

Abstract
Robinow syndrome (RS) is a phenotypically and genetically heterogeneous condition that can be caused by mutations in genes encoding components of the non-canonical Wnt signaling pathway. In contrast, germline mutations that act to increase canonical Wnt signaling lead to distinctive osteosclerotic phenotypes. Here, we identified de novo frameshift mutations in DVL1, a mediator of both canonical and non-canonical Wnt signaling, as the cause of RS-OS, an RS subtype involving osteosclerosis, in three unrelated individuals. The mutations all delete the DVL1 C terminus and replace it, in each instance, with a novel, highly basic sequence. We showed the presence of mutant transcript in fibroblasts from one individual with RS-OS and demonstrated unimpaired protein stability with transfected GFP-tagged constructs bearing a frameshift mutation. In vitro TOPFlash assays, in apparent contradiction to the osteosclerotic phenotype, revealed that the mutant allele was less active than the wild-type allele in the canonical Wnt signaling pathway. However, when the mutant and wild-type alleles were co-expressed, canonical Wnt activity was 2-fold higher than that in the wild-type construct alone. This work establishes that DVL1 mutations cause a specific RS subtype, RS-OS, and that the osteosclerosis associated with this subtype might be the result of an interaction between the wild-type and mutant alleles and thus lead to elevated canonical Wnt signaling.
Main Text
Robinow syndrome (RS [dominant RS, MIM 180700; recessive RS, MIM 268310]) is a genetically and phenotypically heterogeneous skeletal dysplasia characterized by the distinctive facial appearance of midface hypoplasia, hypertelorism, a short nose, and a broad mouth, known collectively as “fetal facies.” Additional but variable features of RS include mesomelic dwarfism, macrocephaly, gingival hypertrophy, dental malocclusion, genital hypoplasia, brachydactyly, bifid thumbs, and segmentation defects (reviewed in Mazzeu et al.1).
Some forms of RS are caused by mutations in genes encoding components of Wnt signaling pathways.2–4 Wnt signaling is a complex pleiotropic network for which more than 19 Wnt ligands and more than 15 Wnt receptors and co-receptors have been described.5 Signaling via these complexes is broadly divided into canonical and non-canonical pathways. Activation of the canonical pathway by Wnt ligands prevents the degradation of β-catenin, which accumulates in the cytoplasm and moves to the nucleus to alter gene expression.5 Germline mutations in the canonical pathway in humans cause a number of defects, the most notable of which is an impact upon bone mineral density (BMD). A high-bone-mass phenotype (MIM 144750) is caused by gain-of-function mutations in LRP5 (MIM 603506),6,7 whereas a reduction in expression or activity of the extracellular soluble Wnt antagonist SOST (MIM 605740) leads to van Buchem disease (MIM 239100)8 and sclerosteosis type 1 (MIM 269500).9 That an increase in canonical Wnt signaling will cause an increase in bone mass, and the converse, is well established through a number of other human10–12 and animal13,14 studies (reviewed in Wang et al.15).
Non-canonical Wnt signaling is more mechanistically heterogeneous in that it involves a number of interrelated signaling cascades. Germline mutations in genes encoding components of one particular non-canonical Wnt signaling pathway, termed the planar cell polarity (PCP)-Wnt pathway, cause some forms of RS.2–4
RS has both dominantly and recessively inherited forms, which differ both by their phenotype and by their mode of inheritance.1 Recessive RS, characterized by severe mesomelia and segmentation defects such as hemivertebrae or rib fusions, is caused by biallelic loss-of-function mutations in the gene encoding the transmembrane Wnt co-receptor ROR2 (receptor tyrosine kinase-like orphan receptor 2).2,3 In a small minority of individuals with dominant RS, characterized by a milder phenotype without rib fusions, the condition is caused by loss-of-function mutations in the gene encoding the classical PCP-Wnt ligand Wnt-5a.4 No other mutations have previously been associated with human RS.
