 induction might not be optimal, such that in our in vitro assays a partial loss of function of the mutant receptor was exacerbated. One caveat to the cell culture studies is that the response of the mutant receptor to endothelins in MC3T3-E1 cells might simply not be representative of the response that occurs in pharyngeal arch cells in vivo. The effects of reduced signaling capacity of the mutant receptor might be restricted to certain regions within the PAs. Specifically, reduced signaling by EDN1 via the mutant receptor could explain, in part, the micrognathia observed in MFDA-affected individuals 1–3. Although Edn3 does appear to be expressed in the mandibular prominence, it is less likely that the micrognathia is due to increased mutant EDNRA signaling induced by EDN1/EDN3, because micrognathia is not observed in EdnraEdn1/+ embryos.7
All MFDA-affected individuals presented with alopecia. To our knowledge a role for endothelin signaling in hair development has not been previously established. EdnraEdn1/+ mice die perinatally, preventing analysis of postnatal hair development. However, it has been demonstrated that the expression of Ednra and Edn2 is regulated by nuclear factor I/B (NFIB) in hair follicle stem cells,36 and transcriptomic analysis suggested high relative expression of EDNRA and EDN3 in the dermal papilla, a mesenchymal signaling center of the hair follicle.37 In a mouse line driving lacz expression from the Ednra locus, we observed strong expression during hair follicle development. Of note, alopecia is observed in mice in which Dlx3 (a target gene of Ednra signaling in the mandible) is conditionally deleted in skin.38 Our observation of Ednralacz expression in the developing eyelids and pinnae is also consistent with a primary defect in these tissues in MFDA and EdnraEdn1/+ mice. Finally, given the presence of tooth agenesis in individual 2, it is worth noting that Ednralacz expression has been previously demonstrated in condensing mesenchyme adjacent to the dental lamina.17 Eyelid and tooth defects have not been reported in Ednra-null mice, suggesting that EDNRA does not play an essential role in those tissues. However, the presence of Ednralacz expression at those sites is consistent with the hypothesis that the MFDA phenotypes in those tissues are caused by a gain of function of EDNRA signaling. Eyelid defects are present in EdnraEdn1/+ mice, emphasizing that EDNRA over-activation within its endogenous expression domain can lead to phenotypes not observed in the loss-of-function situation.
Zechi-Ceide et al.19 suggested that individual 2 might represent a severe form of JMS, a condition characterized by alopecia, hyposomia or anosmia, hypogonadotropic hypogonadism, hearing loss, and ear malformations. Of published JMS-affected individuals, we considered that individual 4 in the present report, originally described by Cushman et al.,18 was the most phenotypically similar to MFDA-affected individuals 1–3. Despite confirmation here that the individual in Cushman et al. has an EDNRA mutation, we remain cautious about the possibility that other JMS-affected individuals in the literature also have EDNRA mutations. Those JMS-affected individuals do not appear to have eyelid or orbital anomalies, nor auricular anomalies of the type observed in our EDNRA-mutated individuals.39–43 Given the phenotypic similarities between individual 4 with the p.Glu303Lys variant and individuals 1–3, we speculate that p.Glu303Lys might also be gain of function, although perhaps ligand independent, given that it falls in an intra-cellular loop of the protein. However, EDNRA harboring p.Glu303Lys failed to rescue the ednra MO phenotype in zebrafish, suggesting that it is loss of function in that assay. Based on the phenotypes of the affected individuals, it is difficult to know whether p.Tyr129Phe and p.Glu303Lys are truly equivalent in signaling output, because the latter was a somatic mosaic mutation and was associated with a milder phenotype. In the ligand-receptor co-misexpression assays, the combination of either ligand plus p.Glu303Lys gave a higher frequency of rostral hypoplasia phenotypes than ligand plus wild-type receptor. One could speculate that this intra-cellular mutation changes the conformation of the receptor molecule in such a way that the ligands bind and/or activate the receptor differently to the wild-type receptor.
