

Abstract
Microphthalmia with linear skin defects (MLS) syndrome is an X-linked male-lethal disorder also known as MIDAS (microphthalmia, dermal aplasia, and sclerocornea). Additional clinical features include neurological and cardiac abnormalities. MLS syndrome is genetically heterogeneous given that heterozygous mutations in HCCS or COX7B have been identified in MLS-affected females. Both genes encode proteins involved in the structure and function of complexes III and IV, which form the terminal segment of the mitochondrial respiratory chain (MRC). However, not all individuals with MLS syndrome carry a mutation in either HCCS or COX7B. The majority of MLS-affected females have severe skewing of X chromosome inactivation, suggesting that mutations in HCCS, COX7B, and other as-yet-unidentified X-linked gene(s) cause selective loss of cells in which the mutated X chromosome is active. By applying whole-exome sequencing and filtering for X-chromosomal variants, we identified a de novo nonsense mutation in NDUFB11 (Xp11.23) in one female individual and a heterozygous 1-bp deletion in a second individual, her asymptomatic mother, and an affected aborted fetus of the subject’s mother. NDUFB11 encodes one of 30 poorly characterized supernumerary subunits of NADH:ubiquinone oxidoreductase, known as complex I (cI), the first and largest enzyme of the MRC. By shRNA-mediated NDUFB11 knockdown in HeLa cells, we demonstrate that NDUFB11 is essential for cI assembly and activity as well as cell growth and survival. These results demonstrate that X-linked genetic defects leading to the complete inactivation of complex I, III, or IV underlie MLS syndrome. Our data reveal an unexpected role of cI dysfunction in a developmental phenotype, further underscoring the existence of a group of mitochondrial diseases associated with neurocutaneous manifestations.
Main Text
The microphthalmia with linear skin defects (MLS) syndrome (MIM 309801) is an X-linked neurocutaneous disorder manifesting exclusively in females, suggesting embryonic lethality in hemizygous males. Affected females typically show uni- or bilateral microphthalmia and/or anophthalmia and linear skin lesions, which are usually restricted to the face and neck and consist of aplastic skin that evolves into healed hyperpigmented areas. Facultative features include a great variety of ocular anomalies, such as microcornea, coloboma, anterior-chamber defects, optic-nerve hypoplasia, retinal abnormalities, congenital glaucoma with peripheral anterior synechia, and cataracts. Other clinical characteristics, such as developmental delay, abnormalities of the central nervous system (CNS), short stature, and cardiac defects have also been observed (see GeneReviews in Web Resources).1–3 Although most affected females show the classical phenotype, a few reported individuals have displayed eye abnormalities in an absence of skin defects,2–7 and in other individuals, skin defects in an absence of ocular anomalies have been described.8–10
MLS syndrome is a genetically heterogeneous disorder given that mutations in two different X-chromosomal genes have been identified to date.3,11 The majority of individuals diagnosed with MLS carry a terminal Xp deletion, detectable by light microscopy. The deletions include HCCS in Xp22.2 (MIM 300056; RefSeq accession number NM_005333.4) (see GeneReviews in Web Resources). HCCS encodes the mitochondrial holocytochrome c-type synthase.12,13 After this initial observation, heterozygous submicroscopic interstitial deletions and intragenic mutations of HCCS were detected in females with the classical MLS phenotype, confirming a pathogenic role for HCCS disruption in MLS.1–3,7,14,15 More recently, heterozygous mutations in COX7B (MIM 300885; RefSeq NM_001866.2), located in chromosome band Xq21.1, have been found in girls who had linear skin defects as the predominant feature but who had no HCCS mutation or microphthalmia.11 COX7B encodes a structural subunit of cytochrome c oxidase, also known as mitochondrial complex IV (cIV).16,17 cIV is one of the five enzymatic complexes (cI–cV) of the mitochondrial respiratory chain (MRC), which carries out oxidative phosphorylation (OXPHOS). Both HCCS and COX7B gene products are indispensable for MRC biogenesis and function. HCCS catalyzes the incorporation of heme moieties in both cytochrome c1, a structural component of cIII, and cytochrome c, the electron shuttle between cIII and IV.12,13 Complementation studies in yeast revealed that HCCS deficiency led to severe OXPHOS defects.18 Similarly, depletion of COX7B impairs cIV assembly and activity, leading to severe OXPHOS failure.11
Because the cIII–cIV MRC segment is impaired in individuals with MLS syndrome, this disorder is by definition a mitochondrial disease. However, whereas typical mitochondrial diseases are usually characterized by postnatal organ failure,19,20 MLS syndrome is a truly developmental phenotype, in that it involves impaired development and malformation of specific organs, such as the eyes, skin, and CNS. Linear skin defects, which are present at birth, are the major clinical criteria of MLS syndrome. The lesions follow the lines of Blaschko, heal with age, and leave scars in some cases (see GeneReviews in Web Resources). Downregulation of hccs or cox7b in medaka has been shown to recapitulate the MLS phenotype; the morphant fishes show microphthalmia and microcephaly, indicating the important roles of cIII and cIV in vertebrate CNS development.11,18
High inter- and intrafamilial phenotypic variabilities have been reported in females with MLS syndrome.3,21–23 The spectrum of clinical features ranges widely, from a neonatal lethal form of MLS syndrome to no apparent clinical sign in carrier females.1–3,7,9,21,22,24 In Table 1 we compare the clinical features described in females positive for the HCCS or COX7B mutation. Somatic mosaicism for the causative mutation and the degree of X chromosome inactivation (XCI) skewing in different tissues have been postulated as explanations for the great clinical variability of MLS.2,3,24,27,28 Positive selection of OXPHOS-proficient cells during development could result in extreme skewing of the XCI ratio in females positive for the HCCS or COX7B mutation.2,11 Indeed, monoallelic expression of the wild-type X chromosome was shown in lymphoblastoid cells of an MLS-affected female with the heterozygous HCCS nonsense mutation c.589C>T (p.Arg197∗).3 These results suggest that, during embryonic development, cells with an active wild-type X chromosome are likely to reproduce faster and eventually outgrow cells expressing the COX7B- or HCCS-mutation-bearing X chromosome and that this faster growth leads to skewed XCI and high clinical variability.2,29 Thus, MLS syndrome is the result of extremely severe MRC disruption combined with inactivation of the X chromosome. Importantly, because cells expressing the mutated X chromosome are filtered out during development, no obvious biochemical defect of MRC activities is detected in the cells of MLS-affected females (see below).
Table 1.