It is well known that there is substantial cross-talk between the canonical and non-canonical pathways (reviewed in Niehrs5). Despite this, there is no indication that the Mendelian conditions discussed above have impacts across more than one Wnt pathway. However, a number of Wnt mediators are shared components of both canonical and non-canonical signaling—the dishevelled family of proteins (DVL) is the primary example.16
Three unrelated individuals with the clinical diagnosis of sporadic RS with atypical dramatic osteosclerosis (RS-OS) were ascertained and described previously (Figures 1A–1G).17,18 These individuals exhibited a clinical presentation of dominant RS with characteristic facial dysmorphism (marked hypertelorism, short nose, broad mouth, and midface hypoplasia), camptodactyly and brachydactyly (in subjects 1 and 2), cleft palate (in subjects 2 and 3), and dental anomalies. Mesomelia, another common finding in RS, ranged from mild to absent in these individuals, but this observation does not preclude a clinical diagnosis of RS.19 Radiographs revealed osteosclerosis of the cranium, which was particularly prominent at the skull base (Figure 1E). In subjects 1 and 2, generalized axial and appendicular osteosclerosis was also documented and was particularly marked in the cortices of the long bones, which were also undertubulated (Figures 1F and 1G). No information on the remainder of the skeleton was available for individual 3. In subjects 1 and 2, the distal phalanges of the hands and feet were hypoplastic, and the terminal phalanx of the thumb was bifid, a pattern typical of RS.20 Dual-energy X-ray absorptiometry scanning of the skeleton revealed markedly elevated BMD (lumbar spine Z scores were +7.4 for subject 1 and +7.6 for subject 2). The parents of all three individuals were clinically unremarkable. Table 1 summarizes these clinical features.
Figure 1.
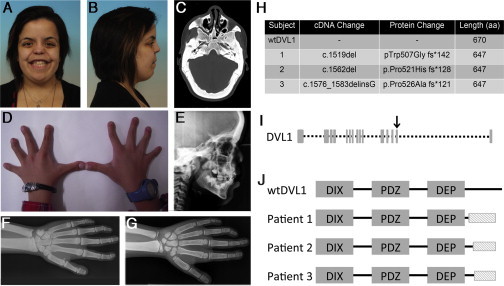
Clinical Presentation of and Mutations in RS-OS
(A–G) Clinical features of RS-OS.
(A and B) Facial appearance of subject 1. Note the midface hypoplasia, flat facial profile, hypertelorism, and broad mouth.
(C) Transverse CT of subject 1. Note the pronounced osteosclerosis of the cranial vault.
(D) Appearance of the hands of subject 2. Note the the camptodactyly and brachydactyly.
(E) Lateral X-ray of the skull of subject 2. Note the thickened calvarium and increased bone density.
(F) X-ray of the left hand, wrist, and forearm of subject 1. Note the osteosclerosis in the cortices of the long bones of the forearm and the camptodactyly, clinodactyly, and bifid thumb. (This image was reproduced with permission from Bunn et al.17)
(G) X-ray of the left hand, wrist, and forearm of subject 1. Note the cortical osteosclerosis of the forearm and the camptodactyly, clinodactyly, and bifid thumb (less obvious than in subject 1).
(H) DVL1 mutations leading to RS-OS.
(I) DVL1 structure showing exons. The arrow indicates exon 14, the location of the three mutations.
(J) An illustration of DVL1 shows the similarity among the altered proteins and their difference from the wild-type. The shaded box indicates the novel shared C-terminal sequence.
Table 1.
Clinical and Radiographic Features of the Three Subjects with RS-OS
| Subject 1 | Subject 2 | Subject 3 | |
|---|---|---|---|
| Hypertelorism | + | + | + |
| Mesomelia | − | − | − |
| Cleft palate | − | + | + |
| Camptodactyly and brachydactyly | + | + | − |
| Gingival hyperplasia | + | + | − |
| Oligodontia | + | + | + |
| Bilateral mixed hearing loss | + | + | + |
| Osteosclerosis of skull | + | + | + |
| Osteosclerosis of long bones | + | + | NA |
| Bifid thumb and great toe | + | + | NA |
Abbreviations are as follows: +, present; −, absent; NA, information not available.