In conclusion, we report four unrelated individuals with a unique combination of MFD and alopecia, ascribed to mutations in EDNRA. The recurrent tyrosine-to-phenylalanine substitution at position 129 leads to loss of selective affinity of EDNRA for EDN1. A tyrosine at position 129 is conserved in EdnrA in lamprey, an agnathan vertebrate that within the PAs displays dorso-ventral restriction of expression of some genes known to be downstream of EDN1-EDNRA in gnathostomes.44,45 Of note, a phenylalanine is found at this position in the Ednr-like gene of amphioxus, a non-vertebrate chordate.2 We speculate that acquisition of a tyrosine at position 129 in an ancestral EDNR gene was a key event in the evolution of selective affinity among endothelin substrates, and in combination with polarized EDN1 expression, might have been important for the evolution of mandibular specification within the first PA in vertebrates.
Acknowledgments
This work was supported by a Université Sorbonne Paris-Cité Pôle de recherche et d’enseignement supérieur (PRES) grant, project number SPC/JFG/2013-031 (to J.A.) and from NIH grants DE014181, DE018899, and DE023050 (to D.E.C.). J.A. and S.L. are supported by funding from the ANR E-Rare grant CRANIRARE. S.L. is supported by the ANR grant EvoDevoMut. N.K. is a Distinguished Brumley Professor. E.C.M. is supported by NIH Training Grant 5T32HD060558-04. The Institut Imagine is supported by an ANR grant ANR-10-IAHU-01. We thank Pierre Corvol, Gerard Couly, and Giovanni Levi for discussions and Holly Buttermore for technical support.
Contributor Information
Christopher T. Gordon, Email: chris.gordon@inserm.fr.
Jeanne Amiel, Email: jeanne.amiel@inserm.fr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
FaceBase, https://www.facebase.org
GATK Best Practices, https://www.broadinstitute.org/gatk/guide/best-practices
ImageJ, http://rsbweb.nih.gov/ij/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Le Douarin N., Kalcheim C. Cambridge University Press; Cambridge: 1999. The Neural Crest. [Google Scholar]
- 2.Braasch I., Schartl M. Evolution of endothelin receptors in vertebrates. Gen. Comp. Endocrinol. 2014;209:21–34. doi: 10.1016/j.ygcen.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 3.Clouthier D.E., Passos-Bueno M.R., Tavares A.L.P., Lyonnet S., Amiel J., Gordon C.T. Understanding the basis of auriculocondylar syndrome: Insights from human, mouse and zebrafish genetic studies. Am. J. Med. Genet. C. Semin. Med. Genet. 2013;163C:306–317. doi: 10.1002/ajmg.c.31376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krystek S.R., Jr., Patel P.S., Rose P.M., Fisher S.M., Kienzle B.K., Lach D.A., Liu E.C., Lynch J.S., Novotny J., Webb M.L. Mutation of peptide binding site in transmembrane region of a G protein-coupled receptor accounts for endothelin receptor subtype selectivity. J. Biol. Chem. 1994;269:12383–12386. [PubMed] [Google Scholar]
- 5.Lee J.A., Elliott J.D., Sutiphong J.A., Friesen W.J., Ohlstein E.H., Stadel J.M., Gleason J.G., Peishoff C.E. Tyr-129 is important to the peptide ligand affinity and selectivity of human endothelin type A receptor. Proc. Natl. Acad. Sci. USA. 1994;91:7164–7168. doi: 10.1073/pnas.91.15.7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clouthier D.E., Hosoda K., Richardson J.A., Williams S.C., Yanagisawa H., Kuwaki T., Kumada M., Hammer R.E., Yanagisawa M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998;125:813–824. doi: 10.1242/dev.125.5.813. [DOI] [PubMed] [Google Scholar]