Clinical Features Reported in MLS-Affected Individuals with a Mutation of HCCS, COX7B, or NDUFB11
| Clinical Features |
HCCSa,bMutation |
COX7BcMutation |
NDUFB11Mutation |
|||
|---|---|---|---|---|---|---|
| Occurence | Occurence | Subject 1 | Subject 2 | Mother of Subject 2 | Aborted Fetus of Subject-2’s Mother | |
| Microphthalmia | 51/63 (81%) | 0/4 | – | – | – | NA |
| Sclerocornea | 29/63 (46%) | 0/4 | – | – | – | NA |
| Other eye abnormalities | 34/63 (54%) | 1/4 | + | + | – | NA |
| Linear skin lesions | 47/63 (75%) | 4/4 | + | + | – | NA |
| Nail dystrophy | 3/55 (5%) | 4/4 | – | – | – | NA |
| Microcephaly, agenesis of the corpus callosum, and other CNS abnormalities | 27/59 (46%) | 3/4 | –d | + | – | + |
| Developmental delay and/or intellectual disability | 11/49 (22%) | 2/4 | NA hypotonia | + hypotonia | – | NA |
| Short stature | 18/42 (43%) | 2/4 | NA | + | – | intrauterine growth retardation |
| Cardiac abnormalities | 22/60 (37%) | 2/4 | + | + | – | + |
| Diaphragmatic hernia | 3/63 (5%) | 1/4 | – | – | – | – |
| Genitourinary abnormalities | 14/63 (22%) | 1/4 | – | – | – | – |
| Other anomalies | NA | NA | thyroid abnormalities (large and multiple sites of oncocytic metaplasia) after autopsy | seizures, epilepsy | – | – |
Clinical features in this table correspond to those in Table 1 in Indrieri et al.11 For each feature listed in the table, the total number of individuals includes only individuals in whom each clinical feature was analyzed and documented. Other anomalies are only given for individuals with an NDUFB11 mutation. Abbreviations are as follows: –, absent; +, present; NA, not applicable.
Mutations in HCCS include intragenic alterations and chromosomal abnormalities that result in monosomy for Xp22.
See GeneReviews in Web Resources and Sharma et al.,25 Kono et al.,6 Zumwalt et al.,26 Vergult et al.,15 and van Rahden et al.2
See Indrieri et al.11
Brain size and morphology were normal; an autopsy showed no corpus callosum agenesis.
In DNA samples taken from 11 MLS-affected females and pooled from two different labs, sequencing and copy-number analysis failed to detect a mutation in either HCCS or COX7B.11 In our own cohort of seven individuals, whole-exome sequencing (WES) revealed heterozygous truncating mutations in NDUFB11 (MIM 300403; RefSeq NM_019056.6), located in chromosomal band Xp11.23, in two unrelated female probands. Both had a highly skewed XCI pattern in peripheral blood cells and typical features of MLS syndrome, except for the absence of microphthalmia and sclerocornea (Table 1). Subject 1 belongs to a simplex family. Subject 2 was a 15-month-old girl whose asymptomatic mother subsequently aborted a female fetus with severe malformations; all three harbored the same heterozygous mutation in NDUFB11. The five remaining individuals in our cohort showed no mutation in NDUFB11, and WES is underway. In all cases, genetic analysis was performed with informed consent, including consent to use photographs in this report, and was approved by the ethics committee of the Medical Chamber of Hamburg (reference numbers PV3585 and PV3802).
Subject 1 was an infant girl born to healthy, non-consanguineous parents (Figure 1A and Table 1). Her birth weight was 3,060 g (10th–25th centile), her length was 50 cm (50th centile), and her occipito-frontal head circumference (OFC) was 36.5 cm (90th centile). Linear skin defects that disappeared in the first few months were noted on her nose, chin, and neck (Figure 1B). Axial hypotonia was present from birth. Failure to thrive was documented at 1 month of age. Ocular examination revealed lacrimal-duct atresia but neither microphthalmia nor sclerocornea. Blood lactate levels were consistently in the normal range. She was admitted to the hospital at 6 months of age for cardiac arrest. She was repeatedly treated for ventricular fibrillation and tachycardia but died a few weeks after admission. An autopsy revealed histiocytoid cardiomyopathy and thyroid abnormalities consisting of large and multiple sites of oncocytic metaplasia. Immunohistochemical staining showed that oncocytic cells were thyroid transcription factor-1 (TTF-1) positive and cytokeratin 19 (CK19) negative. An anti-calcitonin antibody showed several sites of C cell hyperplasia (more than 50 C cells in at least three microscope fields [100× magnification]).
Figure 1.
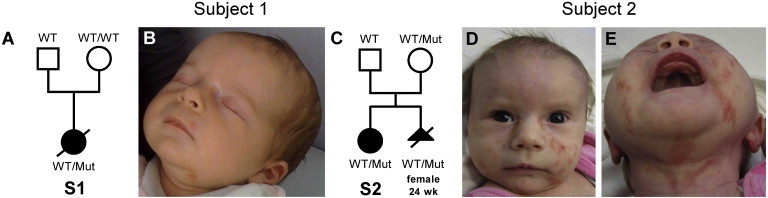
Pictures of Two Subjects with MLS Syndrome and the NDUFB11 Mutation
(A) Pedigree of subject 1’s (S1’s) family and segregation analysis of the NDUFB11 mutation c.262C>T.
(B) At the age of 6 weeks, subject 1 had linear skin defects on her face and ears.
(C) Pedigree of subject 2’s (S2’s) family and segregation analysis of the 1-bp deletion c.402delG in NDUFB11. The mother of subject 2 is a healthy carrier of the NDUFB11 mutation; she aborted a female fetus who had severe brain and heart anomalies and carried the same mutation.
(D and E) Subject 2 (2 months of age) had linear skin defects on the nose, chin, neck, and left ear lobe. Abbreviations are as follows: Mut, NDUFB11 mutation; wk, weeks; WT, wild-type.
Subject 2 was a 15-month-old girl, the first child of German, non-consanguineous, healthy parents (Figure 1C and Table 1). During pregnancy, corpus-callosum agenesis as well as dilated lateral ventricles were detected by ultrasound and confirmed by prenatal magnetic resonance imaging (MRI) of the brain. Measurements at birth (41st week of gestation) were normal and included a weight of 3,570 g (50th–75th centile), a length of 51 cm (25th–50th centile), and an OFC of 34.5 cm (25th–50th centile). After birth, linear and atrophic hyperpigmented streaks on her face and neck (Figures 1D and 1E) as well as on her left index finger were noticed. At 2 months of age, seizures, which responded well to anticonvulsants, occurred. At the same age, dilated cardiomyopathy ensued, prompting a heart transplantation at 6 months of age. At 15 months of age, an ophthalmologic examination revealed myopia, nystagmus, and strabismus, but neither microphthalmia nor sclerocornea. Over time, severe psychomotor developmental delay became evident; she was able to roll at 15 months of age and to sit at 18 months. She started walking at 3 years of age. At 7 years of age, she was able to speak in simple sentences and had sphincter control. At 15 months of age, her height (73.5 cm, −2.1 SD), weight (7.5 kg, −2.7 SD), and OFC (41 cm, −4.4 SD) were below the third centile. Severe muscular hypotonia and delayed dentition were present. A brainstem auditory evoked response demonstrated normal hearing. Conventional cytogenetic analysis revealed normal results. At 2.5 years of age, MRI of the brain confirmed agenesis of corpus callosum and enlarged ventricles. Blood lactate was measured at the ages of 6 months and 4.5 years and was in the normal range both times.
In the next pregnancy of subject-2’s mother, an ultrasound examination of the female fetus at the 24th week of gestation showed a thickened myocardium and pericardial effusion, corpus-callosum dysgenesis, a small cerebellum, and a connection between a lateral ventricle and the cavum septum pellucidum, as well as intrauterine growth retardation (Table 1). The pregnancy was terminated (Figure 1C).