Elevated BMD occurring in the context of sporadic RS in three unrelated individuals of different ethnic backgrounds and without relevant family history, consanguinity, or sibling recurrence led us to hypothesize that this phenotype was underpinned by rare de novo mutations at a previously unidentified locus. All subjects participated after informed consent was obtained in keeping with the principles of the Declaration of Helsinki. Ethical approval for this work was obtained from the Southern Health and Disability Ethics Committee of New Zealand (reference no. 13/STH/56). Accordingly, we performed whole-exome sequencing and employed a parent-proband-trio design to identify de novo variants in subject 1. DNA was extracted from blood leukocytes and captured with an Agilent SureSelect All Exon V4 Kit and sequenced on an Illumina HiSeq2000 with paired-end 100-bp reads. Data were processed for alignment, and variants were called according to the current best-practice guidelines from the Broad Institute.21 Reads were aligned to the human genome (GRCh37) with the Burrows-Wheeler Aligner (BWA)-MEM,22 base quality scores were recalibrated with the Genome Analysis Toolkit (GATK), and variants were called with the GATK HaplotypeCaller. Each exome had an average read depth of >30×. Variants were annotated with SnpEff,23 and SnpSift was used to identify de novo mutations in subject 1.24 Novelty was determined by the exclusion of variants identified in 400 in-house exomes and of variants found in the NHLBI Exome Sequencing Project Exome Variant Server (ESP6500). Variants were filtered for quality and for a read depth of >5× across the trio. Annotation with SnpEff was used to filter for effects likely to disrupt protein sequences. All apparently de novo single-base substitutions and small insertions or deletions were validated by Sanger sequencing of the parents and proband.
Two coding variants, a frameshift (c.1519del [p.Trp507Glyfs∗142]; RefSeq accession number NM_004421.2) in DVL1 (MIM 601365) and a single-base substitution (c.2753C>T [p.Pro918Leu]; RefSeq NM_014494.2) in TNRC6A (MIM 610739), satisfied these criteria as the only functional de novo mutations found in this individual. TNRC6A encodes a component of a cytoplasmic ribonucleoprotein complex that regulates mRNA silencing, stability, and translation. PolyPhen-2,25 which uses physical properties and evolutionary conservation to predict deleterious effects of coding variants, predicted that the p.Pro918Leu substitution is likely to be benign. No novel sequence variants in TNRC6A were observed in subject 2.
DVL1 is a central mediator of Wnt signal transduction. The c.1519del frameshift falls within the penultimate exon of the 15-exon DVL1. This mutation is predicted to remove almost the entirety of the C-terminal tail of DVL1 and replace this sequence with a 142-amino-acid-long, novel, highly basic sequence (the predicted pI of the C-terminal section rises from 9.5 to 12.526). Direct sequencing of the relevant exons and intron-exon boundaries of DVL1 in individuals 2 and 3 identified two similar de novo frameshift mutations in DVL1: c.1562del (p.Pro521Hisfs∗128) and c.1576_1583delinsG (p.Pro526Alafs∗121), respectively. Each DVL1 mutation leads to a frameshift in the same reading frame. All three DVL1 mutations are predicted to lead to a protein product 23 residues shorter than wild-type DVL1 (Figures 1H–1J). The same novel C-terminal sequence, which ranges from 121 to 142 residues in length and differs only in the extent of its N terminus, is predicted to be appended to the DVL1 proteins produced from these mutant alleles (Figures 1I and 1J).