- 7.Sato T., Kurihara Y., Asai R., Kawamura Y., Tonami K., Uchijima Y., Heude E., Ekker M., Levi G., Kurihara H. An endothelin-1 switch specifies maxillomandibular identity. Proc. Natl. Acad. Sci. USA. 2008;105:18806–18811. doi: 10.1073/pnas.0807345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon C.T., Petit F., Kroisel P.M., Jakobsen L., Zechi-Ceide R.M., Oufadem M., Bole-Feysot C., Pruvost S., Masson C., Tores F. Mutations in endothelin 1 cause recessive auriculocondylar syndrome and dominant isolated question-mark ears. Am. J. Hum. Genet. 2013;93:1118–1125. doi: 10.1016/j.ajhg.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rieder M.J., Green G.E., Park S.S., Stamper B.D., Gordon C.T., Johnson J.M., Cunniff C.M., Smith J.D., Emery S.B., Lyonnet S. A human homeotic transformation resulting from mutations in PLCB4 and GNAI3 causes auriculocondylar syndrome. Am. J. Hum. Genet. 2012;90:907–914. doi: 10.1016/j.ajhg.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wieczorek D. Human facial dysostoses. Clin. Genet. 2013;83:499–510. doi: 10.1111/cge.12123. [DOI] [PubMed] [Google Scholar]
- 11.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westerfield M. University of Oregon Press; Eugene: 1995. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio) [Google Scholar]
- 14.Javidan Y., Schilling T.F. Development of cartilage and bone. Methods Cell Biol. 2004;76:415–436. doi: 10.1016/s0091-679x(04)76018-5. [DOI] [PubMed] [Google Scholar]
- 15.Nair S., Li W., Cornell R., Schilling T.F. Requirements for Endothelin type-A receptors and Endothelin-1 signaling in the facial ectoderm for the patterning of skeletogenic neural crest cells in zebrafish. Development. 2007;134:335–345. doi: 10.1242/dev.02704. [DOI] [PubMed] [Google Scholar]
- 16.Niederriter A.R., Davis E.E., Golzio C., Oh E.C., Tsai I.-C., Katsanis N. In vivo modeling of the morbid human genome using Danio rerio. J. Vis. Exp. 2013;78:e50338. doi: 10.3791/50338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato T., Kawamura Y., Asai R., Amano T., Uchijima Y., Dettlaff-Swiercz D.A., Offermanns S., Kurihara Y., Kurihara H. Recombinase-mediated cassette exchange reveals the selective use of Gq/G11-dependent and -independent endothelin 1/endothelin type A receptor signaling in pharyngeal arch development. Development. 2008;135:755–765. doi: 10.1242/dev.012708. [DOI] [PubMed] [Google Scholar]
- 18.Cushman L.J., Torres-Martinez W., Weaver D.D. Johnson-McMillin syndrome: report of a new case with novel features. Birth Defects Res. A Clin. Mol. Teratol. 2005;73:638–641. doi: 10.1002/bdra.20178. [DOI] [PubMed] [Google Scholar]
- 19.Zechi-Ceide R.M., Guion-Almeida M.L., Jehee F.S., Rocha K., Passos-Bueno M.R. Mandibulofacial dysostosis, severe lower eyelid coloboma, cleft palate, and alopecia: A new distinct form of mandibulofacial dysostosis or a severe form of Johnson-McMillin syndrome? Am. J. Med. Genet. A. 2010;152A:1838–1840. doi: 10.1002/ajmg.a.33477. [DOI] [PubMed] [Google Scholar]
- 20.Schön M.P., Arya A., Murphy E.A., Adams C.M., Strauch U.G., Agace W.W., Marsal J., Donohue J.P., Her H., Beier D.R. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J. Immunol. 1999;162:6641–6649. [PubMed] [Google Scholar]