High-resolution molecular karyotyping with the Agilent 180K Oligo Array (Agilent Technologies) revealed no disease-associated copy-number variant (CNV) in subject 1. Array comparative genomic hybridization (CGH) with the Agilent Human Genome Microarray Kit 244K was performed on the DNA sample obtained from the aborted fetus of subject-2’s mother and detected a heterozygous deletion of at least 70 kb at 2p16.3, containing part of NRXN1 (data not shown). Quantitative real-time PCR confirmed the microdeletion in the fetal DNA and identified the same CNV in both subject 2 and her mother but excluded this CNV in the father (data not shown). Heterozygous intragenic mutations and deletions of NRXN1 are enriched in cohorts of persons with autism spectrum disorder and neurodevelopmental disorders when these individuals are compared to control individuals. Incomplete penetrance has been described; some healthy parents carry the same deletion as the affected child.30–32 Therefore, we cannot exclude the possibility that the NRXN1 deletion might have contributed to the neurological phenotype in subject 2 (for instance, by worsening developmental delay). However, this copy-number loss is highly unlikely to be causative for the multiple congenital anomalies observed in this girl.
We next determined the XCI pattern by quantitative examination of the methylation status at the AR (androgen receptor) locus.2,33 Leukocyte-derived DNA of subject 1 showed a highly skewed XCI pattern with a 10:90 ratio, whereas her healthy mother had a less skewed XCI ratio (20:80). In peripheral blood cells of subject 2 and her healthy mother, we detected complete skewing (100:0). Likewise, an XCI ratio of 99:1 was found in DNA from the aborted fetus of subject-2’s mother (data not shown). Given that complete or highly skewed XCI was observed in COX7B- or HCCS-mutation-positive females,2,3,7,11 these findings strongly suggest that subjects 1 and 2 harbored mutations affecting a currently unidentified X-linked gene.
We then performed WES of peripheral blood DNA from subject 1 and her parents as well as from subject 2 and her mother by using the Illumina TruSeq Exome Enrichment Kit and an Illumina HiSeq 2500 sequencer. We aligned the sequence to the human genome reference sequence (UCSC Genome Browser hg19) by using the Burrows-Wheeler Aligner (BWA).34 We used SAMtools to remove potential PCR duplicates from the alignments and the Genome Analysis Toolkit (GATK) for calling single-nucleotide variants (SNVs) and indels.35 In addition, SAMtools was used for calling a second set of variants from the deduplicated alignments.36 Variant effects were predicted with SnpEff.37 For each individual, the two sets of variant calls were used for variant filtration and prioritization. In both families, we identified common variants (minor-allele frequency ≥1%) by filtering exome data against data in dbSNP version 137, the NHLBI Exome Variant Server, and the ExAC Browser. A detailed description of WES can be found elsewhere.38 Exome read-depth metrics are summarized in Table S1.
Highly skewed XCI in subject 1 and slight skewing in her mother suggested the presence of an X-chromosomal de novo mutation in subject 1. In contrast, severely skewed XCI in subject 2, her asymptomatic mother, and the severely affected fetus led us to hypothesize that the mother carried an X-chromosomal variant that was inherited by both of her children. Indeed, carrier females with an HCCS mutation or terminal Xp deletion but with no clinical features of MLS syndrome have been described.2,3,24 By applying this filter strategy, we identified the heterozygous de novo variant c.262C>T in NDUFB11 (Xp11.23) in subject 1. This variant is predicted to lead to the introduction of a premature stop codon (p.Arg88∗) (Figure 2A and Table S2). Notably, the mutated base thymine was present in 42 sequence reads and the wild-type base cytosine in 84, suggesting that the mutation was present in mosaic state in subject-1’s lymphocytes (data not shown). In subject 2 and her healthy mother, three heterozygous X-chromosomal sequence variants were identified: two missense variants, c.1496A>G (p.Asn499Ser) in ARSH (MIM 300586; RefSeq NM_001011719.1) and c.3498G>T (p.Lys1166Asn) in DGKK (MIM 300837; RefSeq NM_001013742.3), and the frameshift mutation c.402delG (p.Arg134Serfs∗3) in NDUFB11 (Figure 2B and Table S2). The variants in NDUFB11 and ARSH were absent in dbSNP, 1000 Genomes, the NHLBI Exome Variant Server, and the ExAC Browser, whereas the rare DGKK variant was annotated (Table S2). Validation of all variants was achieved with standard bidirectional Sanger methods (primer sequences are available on request). Further evidence of mosaicism of the NDUFB11 c.262C>T mutation came from Sanger sequencing of DNA isolated from leukocytes and fibroblast cells of subject 1; the two different sequence profiles showed a low signal for the mutant variant (thymine) superimposed on the wild-type sequence (cytosine) (Figure 2A). Sanger sequencing in the family of subject 2 excluded the three X-chromosomal variants in the subject’s father (paternity confirmed; Table S2). The two missense variants in ARSH and DGKK are predicted by SIFT, SNAP, PolyPhen-2, and/or MutationTaster to not interfere with protein function (Table S2). Together, the identification of two unrelated individuals who had typical features of MLS syndrome and each carried a heterozygous loss-of-function mutation in NDUFB11 indicated pathogenicity of the sequence changes c.262C>T (p.Arg88∗) and c.402delG (p.Arg134Serfs∗3). This conclusion is further supported by the presence of the 1-bp deletion c.402delG in NDUFB11 in DNA isolated from the severely malformed fetus of subject-2’s mother (Table 1 and Figures 1D and 2B). The NDUFB11 gene product is the 17.3-kDa subunit 11 of the NADH dehydrogenase (ubiquinone) 1 beta subcomplex and is part of the MRC cI. NDUFB11 comprises three exons; exon 2 is alternatively spliced.39 The c.262C>T transition is located in exon 2, upstream of the alternatively used splice donor site (last 30 nucleotides), and the c.402delG mutation is located in exon 3. The longer transcript encodes a 163 amino acid (aa) protein, while the shorter one encodes a more abundant protein consisting of 153 aa.39 Human NDUFB11, also called NP17.3 (neuronal protein 17.3), corresponds to the ESSS protein subunit in the nomenclature of bovine heart cI.40,41 Complex I, or NADH:ubiquinone oxidoreductase, is the largest enzyme of the mitochondrial electron-transport chain and has two critical functions: NADH oxidation and formation of membrane potential via proton