DVL proteins are widely conserved Wnt signaling mediators throughout evolution.27 Mammals have three DVL1 paralogs (DVL1–DVL3), all of which share a high level of sequence homology. They encode intracellular scaffolding proteins acting directly downstream from the transmembrane Wnt receptors (frizzled family). At DVL, the Wnt pathways functionally diverge into the canonical and non-canonical cascades; which way they diverge is dependent upon which downstream mediators DVL interacts with (reviewed in Gao and Chen16). The three DVL paralogs have separate but overlapping roles, which are elegantly shown in a series of mouse knockouts.28 Of the three Dvl knockouts, loss of Dvl1 has the least phenotypic impact; null mice display no dysmorphic phenotype.29 In contrast, Dvl2-null mice have cardiac, segmentation, and neural-tube defects.30 Dvl3-null mice show pathologies similar to those of Dvl2-null mice, and additional cochlear anomalies, and rarely survive until adulthood.31 DVL proteins have three well-defined functional domains: the N-terminal DIX (dishevelled and axin), the PDZ (postsynaptic density 95, disc large, and zonula occludens 1), and the C-terminal DEP (dishevelled, Egl-10, and pleckstrin) domains (Figure 1J). To date, no mutations at any of the DVL loci have been associated with clinically apparent defects in human Wnt signaling.
The three clustered mutations in the individuals described in this study fall outside the three established domains of DVL and thus introduce C-terminal frameshifts after the DEP domain. However, a number of conserved C-terminal regions, including sites that are subject to phosphorylation,32 binding sites for ubiquitin ligases,33–35 and enzymes that mediate DVL degradation,36 are predicted to be removed by these mutations. Additionally, the mutations delete a site necessary for the function of a deubiquitination enzyme,37 the binding site for IQGAP1 (which controls DVL nuclear localization38), and a frizzled interacting domain.39Considering the lack of malformations in Dvl1−/− mice, the lack of phenotypic effects similar to RS-OS in individuals with deletions encompassing DVL1,40 and our observation that all three frameshift mutations replace functional domains with the same, highly basic novel C terminus, these DVL1 alleles seem to confer the RS-OS phenotype by a specific mechanism distinct from simple haploinsufficiency. Thus, the focus of this work was to understand the mechanistic basis of the osteosclerotic aspect of the RS-OS phenotype, given that osteosclerosis is the main phenotypic feature that differentiates these individuals from other subjects with RS.
The three DVL1 mutations all occur within a confined region of DVL1 and could be acting through either a loss or gain of function. All three fall in the penultimate exon, where the novel reading frame does not encounter a premature stop codon before entering the last exon (Figures 1I and 1J). This observation presents the possibility that these mutation-bearing transcripts could escape nonsense-mediated decay (NMD). To investigate, we prepared cDNA from dermal fibroblasts obtained from subject 1 and performed RT-PCR to amplify a product that spanned parts of exons 14 and 15, including the site of the mutation. After this product was sequenced, both mutant and wild-type alleles were observed on the Sanger chromatogram, suggesting that the mutant allele was not subject to NMD (data not shown). To confirm this, we digested these same RT-PCR products with BstN1, which specifically cuts at a site that is destroyed by the mutation. A substantial proportion of the product derived from the subject-1 cDNA was refractory to digestion, whereas a control was digested to completion (Figure 2A).
Figure 2.
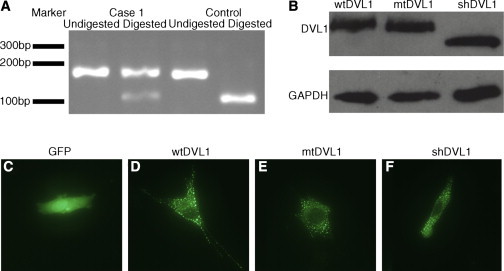
Expression and Localization of Frameshifted and Truncated DVL1
(A) The DVL1 PCR product was digested with the restriction enzyme BstN1 for 4 hr at 60°C. The digest shows the presence of mutant transcript (which is refractory to digestion) alongside wild-type transcript in fibroblasts obtained from subject 1.