- 21.Liu B., Wu D. The first inner loop of endothelin receptor type B is necessary for specific coupling to Galpha 13. J. Biol. Chem. 2003;278:2384–2387. doi: 10.1074/jbc.M208683200. [DOI] [PubMed] [Google Scholar]
- 22.Breu V., Hashido K., Broger C., Miyamoto C., Furuichi Y., Hayes A., Kalina B., Löffler B.M., Ramuz H., Clozel M. Separable binding sites for the natural agonist endothelin-1 and the non-peptide antagonist bosentan on human endothelin-A receptors. Eur. J. Biochem. 1995;231:266–270. [PubMed] [Google Scholar]
- 23.Brand M., Le Moullec J.M., Corvol P., Gasc J.M. Ontogeny of endothelins-1 and -3, their receptors, and endothelin converting enzyme-1 in the early human embryo. J. Clin. Invest. 1998;101:549–559. doi: 10.1172/JCI524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nataf V., Amemiya A., Yanagisawa M., Le Douarin N.M. The expression pattern of endothelin 3 in the avian embryo. Mech. Dev. 1998;73:217–220. doi: 10.1016/s0925-4773(98)00048-3. [DOI] [PubMed] [Google Scholar]
- 25.Brunskill E.W., Potter A.S., Distasio A., Dexheimer P., Plassard A., Aronow B.J., Potter S.S. A gene expression atlas of early craniofacial development. Dev. Biol. 2014;391:133–146. doi: 10.1016/j.ydbio.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller C.T., Schilling T.F., Lee K., Parker J., Kimmel C.B. sucker encodes a zebrafish Endothelin-1 required for ventral pharyngeal arch development. Development. 2000;127:3815–3828. doi: 10.1242/dev.127.17.3815. [DOI] [PubMed] [Google Scholar]
- 27.Morimoto H., Shimadzu H., Kushiyama E., Kawanishi H., Hosaka T., Kawase Y., Yasuda K., Kikkawa K., Yamauchi-Kohno R., Yamada K. Potent and selective ET-A antagonists. 1. Syntheses and structure-activity relationships of N-(6-(2-(aryloxy)ethoxy)-4-pyrimidinyl)sulfonamide derivatives. J. Med. Chem. 2001;44:3355–3368. doi: 10.1021/jm0102304. [DOI] [PubMed] [Google Scholar]
- 28.Kurihara Y., Kurihara H., Suzuki H., Kodama T., Maemura K., Nagai R., Oda H., Kuwaki T., Cao W.H., Kamada N. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368:703–710. doi: 10.1038/368703a0. [DOI] [PubMed] [Google Scholar]
- 29.Maemura K., Kurihara H., Kurihara Y., Oda H., Ishikawa T., Copeland N.G., Gilbert D.J., Jenkins N.A., Yazaki Y. Sequence analysis, chromosomal location, and developmental expression of the mouse preproendothelin-1 gene. Genomics. 1996;31:177–184. doi: 10.1006/geno.1996.0029. [DOI] [PubMed] [Google Scholar]
- 30.Gordon C.T., Cunniff C.M., Green G.E., Zechi-Ceide R.M., Johnson J.M., Henderson A., Petit F., Kokitsu-Nakata N.M., Guion-Almeida M.L., Munnich A. Clinical evidence for a mandibular to maxillary transformation in Auriculocondylar syndrome. Am. J. Med. Genet. A. 2014;164A:1850–1853. doi: 10.1002/ajmg.a.36505. [DOI] [PubMed] [Google Scholar]
- 31.Kimmel C.B., Walker M.B., Miller C.T. Morphing the hyomandibular skeleton in development and evolution. J. Exp. Zoolog. B Mol. Dev. Evol. 2007;308:609–624. doi: 10.1002/jez.b.21155. [DOI] [PubMed] [Google Scholar]
- 32.Gordon C.T., Vuillot A., Marlin S., Gerkes E., Henderson A., AlKindy A., Holder-Espinasse M., Park S.S., Omarjee A., Sanchis-Borja M. Heterogeneity of mutational mechanisms and modes of inheritance in auriculocondylar syndrome. J. Med. Genet. 2013;50:174–186. doi: 10.1136/jmedgenet-2012-101331. [DOI] [PubMed] [Google Scholar]