translocation. Bovine, and presumably human, cI is composed of 45 subunits, seven of which are encoded by the mitochondrial DNA.42 Only 14 conserved core subunits are essential for energy transduction.43 The function of the 30 supernumerary subunits, including NDUFB11, is still poorly understood. They might participate in complex stabilization, protection of the core subunits against oxidative stress, assembly of the complex, and/or regulation of complex activity.44,45 Together, the products of HCCS and COX7B, the two other genes with a known association to MLS, as well as the products of NDUFB11, are all involved in OXPHOS system biogenesis,12,13,16,17,40,46 underscoring the existence of a new group of mitochondrial diseases with neurodevelopmental and skin defects as the major clinical features. To test the idea that the extremely skewed XCI found in our MLS-affected subjects was due to strong selection against OXPHOS-faulty cells expressing mutant NDUFB11, we performed RT-PCR analysis followed by Sanger sequencing of reverse-transcribed total RNA. As expected, we identified only wild-type NDUFB11 transcripts in fibroblast cells of subject 1 (Figure 2A) as well as in leukocytes of subject 2 and her mother (Figure 2B and data not shown). In concordance with these data, cells from an affected individual do not show a cI defect (Table S3) because NDUFB11 is exclusively expressed by the normal gene present on the wild-type X chromosome. Consequently, fibroblasts from affected females are unsuitable for use in investigations of the biochemical consequences of defective NDUFB11. To overcome this problem, we decided to silence NDUFB11 in HeLa cells by stably expressing each of five different shRNAs (shRNA-1 to shRNA-5), all targeting the NDUFB11 mRNA. HeLa cells transduced with the empty pLKO.1 vector were used as a control. The degree of NDUFB11 silencing was evaluated with immunoblot analysis of the corresponding protein. The amount of NDUFB11 was decreased in all five NDUFB11-depleted cell lines; the lowest amount was achieved in the shRNA-1-expressing cells, where NDUFB11 was virtually undetectable, and shRNA-2-expressing cells, which showed a residual protein amount of ∼5% of the amounts in controls (Figure 3A). Interestingly, the amount of another cI membrane-arm subunit, NDUFB8, was as decreased as NDUFB11 in the five NDUFB11-knockdown cell lines (shRNA-1 to shRNA-5) (Figure 3A). The low amount of NDUFB8 in the knockdown cells was not due to reduced mRNA levels, given that these were even slightly higher than mRNA levels in control cells (Figure S1). This result excludes an off-target effect of the shRNA silencing NDUFB11, suggesting that the absence of the NDUFB11 subunit might destabilize the region where both NDUFB11 and NDUFB8 subunits are located in the cI membrane arm.42 This is further supported by the observation that NDUFS3, a cI subunit that takes part in the peripheral arm of the complex, was detected in a normal amount in shRNA-transduced cells (Figure 3A). Likewise, cIII and cIV were not affected by NDUFB11 silencing, given that the amount of both UQCRC2 (ubiquinol-cytochrome c reductase core protein II, part of cIII) and MTCO1 (mtDNA-encoded cytochrome c oxidase subunit 1, part of cIV) was the same in all tested cell lines (Figure 3A). These results suggest that NDUFB11 is necessary for the assembly of the membrane arm of cI, and thus for the assembly of the cI holocomplex as well.
Figure 2.
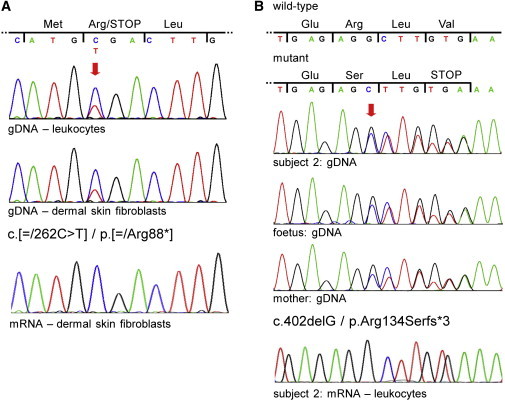
NDUFB11 Mutations in Subjects 1 and 2
(A) Sequence electropherograms of part of NDUFB11 exon 2 from subject 1’s genomic DNA (gDNA) isolated from blood (upper panel) and gDNA and mRNA (cDNA) of dermal skin fibroblasts (middle and lower panels, respectively). Nucleotide triplets and encoded amino acids (three-letter code) are indicated above the electropherograms. The red arrow points to the double peak in the two upper electropherograms; the lower peak of the mutant base (T) is superimposed on the higher peak of the wild-type base (C). Subject 1 carries the mosaic NDUFB11 mutation c.[ = /262C>T] (p.[ = /Arg88∗]).
(B) Sequence electropherograms of part of NDUFB11 exon 3 from gDNA of subject 2 (upper panel), the aborted fetus (second from the top panel), and their healthy mother (second from the bottom panel). Nucleotide triplets and encoded amino acids of the wild-type and mutated allele are indicated above the electropherograms. The red arrow points to the first double peak in the electropherograms and indicates the start of the frameshift. The three individuals are heterozygous for the NDUFB11 mutation c.402delG (p.Arg134Serfs∗3). The sequence electropherogram from leukocyte-derived mRNA (cDNA) of subject 2 shows only wild-type NDUFB11 transcripts (bottom panel).
Figure 3.
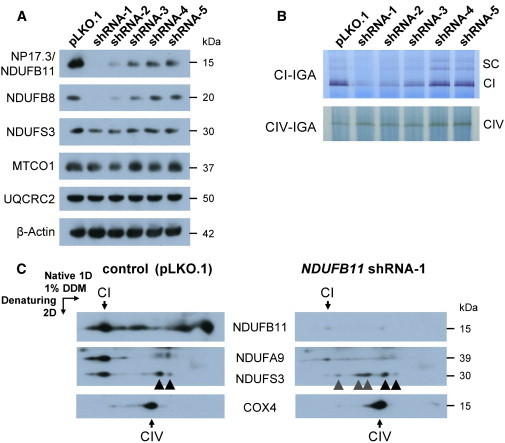
Impaired Assembly and Activity of CI in NDUFB11-Depleted HeLa Cells
(A) HeLa cells were stably transfected with one of five different MISSION shRNA1-5 clones (TRCN0000107101–105, Sigma-Aldrich) targeting the NDUFB11 mRNA or with the empty vector pLKO.1. Immunoblotting with an anti-NDUFB11 antibody (Proteintech) allowed NDUFB11 depletion to be monitored. The antibodies against NDUFB8, NDUFS3, MTCO1 (cytochrome c oxidase subunit 1; theoretical molecular mass of 57 kDa, but migrating at an apparent molecular mass of 37 kDa in SDS-PAGE), and cytochrome b-c1 complex subunit 2 (UQCRC2) were from Abcam (formerly Mitosciences). An anti-β-actin antibody (Sigma-Aldrich) was used as the loading control.
(B) IGA of cI and cIV on 1D-BNGE47 in the five NDUFB11-knockdown cell lines (shRNA1-5) and the control (pLKO.1). 1D-BNGE was performed on mitoplasts solubilized with 1% DDM (n-Dodecyl β-D-maltoside).48 The intensity of the cI-specific signals representing the active monomer (CI) and the active cI associated with the other MRC complexes in the form of supercomplexes (SCs) was markedly reduced in HeLa cells stably expressing NDUFB11 shRNA-1 or -2 (upper panel). In contrast, cIV-IGA was comparable in all the samples.
(C) Immunodetection of cI-assembly intermediates on 2D-BNGE immunoblot with antibodies against NDUFB11, NDUFA9 (Abcam), and NDUFS3. The NDUFS3-containing assembly intermediates detected in both control (pLKO.1) and NDUFB11 shRNA-1-depleted cells are marked by black arrowheads; the NDUFS3-containing subcomplexes of the cI peripheral arm accumulated only in the shRNA-1 cells are marked by gray arrowheads. An anti-COX4 antibody (Abcam; theoretical molecular mass of 19.5 kDa but migrating at an apparent molecular mass of 15 kDa in SDS-PAGE) was used as a loading control.