(B) Chemiluminescent immunoblot of C2C12 cells (40,000/well) transiently transfected (0.6 μl/well of Lipofectamine2000, Life Technologies) with EGFP-tagged DVL1 constructs (100 ng/well,incubated for 24 hr) demonstrates comparable protein levels (anti-GFP, A6455, Life Technologies; anti-GAPDH, G8795, Sigma). Note the size difference between the truncated DVL and the other constructs.
(C–F) Representative images from fluorescent microscopy of C2C12 cells transiently transfected with EGFP or the EGFP-DVL1 constructs (100 ng/well) show that p.Trp507∗ DVL1 (shDVL1) and mtDVL1 retain the ability to form puncta. shDVL1 is a construct that leads to a truncated protein terminating at the site of the frameshift mutation (c.1519insTAA [p.Trp507∗]) found in subject 1.
To ascertain whether the frameshift-containing protein produced from the c.1519del allele found in individual 1 demonstrated significant instability, we performed an immunoblot on skin fibroblast lysate, but this failed to detect DVL1 in either subject or control cultures. To circumvent this inability to detect endogenous DVL1, we cloned an N-terminal GFP-tagged full-length DVL1 construct (GFP-wtDVL1) and introduced the c.1519del mutation by site-directed mutagenesis (GFP-mtDVL1). This clone was transfected into C2C12 cells, and immunodetection was performed with anti-GFP antibodies with band intensities normalized to endogenous GAPDH. These results indicated no apparent difference in abundance between the altered and wild-type DVL1 (Figure 2B). This was confirmed by infrared immunofluorescence, normalized to α-tubulin (n = 3; Figure S1). These data indicate that mutation-bearing alleles that lead to RS-OS are transcribed and translated into stable proteins and consequently hold the potential to exert novel biological functions or interfere with endogenous DVL1-mediated activities.
Previous work has found that loss of the DVL1 C terminus reduces the protein’s ability to signal in the canonical Wnt pathway and is mediated by either a loss of interaction with the frizzled receptor39 or a deubiquitinating enzyme.37 The osteosclerosis observed in the three RS-OS-affected individuals studied here is phenotypically reminiscent of a LRP6-related high-bone-mass phenotype, van Buchem disease, and sclerosteosis, which are all caused by increases in canonical Wnt signaling, albeit via differing mechanisms.6–9,41 We reasoned that, although previous genes linked to RS signal through the PCP-Wnt pathway, DVL1 mediates both canonical and PCP-Wnt signaling, leaving open the possibility that the osteosclerotic aspects of the RS-OS phenotype could be caused by a defect in the canonical pathway. We therefore investigated the impact of one of the DVL1 mutations on signaling through the canonical Wnt pathway.
We employed a transient transfection system utilizing the TOPFlash reporter42 to investigate the impact of the DVLI mutation on canonical Wnt signaling. This transcriptional reporter is composed of three β-catenin-responsive TCF promoter elements driving firefly luciferase gene transcription.42 C2C12 cells were transfected with plasmids containing different forms of DVL1: wild-type DVL1 (wtDVL1), a construct containing the variant found in subject 1 (c.1519del [p.Trp507Glyfs∗142]; mtDVL1), and a short form encoding a protein truncated at the site affected by the frameshift mutation in individual 1 (c.1519insTAA [p.Trp507∗]; shDVL1). We used the last construct to investigate any detectable differences between the activity of DVL1 proteins without the wild-type C terminus and the activity of the proteins that had C termini but were encoded by the frameshifted sequence that was 3′ to each of the three mutations.
Typically, wild-type DVL1 oligomerizes to form focal puncta in cells.43 The ability to form supramolecular complexes (seen as puncta) is a necessary property for canonical Wnt signaling.43,44 We therefore questioned whether the truncated or frameshifted DVL1 proteins demonstrated altered subcellular distribution in comparison to that of wild-type proteins. C2C12 cells were transfected with constructs specifying N-terminal, GFP-tagged versions of each DVL1 variant. Fluorescent microscopy of transfected cells yielded a punctate pattern of expression and no evident differences among frameshifted, truncated, and wild-type proteins (Figures 2D–2F).