- 33.Romanelli Tavares V.L., Gordon C.T., Zechi-Ceide R.M., Kokitsu-Nakata N.M., Voisin N., Tan T.Y., Heggie A.A., Vendramini-Pittoli S., Propst E.J., Papsin B.C. Novel variants in GNAI3 associated with auriculocondylar syndrome strengthen a common dominant negative effect. Eur. J. Hum. Genet. 2014 doi: 10.1038/ejhg.2014.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruest L.B., Xiang X., Lim K.C., Levi G., Clouthier D.E. Endothelin-A receptor-dependent and -independent signaling pathways in establishing mandibular identity. Development. 2004;131:4413–4423. doi: 10.1242/dev.01291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Webb M.L., Patel P.S., Rose P.M., Liu E.C., Stein P.D., Barrish J., Lach D.A., Stouch T., Fisher S.M., Hadjilambris O. Mutational analysis of the endothelin type A receptor (ETA): interactions and model of selective ETA antagonist BMS-182874 with putative ETA receptor binding cavity. Biochemistry. 1996;35:2548–2556. doi: 10.1021/bi951836v. [DOI] [PubMed] [Google Scholar]
- 36.Chang C.Y., Pasolli H.A., Giannopoulou E.G., Guasch G., Gronostajski R.M., Elemento O., Fuchs E. NFIB is a governor of epithelial-melanocyte stem cell behaviour in a shared niche. Nature. 2013;495:98–102. doi: 10.1038/nature11847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohyama M., Kobayashi T., Sasaki T., Shimizu A., Amagai M. Restoration of the intrinsic properties of human dermal papilla in vitro. J. Cell Sci. 2012;125:4114–4125. doi: 10.1242/jcs.105700. [DOI] [PubMed] [Google Scholar]
- 38.Hwang J., Mehrani T., Millar S.E., Morasso M.I. Dlx3 is a crucial regulator of hair follicle differentiation and cycling. Development. 2008;135:3149–3159. doi: 10.1242/dev.022202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson V.P., McMillin J.M., Aceto T., Jr., Bruins G. A newly recognized neuroectodermal syndrome of familial alopecia, anosmia, deafness, and hypogonadism. Am. J. Med. Genet. 1983;15:497–506. doi: 10.1002/ajmg.1320150316. [DOI] [PubMed] [Google Scholar]
- 40.Johnston K., Golabi M., Hall B., Ito M., Grix A. Alopecia-anosmia-deafness-hypogonadism syndrome revisited: report of a new case. Am. J. Med. Genet. 1987;26:925–927. doi: 10.1002/ajmg.1320260422. [DOI] [PubMed] [Google Scholar]
- 41.Hennekam R.C., Holtus F.J. Johnson-McMillin syndrome: report of another family. Am. J. Med. Genet. 1993;47:714–716. doi: 10.1002/ajmg.1320470526. [DOI] [PubMed] [Google Scholar]
- 42.Schweitzer D.N., Yano S., Earl D.L., Graham J.M., Jr. Johnson-McMillin syndrome, a neuroectodermal syndrome with conductive hearing loss and microtia: report of a new case. Am. J. Med. Genet. A. 2003;120A:400–405. doi: 10.1002/ajmg.a.20085. [DOI] [PubMed] [Google Scholar]
- 43.De Metsenaere F., Mortier G., Dhont M. Hypogonadotropic hypogonadism in a female with the Johnson-McMillin syndrome. Am. J. Obstet. Gynecol. 2004;191:1728–1729. doi: 10.1016/j.ajog.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 44.Cerny R., Cattell M., Sauka-Spengler T., Bronner-Fraser M., Yu F., Medeiros D.M. Evidence for the prepattern/cooption model of vertebrate jaw evolution. Proc. Natl. Acad. Sci. USA. 2010;107:17262–17267. doi: 10.1073/pnas.1009304107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuraku S., Takio Y., Sugahara F., Takechi M., Kuratani S. Evolution of oropharyngeal patterning mechanisms involving Dlx and endothelins in vertebrates. Dev. Biol. 2010;341:315–323. doi: 10.1016/j.ydbio.2010.02.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.