We next assessed cI in-gel activity (cI-IGA) by performing one-dimensional blue-native gel electrophoresis (1D-BNGE) in mitochondrial extracts from the five NDUFB11-knockdown cell lines and the pLKO.1 control cell line. The specific cI-reactive band was markedly reduced in HeLa cells expressing shRNA-1 and shRNA-2 and was reduced but clearly visible in shRNA-3 cells, whereas the signal detected in shRNA-4 and shRNA-5 cell lines was comparable to that of the pLKO.1 control cell line (Figure 3B). In contrast, comparable cIV in-gel activity was observed in control and all NDUFB11-depleted cell lines (Figure 3B). Accordingly, the cI enzymatic activity was extremely low in the shRNA-1 cells with respect to the pLKO.1 control cells, but the other complex activities were comparable to the control values (Table S4). These results demonstrate a direct relationship between the decreased NDUFB11 amount and the catalytic activity of the cI holocomplex.
To further investigate how the knockdown of NDUFB11 affects cI assembly, we performed second-dimensional (2D) BNGE on shRNA-1 and pLKO.1 cell lines (Figure 3C). Immunoblot analysis was carried out with antibodies that probed three topologically distinct cI subunits (α-NDUFB11, α-NDUFA9, and α-NDUFS3). α-NDUFB11 revealed a barely detectable signal in shRNA-1 cells (right panel in Figure 3C), whereas pLKO.1 control cells displayed a strong reactive band corresponding to the fully assembled cI and to different membrane-arm subcomplexes (left panel in Figure 3C). NDUFA9 is a cI subunit that is incorporated late during assembly and is located at the joint connecting the membrane and the peripheral arms.49 The NDUFA9-specific signal was weak and non-focalized in shRNA-1 cells (right panel in Figure 3C), whereas it was readily detected in pLKO.1 control cells (left panel in Figure 3C). NDUFS3 is incorporated early in the assembly pathway into sub-assembled species that eventually form the peripheral arm.50 Although the amount of NDUFS3-specific signal was comparable in both shRNA-1 and pLKO.1 control cells (Figure 3A), 2D-BNGE immunoblotting clearly showed the presence of several aberrant sub-assembly intermediates containing NDUFS3 in the shRNA-1 cells but hardly any cI holocomplex. On the contrary, most of α-NDUFS3 was present in the cI holocomplex in pLKO.1 control cells (left panel in Figure 3C). Normal assembly of the cIV complex was demonstrated in both shRNA-1 and pLKO.1 control cells (Figure 3C).
Finally, we tested whether defective cI assembly and activity upon NDUFB11 knockdown could impair mitochondrial respiration. Endogenous, leak (or state 4), and uncoupled (maximal rate) respiration were sequentially determined with high-resolution respirometry (Oroboros). That is, by measuring oxygen consumption without any external additions; in the presence of oligomycin, an inhibitor of the mitochondrial ATP-synthase (cV); and finally in the presence of the OXPHOS-uncoupler FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone)..51,52 Compared to respiration-rate values in pLKO.1 control cells, respiration-rate values were reduced 4-fold in shRNA-1 NDUFB11-depleted cells (Figure 4). In HeLa cells expressing shRNA-2, oxygen consumption was also decreased, but not as profoundly as in shRNA-1 cells (Figure 4). This is in accordance with the drastically reduced amount of NDUFB11 in shRNA-1 compared to shRNA-2 knockdown HeLa cells (Figure 3A). Together, these data demonstrate that the NDUFB11 subunit is indispensable for assembly of the cI membrane arm, for maturation of the cI holocomplex, and for cI-dependent mitochondrial respiration.
Figure 4.
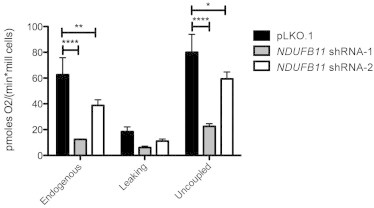
NDUFB11 Is Necessary for Respiratory Activity
The oxygen consumption rate was measured in NDUFB11-knockdown (shRNA-1 and -2) and control (pLKO.1) cells with the high-resolution oxygraph OROBOROS (Oroboros Instrument) as described previously.53 Cellular respiration was measured in basal conditions (endogenous) in the presence of oligomycin (leaking) and in the presence of FCCP (uncoupled). For calculation of the respiratory rates, the oxygen consumption per minute was normalized to the cell number. Statistical significance was determined by ANOVA and is indicated as follows: ∗p<0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001; n = 3. The plotted values correspond to the mean ± SD.
Our data clearly demonstrate the importance of NDUFB11 for cI biogenesis and mitochondrial respiration, suggesting that severe cI deficiency underlies the developmental defects in MLS-affected individuals with the NDUFB11 mutation. A combined effect of OXPHOS defects and enhanced cell death has been shown to underlie the brain and eye abnormalities observed in a hccs-deficient medaka fish model.18 In order to investigate whether NDUFB11-knockdown cells have a proliferative disadvantage in comparison to control cells, we monitored cell growth of the shRNA-transduced versus control HeLa cells and demonstrated that cells in which the knockdown of NDUFB11 was most severe (shRNA-1) showed significantly reduced growth (Figure S2). To determine whether the reduced growth of NDUFB11 knockdown cells correlates with increased apoptosis in these cells, we quantified an early marker of apoptosis (i.e., externalization of phosphatidylserine in the cell membrane) by using the specific apoptosis marker Annexin V (FITC conjugated to Annexin V). As compared to that of pLKO.1 cells, the proportion of shRNA-1 NDUFB11-depleted cells positive for Annexin V was significantly increased (Figure 5). In conclusion, a reduced amount of NDUFB11 is associated with slower cell growth and increased apoptosis, suggesting that cells expressing the NDUFB11 mutant allele are likely to be offset by those expressing the normal allele and eventually to be selected out.
Figure 5.
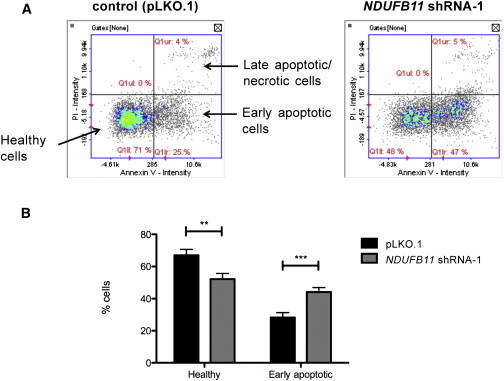
NDUFB11-Depleted HeLa Cells Show Enhanced Apoptosis
(A) The data were generated by the NucleoCounter NC-3000 system (ChemoMetec). This assay uses Hoechst 33342 to stain DNA and detect the total amount of cells. Staining cells with a conjugate of Annexin V and FITC allowed detection of cells in which phosphatidylserine is flipped inside out and exposed to the outer layer of the plasma membrane, a phenomenon occurring in the early execution phase of apoptosis. Cells were also stained with propidium iodide (PI) so that the non-viable (i.e., necrotic and late apoptotic) ones could be detected. The scatter plots show the intensity of Annexin-V-FITC versus PI fluorescence. Low Annexin-V-FITC/low PI intensity (lower left quadrant) corresponds to normal cells; high Annexin-V-FITC/low PI intensity (lower right quadrant) corresponds to cells in early apoptosis.