Increasing amounts of untagged constructs were transfected into C2C12 cells, and after an overnight incubation, their impact on canonical signaling was measured through TOPFlash activation (Figure 3A). Transfection of wtDVL1 led canonical Wnt activity to increase up to 29-fold (at 32 ng/well) higher than that of an empty vector control. Consistent with the previously published data, the shDVL1 construct was less potent in increasing signaling activity: at high levels of expression, the DVL1 ΔC terminus was less active in the canonical pathway than full-length wtDVL1.37,39 The mtDVL1 construct only raised signaling activity to a maximum level of 3-fold higher than that of empty vector controls. A log-transformed two-way ANOVA found that there was a significant difference between the mtDVL1 construct and the other two constructs (p < 0.001). The most remarkable and unanticipated finding from these data was the substantially reduced activity of the mtDVL1 construct. These results are in apparent conflict with the hypothesis that the osteosclerotic phenotype of individuals with RS-OS is attributable to over-activity in the canonical Wnt pathway.
Figure 3.
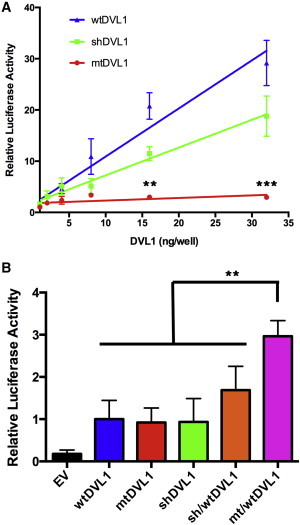
Impact of DVL1 Constructs on Canonical Wnt Signaling
(A) TOPFlash reporter assay in C2C12 cells. Cells were transiently transfected with 80 ng/well of TOPFlash reporter, 20 ng/well of a constitutively active β-galactosidase construct, and a variable amount of a DVL1 construct and incubated for 18 hr. Combined luciferase activities of three independent experiments normalized to the β-galactosidase activity are depicted and reported as a relative increase over that of an empty vector (n = 3). Error bars represent the SEM. Log-transformed two-way ANOVA found a significant difference between the mutant and every other construct (p < 0.001). Individual p values were calculated with Tukey HSD tests and denote the difference between mtDVL1 and every other construct.
(B) C2C12 cells were transiently transfected with a fixed amount of each DVL1 construct (4 ng/well) or with a 1:1 stoichiometric ratio of two constructs with the same total amount of DVL1 and incubated for 18 hr. The same reporters and processing were used as above, and the luciferase activity is expressed as a proportion of the wtDVL1 activation (n = 5). Error bars represent the SEM. P values were calculated with Tukey HSD tests (∗p 0.05, ∗∗p = 0.01, ∗∗∗p = 0.001).
To further investigate this issue, we focused on the stimulatory capabilities of these constructs at lower levels of transfection because the concentrations of transfected plasmid that yielded maximal stimulation in the foregoing experiments are unlikely to reflect physiological concentrations of DVL1 either in vivo or in C2C12 cells, which are known to express this gene.45 Given that the individuals studied here are heterozygous for the disease-causing mutation and therefore express a wtDVL1 allele in trans with the mutant, we hypothesized that a stoichiometric balance between these two alleles would more closely reflect the in vivo context. We co-transfected mtDVL1 and wtDVL1 in a 1:1 ratio while maintaining the same total amount of DVL1 as each construct alone (4 ng/well). A similar 1:1 ratio of shDVL1:wtDVL1 was also examined under the same conditions. After a 24-hr incubation, the mtDVL1:wtDVL1 co-transfection elevated canonical activity to 2-fold higher than that of wtDVL1 alone. A significant difference was found between the mtDVL1:wtDVL1 combination and all other constructs and construct combinations (p < 0.01, Tukey honest significant difference (HSD) tests; Figure 3B). This same pattern was also found in the presence of a Wnt stimulus in the form of Wnt-3a-conditioned media (Figure S2). These experiments suggest that the protein produced by this frameshift mutation, which results in RS-OS, exerts a stimulatory effect on canonical Wnt signaling when it is co-expressed with the wild-type protein. This stimulatory activity was not evident, even at supraphysiological levels of expression, when mtDVL1 was transfected alone. Furthermore, the statistical difference between co-transfections of shDVL1:wtDVL1 and mtDVL1:wtDVL1 also suggests that the stimulatory effect of the mtDVL1 allele is not solely attributable to the truncation of the C terminus of DVL1.