(B) Quantification plot of the percentage of healthy and early apoptotic cells in control (pLKO.1) and NDUFB11 knockdown (shRNA-1) cells. Statistical significance was determined by ANOVA and is indicated as follows: ∗∗p < 0.01; ∗∗∗p < 0.001; n = 3. The plotted values correspond to the mean ± SD.
Our results show that X-linked gene mutations leading to severe MRC impairment, including severe cI deficiency, in addition to already documented defects of cIII and cIV, underlie MLS syndrome. cI is the biggest MRC enzyme and a major entry point of respiration. cI strips two electrons from NADH and transfers them to ubiquinone by using the redox potential to pump four protons across the inner mitochondrial membrane and thus contributing to the maintenance of the electrochemical gradient.43 cI deficiency is the most common respiratory-chain defect in human mitochondrial disorders. To date, mutations in genes coding for all 14 core subunits, including seven mtDNA-encoded subunits, have been reported to cause isolated cI deficiency. In addition, mutations in 11 genes encoding supernumerary subunits, including mutations reported here in NDUFB11, have been found in individuals with cI deficiency, indicating that these genes play an essential role in cI assembly and/or stability.45,54 The phenotypic spectrum associated with cI deficiency ranges broadly, from fatal neonatal lactic acidosis and infantile-onset Leigh syndrome to childhood-onset mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. Single organ involvement has also been reported; examples include isolated hypertrophic cardiomyopathy, isolated skeletal myopathy, and Leber’s hereditary optic neuropathy (LHON).45 Remarkably, early-onset cardiomyopathy was present in subjects 1 and 2 as well as in the aborted female fetus from family 2. After autopsy, histiocytoid cardiomyopathy was diagnosed in subject 1. Hypertrophic and oncocytic (histiocytoid) cardiomyopathy has been reported in a few individuals with MLS syndrome.8,9,55 In particular, in one deceased girl with an HCCS nonsense mutation, histiocytoid cardiomyopathy was found after a post-mortem examination.2 Interestingly, dilated or hypertrophic cardiomyopathy was observed in a proportion of female mice with heart-specific inactivation of one Hccs copy.29 Our results, combined with the marked expression of Ndufb11 observed in the mouse heart (BioGPS database),56 suggest that NDUFB11 is an attractive candidate for involvement in isolated histiocytoid cardiomyopathy.57
HCCS, COX7B, and NDUFB11, which are involved in cellular respiration, are ubiquitously expressed (BioGPS database),56 but developmental defects in MLS-affected individuals are mainly restricted to the skin, eyes, and CNS (Table 1). Given that skin involvement is uncommon in canonical mitochondrial disorders, these findings imply the existence of a new pathogenetic mechanism affecting skin development in severe, X-linked forms of OXPHOS failure. Our data suggest that severe cI deficiency in specific cell types of females with a heterozygous NDUFB11 mutation might cause increased cell death and underlie the developmental defects in individuals with MLS syndrome. A proliferative advantage of cells with an active wild-type X chromosome might compensate for the loss of NDUFB11-deficient cells, as demonstrated in mice with heart-specific Hccs inactivation.29 In support of this idea, we showed that NDUFB11 knockdown also caused slower cell growth and increased apoptosis, although the knockdown experiments in cell culture cannot perfectly recapitulate functional mosaicism in female subjects as a result of random X-chromosome inactivation. The regenerative capacity and compensatory proliferation of healthy cells during embryonic development might lead to the most favorable outcome (i.e., no apparent sign of MLS syndrome). This possibility is supported by incomplete penetrance in oligo-symptomatic individuals as well as in apparently healthy female carriers,2 including the NDUFB11-mutation-positive mother of subject 2.
In summary, MLS syndrome is a developmental disorder caused by the complex interplay between X inactivation and OXPHOS defects of cIII, cIV, or, as shown here, cI. Because of the primary OXPHOS failure, it is possible that marked ATP depletion, abnormal reactive oxygen species (ROS) generation, and increased cell death are interrelated mechanisms contributing to the pathogenesis of MLS syndrome. Indeed, MRC impairment and increased production of ROS have been shown to trigger an increase in caspase-dependent apoptosis in the eyes and brains of hccs-deficient medaka embryos, suggesting that the developmental defects seen in individuals with MLS syndrome are caused by a combined effect of OXPHOS defects and enhanced cell death, possibly through the synergic contribution of decreased ATP and increased ROS production.18 Although more work is warranted to test these hypotheses mechanistically in in vivo models of NDUFB11 deficiency, our results in shRNA-1 HeLa cells do suggest that apoptosis plays a relevant pathogenetic role in NDUFB11-associated MLS syndrome.
Acknowledgments
We are grateful to the individuals with MLS syndrome and their families who contributed to this study. We thank Dr. Luitgard Graul-Neumann for caring for subject 2 and her parents, Mrs. Inka Jantke and Mrs. Inga Ziemer for skilful technical assistance, Prof. Eva Klopocki for array CGH analysis, Dr. Isabella Rau for XCI analysis, Dr. Nicole Sekarski for clinical referral, and Dr. Pu Yan for pathological analysis of subject 1. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (KU 1240/6-1 to K.K.) and an ERC-FP7 advanced grant (322424 to M.Z.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
BioGPS, http://biogps.org/
ExAC Browser, http://exac.broadinstitute.org/
GeneReviews, Morleo, M., and Franco, B. (2009). Microphthalmia with Linear Skin Defects Syndrome, http://www.ncbi.nlm.nih.gov/books/NBK7041
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Morleo M., Pramparo T., Perone L., Gregato G., Le Caignec C., Mueller R.F., Ogata T., Raas-Rothschild A., de Blois M.C., Wilson L.C. Microphthalmia with linear skin defects (MLS) syndrome: clinical, cytogenetic, and molecular characterization of 11 cases. Am. J. Med. Genet. A. 2005;137:190–198. doi: 10.1002/ajmg.a.30864. [DOI] [PubMed] [Google Scholar]
- 2.van Rahden V.A., Rau I., Fuchs S., Kosyna F.K., de Almeida H.L., Jr., Fryssira H., Isidor B., Jauch A., Joubert M., Lachmeijer A.M. Clinical spectrum of females with HCCS mutation: from no clinical signs to a neonatal lethal form of the microphthalmia with linear skin defects (MLS) syndrome. Orphanet J. Rare Dis. 2014;9:53. doi: 10.1186/1750-1172-9-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wimplinger I., Morleo M., Rosenberger G., Iaconis D., Orth U., Meinecke P., Lerer I., Ballabio A., Gal A., Franco B., Kutsche K. Mutations of the mitochondrial holocytochrome c-type synthase in X-linked dominant microphthalmia with linear skin defects syndrome. Am. J. Hum. Genet. 2006;79:878–889. doi: 10.1086/508474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cape C.J., Zaidman G.W., Beck A.D., Kaufman A.H. Phenotypic variation in ophthalmic manifestations of MIDAS syndrome (microphthalmia, dermal aplasia, and sclerocornea) Arch. Ophthalmol. 2004;122:1070–1074. doi: 10.1001/archopht.122.7.1070. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi M., Kiyosawa M., Toyoura T., Tokoro T. An XX male with microphthalmos and sclerocornea. J. Pediatr. Ophthalmol. Strabismus. 1998;35:122–124. doi: 10.3928/0191-3913-19980301-15. [DOI] [PubMed] [Google Scholar]
- 6.Kono T., Migita T., Koyama S., Seki I. Another observation of microphthalmia in an XX male: microphthalmia with linear skin defects syndrome without linear skin lesions. J. Hum. Genet. 1999;44:63–68. doi: 10.1007/s100380050110. [DOI] [PubMed] [Google Scholar]
- 7.Wimplinger I., Shaw G.M., Kutsche K. HCCS loss-of-function missense mutation in a female with bilateral microphthalmia and sclerocornea: a novel gene for severe ocular malformations? Mol. Vis. 2007;13:1475–1482. [PubMed] [Google Scholar]
- 8.Bird L.M., Krous H.F., Eichenfield L.F., Swalwell C.I., Jones M.C. Female infant with oncocytic cardiomyopathy and microphthalmia with linear skin defects (MLS): a clue to the pathogenesis of oncocytic cardiomyopathy? Am. J. Med. Genet. 1994;53:141–148. doi: 10.1002/ajmg.1320530205. [DOI] [PubMed] [Google Scholar]
- 9.Kutsche K., Werner W., Bartsch O., von der Wense A., Meinecke P., Gal A. Microphthalmia with linear skin defects syndrome (MLS): a male with a mosaic paracentric inversion of Xp. Cytogenet. Genome Res. 2002;99:297–302. doi: 10.1159/000071607. [DOI] [PubMed] [Google Scholar]
- 10.Zvulunov A., Kachko L., Manor E., Shinwell E., Carmi R. Reticulolinear aplasia cutis congenita of the face and neck: a distinctive cutaneous manifestation in several syndromes linked to Xp22. Br. J. Dermatol. 1998;138:1046–1052. doi: 10.1046/j.1365-2133.1998.02277.x. [DOI] [PubMed] [Google Scholar]
- 11.Indrieri A., van Rahden V.A., Tiranti V., Morleo M., Iaconis D., Tammaro R., D’Amato I., Conte I., Maystadt I., Demuth S. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am. J. Hum. Genet. 2012;91:942–949. doi: 10.1016/j.ajhg.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaefer L., Ballabio A., Zoghbi H.Y. Cloning and characterization of a putative human holocytochrome c-type synthetase gene (HCCS) isolated from the critical region for microphthalmia with linear skin defects (MLS) Genomics. 1996;34:166–172. doi: 10.1006/geno.1996.0261. [DOI] [PubMed] [Google Scholar]
- 13.Schwarz Q.P., Cox T.C. Complementation of a yeast CYC3 deficiency identifies an X-linked mammalian activator of apocytochrome c. Genomics. 2002;79:51–57. doi: 10.1006/geno.2001.6677. [DOI] [PubMed] [Google Scholar]
- 14.Alberry M.S., Juvanic G., Crolla J., Soothill P., Newbury-Ecob R. Pseudotail as a feature of microphthalmia with linear skin defects syndrome. Clin. Dysmorphol. 2011;20:111–113. doi: 10.1097/MCD.0b013e328342eb66. [DOI] [PubMed] [Google Scholar]
- 15.Vergult S., Leroy B., Claerhout I., Menten B. Familial cases of a submicroscopic Xp22.2 deletion: genotype-phenotype correlation in microphthalmia with linear skin defects syndrome. Mol. Vis. 2013;19:311–318. [PMC free article] [PubMed] [Google Scholar]
- 16.Fornuskova D., Stiburek L., Wenchich L., Vinsova K., Hansikova H., Zeman J. Novel insights into the assembly and function of human nuclear-encoded cytochrome c oxidase subunits 4, 5a, 6a, 7a and 7b. Biochem. J. 2010;428:363–374. doi: 10.1042/BJ20091714. [DOI] [PubMed] [Google Scholar]
- 17.Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H., Shinzawa-Itoh K., Nakashima R., Yaono R., Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 18.Indrieri A., Conte I., Chesi G., Romano A., Quartararo J., Tatè R., Ghezzi D., Zeviani M., Goffrini P., Ferrero I. The impairment of HCCS leads to MLS syndrome by activating a non-canonical cell death pathway in the brain and eyes. EMBO Mol. Med. 2013;5:280–293. doi: 10.1002/emmm.201201739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schapira A.H. Mitochondrial diseases. Lancet. 2012;379:1825–1834. doi: 10.1016/S0140-6736(11)61305-6. [DOI] [PubMed] [Google Scholar]
- 20.Zeviani M., Di Donato S. Mitochondrial disorders. Brain. 2004;127:2153–2172. doi: 10.1093/brain/awh259. [DOI] [PubMed] [Google Scholar]
- 21.Allanson J., Richter S. Linear skin defects and congenital microphthalmia: a new syndrome at Xp22.2. J. Med. Genet. 1991;28:143–144. doi: 10.1136/jmg.28.2.143-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindsay E.A., Grillo A., Ferrero G.B., Roth E.J., Magenis E., Grompe M., Hultén M., Gould C., Baldini A., Zoghbi H.Y. Microphthalmia with linear skin defects (MLS) syndrome: clinical, cytogenetic, and molecular characterization. Am. J. Med. Genet. 1994;49:229–234. doi: 10.1002/ajmg.1320490214. [DOI] [PubMed] [Google Scholar]
- 23.Mücke J., Happle R., Theile H. MIDAS syndrome respectively MLS syndrome: a separate entity rather than a particular lyonization pattern of the gene causing Goltz syndrome. Am. J. Med. Genet. 1995;57:117–118. doi: 10.1002/ajmg.1320570123. [DOI] [PubMed] [Google Scholar]
- 24.Wimplinger I., Rauch A., Orth U., Schwarzer U., Trautmann U., Kutsche K. Mother and daughter with a terminal Xp deletion: implication of chromosomal mosaicism and X-inactivation in the high clinical variability of the microphthalmia with linear skin defects (MLS) syndrome. Eur. J. Med. Genet. 2007;50:421–431. doi: 10.1016/j.ejmg.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 25.Sharma V.M., Ruiz de Luzuriaga A.M., Waggoner D., Greenwald M., Stein S.L. Microphthalmia with linear skin defects: a case report and review. Pediatr. Dermatol. 2008;25:548–552. doi: 10.1111/j.1525-1470.2008.00724.x. [DOI] [PubMed] [Google Scholar]
- 26.Zumwalt J., Moorhead C., Golkar L. Fourteen-month-old girl with facial skin thinning. Pediatr. Dermatol. 2012;29:217–218. doi: 10.1111/j.1525-1470.2011.01632.x. [DOI] [PubMed] [Google Scholar]
- 27.Van den Veyver I.B. Skewed X inactivation in X-linked disorders. Semin. Reprod. Med. 2001;19:183–191. doi: 10.1055/s-2001-15398. [DOI] [PubMed] [Google Scholar]