The components in common between the RS-OS phenotype and other forms of RS are also likely to relate to perturbations in Wnt signaling. Mutations in effectors of PCP-Wnt signaling, such as ROR2 and WNT5A, lead to RS phenotypes2–4 but do not lead to florid alterations in BMD, such as those described here. DVL1 is common to all described Wnt signaling receptor complexes, and so it is plausible that a deficiency in its action within the PCP-Wnt pathway could lead to the RS components of the RS-OS phenotype. A possible mediator of this effect might be the DVL-interacting protein PRICKLE1, which has been shown to bind the C-terminal half of DVL.46 Loss of Prickle1 disrupts the PCP-Wnt-induced DVL gradient in mice and Drosophila,46,47 and mice with hypomorphic Prickle1 alleles appear phenotypically similar to mice with RS.47
The mechanism by which canonical Wnt signaling is upregulated in a manner that is dependent on the presence of a wild-type allele is unresolved. A truncation of even just the final 16 C-terminal residues of DVL1 reduces canonical Wnt signaling as a result of a weaker affinity for the Wnt receptor frizzled.39 The C terminus also contains binding sites for a deubiquitination enzyme required to activate DVL, and specific mutagenesis of residues mediating this interaction also reduces canonical Wnt activity.37 Additionally, a DVL construct that lacked the native C terminus failed to interact with IQGAP1, a protein that facilitates canonical Wnt signaling by controlling DVL nuclear localization.38 These proposed mechanisms perhaps explain the observed reductions of in vitro activity at high levels of expression (Figure 3A) but conflict with the RS-OS osteosclerotic phenotype, which suggests that canonical signaling is elevated.
A partial explanation might relate to the effect of the loss of the wild-type C terminus on the stability of DVL1. The C terminus of DVL1 contains a number of proline-rich clusters that are deleted by the frameshift mutations leading to RS-OS. These regions bind the E3 ubiquitin ligases HECW1 (previously known as NEDL1), ITCH, and NEDD4L, which mediate the degradation of DVL.33–35 Additionally, guanine nucleotide-binding protein β2 (Gβ2) interacts with the DEP-C region of DVL (some of which is deleted by these mutations) to degrade DVL.36 ITCH, NEDD4L, and Gβ2 have been directly shown to inhibit canonical Wnt activity through their interaction with DVL.34–36 This is, however, insufficient to fully explain our observations given that a high level of mtDVL1 dramatically underperformed in comparison to the wtDVL1 construct in our TOPFlash transfection assays (Figure 3A). Simply an increase in stability, and thus amount, of DVL1 is insufficient to explain the RS-OS phenotype.
Phosphorylation of DVL, which requires key Ser and Thr residues in the C terminus, negatively regulates canonical Wnt signaling.32 The phosphorylated DVL disrupts the formation of DVL puncta,48 which we have shown can still be formed by p.Trp507Glyfs∗142 DVL1 (mtDVL1) (Figures 2D–2F) and are essential for β-catenin-dependent signaling.44 Thus, the absence of these key sites in the altered proteins might lend a resistance to phosphorylation and thus stabilize these signaling puncta. Our observation of a significant difference between the activity of mtDVL1 and shDVL1, both at high expression and in trans with wtDVL1 (Figure 3B), suggests that the novel, highly basic, C-terminal region introduced by all three mutant alleles lends additional influence to the signaling capabilities of these protein complexes.