- 28.Morleo M., Franco B. Dosage compensation of the mammalian X chromosome influences the phenotypic variability of X-linked dominant male-lethal disorders. J. Med. Genet. 2008;45:401–408. doi: 10.1136/jmg.2008.058305. [DOI] [PubMed] [Google Scholar]
- 29.Drenckhahn J.D., Schwarz Q.P., Gray S., Laskowski A., Kiriazis H., Ming Z., Harvey R.P., Du X.J., Thorburn D.R., Cox T.C. Compensatory growth of healthy cardiac cells in the presence of diseased cells restores tissue homeostasis during heart development. Dev. Cell. 2008;15:521–533. doi: 10.1016/j.devcel.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 30.Chen X., Shen Y., Zhang F., Chiang C., Pillalamarri V., Blumenthal I., Talkowski M., Wu B.L., Gusella J.F. Molecular analysis of a deletion hotspot in the NRXN1 region reveals the involvement of short inverted repeats in deletion CNVs. Am. J. Hum. Genet. 2013;92:375–386. doi: 10.1016/j.ajhg.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dabell M.P., Rosenfeld J.A., Bader P., Escobar L.F., El-Khechen D., Vallee S.E., Dinulos M.B., Curry C., Fisher J., Tervo R. Investigation of NRXN1 deletions: clinical and molecular characterization. Am. J. Med. Genet. A. 2013;161A:717–731. doi: 10.1002/ajmg.a.35780. [DOI] [PubMed] [Google Scholar]
- 32.Kim H.G., Kishikawa S., Higgins A.W., Seong I.S., Donovan D.J., Shen Y., Lally E., Weiss L.A., Najm J., Kutsche K. Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen R.C., Zoghbi H.Y., Moseley A.B., Rosenblatt H.M., Belmont J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 34.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cingolani P., Platts A., Wang L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdollahpour H., Alawi M., Kortüm F., Beckstette M., Seemanova E., Komárek V., Rosenberger G., Kutsche K. An AP4B1 frameshift mutation in siblings with intellectual disability and spastic tetraplegia further delineates the AP-4 deficiency syndrome. Eur. J. Hum. Genet. 2015;23:256–259. doi: 10.1038/ejhg.2014.73. Published online April 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petruzzella V., Tessa A., Torraco A., Fattori F., Dotti M.T., Bruno C., Cardaioli E., Papa S., Federico A., Santorelli F.M. The NDUFB11 gene is not a modifier in Leber hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 2007;355:181–187. doi: 10.1016/j.bbrc.2007.01.140. [DOI] [PubMed] [Google Scholar]
- 40.Carroll J., Shannon R.J., Fearnley I.M., Walker J.E., Hirst J. Definition of the nuclear encoded protein composition of bovine heart mitochondrial complex I. Identification of two new subunits. J. Biol. Chem. 2002;277:50311–50317. doi: 10.1074/jbc.M209166200. [DOI] [PubMed] [Google Scholar]
- 41.Carroll J., Fearnley I.M., Shannon R.J., Hirst J., Walker J.E. Analysis of the subunit composition of complex I from bovine heart mitochondria. Mol. Cell. Proteomics. 2003;2:117–126. doi: 10.1074/mcp.M300014-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Vinothkumar K.R., Zhu J., Hirst J. Architecture of mammalian respiratory complex I. Nature. 2014;515:80–84. doi: 10.1038/nature13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirst J. Mitochondrial complex I. Annu. Rev. Biochem. 2013;82:551–575. doi: 10.1146/annurev-biochem-070511-103700. [DOI] [PubMed] [Google Scholar]
- 44.Angerer H., Zwicker K., Wumaier Z., Sokolova L., Heide H., Steger M., Kaiser S., Nübel E., Brutschy B., Radermacher M. A scaffold of accessory subunits links the peripheral arm and the distal proton-pumping module of mitochondrial complex I. Biochem. J. 2011;437:279–288. doi: 10.1042/BJ20110359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fassone E., Rahman S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012;49:578–590. doi: 10.1136/jmedgenet-2012-101159. [DOI] [PubMed] [Google Scholar]
- 46.Bernard D.G., Gabilly S.T., Dujardin G., Merchant S., Hamel P.P. Overlapping specificities of the mitochondrial cytochrome c and c1 heme lyases. J. Biol. Chem. 2003;278:49732–49742. doi: 10.1074/jbc.M308881200. [DOI] [PubMed] [Google Scholar]
- 47.Zerbetto E., Vergani L., Dabbeni-Sala F. Quantification of muscle mitochondrial oxidative phosphorylation enzymes via histochemical staining of blue native polyacrylamide gels. Electrophoresis. 1997;18:2059–2064. doi: 10.1002/elps.1150181131. [DOI] [PubMed] [Google Scholar]
- 48.Nijtmans L.G., Henderson N.S., Holt I.J. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods. 2002;26:327–334. doi: 10.1016/S1046-2023(02)00038-5. [DOI] [PubMed] [Google Scholar]
- 49.Pitceathly R.D., Rahman S., Wedatilake Y., Polke J.M., Cirak S., Foley A.R., Sailer A., Hurles M.E., Stalker J., Hargreaves I., UK10K Consortium NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013;3:1795–1805. doi: 10.1016/j.celrep.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vogel R.O., Dieteren C.E., van den Heuvel L.P., Willems P.H., Smeitink J.A., Koopman W.J., Nijtmans L.G. Identification of mitochondrial complex I assembly intermediates by tracing tagged NDUFS3 demonstrates the entry point of mitochondrial subunits. J. Biol. Chem. 2007;282:7582–7590. doi: 10.1074/jbc.M609410200. [DOI] [PubMed] [Google Scholar]
- 51.Brand M.D., Nicholls D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Invernizzi F., D’Amato I., Jensen P.B., Ravaglia S., Zeviani M., Tiranti V. Microscale oxygraphy reveals OXPHOS impairment in MRC mutant cells. Mitochondrion. 2012;12:328–335. doi: 10.1016/j.mito.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gómez-Durán A., Pacheu-Grau D., López-Gallardo E., Díez-Sánchez C., Montoya J., López-Pérez M.J., Ruiz-Pesini E. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum. Mol. Genet. 2010;19:3343–3353. doi: 10.1093/hmg/ddq246. [DOI] [PubMed] [Google Scholar]
- 54.Mimaki M., Wang X., McKenzie M., Thorburn D.R., Ryan M.T. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta. 2012;1817:851–862. doi: 10.1016/j.bbabio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 55.Happle R., Daniëls O., Koopman R.J. MIDAS syndrome (microphthalmia, dermal aplasia, and sclerocornea): an X-linked phenotype distinct from Goltz syndrome. Am. J. Med. Genet. 1993;47:710–713. doi: 10.1002/ajmg.1320470525. [DOI] [PubMed] [Google Scholar]
- 56.Gurok U., Bork K., Nuber U., Spörle R., Nöhring S., Horstkorte R. Expression of Ndufb11 encoding the neuronal protein 15.6 during neurite outgrowth and development. Gene Expr. Patterns. 2007;7:370–374. doi: 10.1016/j.modgep.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 57.Finsterer J. Histiocytoid cardiomyopathy: a mitochondrial disorder. Clin. Cardiol. 2008;31:225–227. doi: 10.1002/clc.20224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.