This report adds to the genetic heterogeneity of RS and places mutations in DVL1 alongside those in ROR2 and WNT5A as causative of a subtype of the disorder, RS-OS. The biochemical mechanisms behind this phenotype remain largely mysterious; however, the mutations reported in this study appear to simultaneously affect both canonical and PCP-Wnt signaling pathways. The nature of the impact upon the canonical Wnt pathway is partially explained by this work, and intriguingly, the effect seems to be mediated by the co-expression of both mutant and wild-type alleles, suggesting that these mutations exert their effects through an interaction with wild-type DVL within the Wnt signalosome.
Acknowledgments
We gratefully acknowledge the participation of the families in this research, A. Al-Ani and M. Farella for images and clinical assistance, A. Carne for assistance with the protein analysis, and A.O.M. Wilkie for discussions. This work used data from the Decipher Consortium and the NHLBI Exome Sequencing Project ESP6500 dataset. This work was funded through support from Cure Kids New Zealand (to S.R.) and from the Maurice and Phyllis Paykel Trust (to K.B.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
DECIPHER, http://decipher.sanger.ac.uk/
ExPASy pI Calculator, http://web.expasy.org/compute_pi/
GATK, https://www.broadinstitute.org/gatk/guide/best-practices
OMIM, http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
References
- 1.Mazzeu J.F., Pardono E., Vianna-Morgante A.M., Richieri-Costa A., Ae Kim C., Brunoni D., Martelli L., de Andrade C.E., Colin G., Otto P.A. Clinical characterization of autosomal dominant and recessive variants of Robinow syndrome. Am. J. Med. Genet. A. 2007;143:320–325. doi: 10.1002/ajmg.a.31592. [DOI] [PubMed] [Google Scholar]
- 2.Afzal A.R., Rajab A., Fenske C.D., Oldridge M., Elanko N., Ternes-Pereira E., Tüysüz B., Murday V.A., Patton M.A., Wilkie A.O., Jeffery S. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat. Genet. 2000;25:419–422. doi: 10.1038/78107. [DOI] [PubMed] [Google Scholar]
- 3.van Bokhoven H., Celli J., Kayserili H., van Beusekom E., Balci S., Brussel W., Skovby F., Kerr B., Percin E.F., Akarsu N., Brunner H.G. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat. Genet. 2000;25:423–426. doi: 10.1038/78113. [DOI] [PubMed] [Google Scholar]
- 4.Person A.D., Beiraghi S., Sieben C.M., Hermanson S., Neumann A.N., Robu M.E., Schleiffarth J.R., Billington C.J., Jr., van Bokhoven H., Hoogeboom J.M. WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev. Dyn. 2010;239:327–337. doi: 10.1002/dvdy.22156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niehrs C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012;13:767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- 6.Little R.D., Carulli J.P., Del Mastro R.G., Dupuis J., Osborne M., Folz C., Manning S.P., Swain P.M., Zhao S.C., Eustace B. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet. 2002;70:11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyden L.M., Mao J., Belsky J., Mitzner L., Farhi A., Mitnick M.A., Wu D., Insogna K., Lifton R.P. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 8.Balemans W., Patel N., Ebeling M., Van Hul E., Wuyts W., Lacza C., Dioszegi M., Dikkers F.G., Hildering P., Willems P.J. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002;39:91–97. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brunkow M.E., Gardner J.C., Van Ness J., Paeper B.W., Kovacevich B.R., Proll S., Skonier J.E., Zhao L., Sabo P.J., Fu Y. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 2001;68:577–589. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gong Y., Slee R.B., Fukai N., Rawadi G., Roman-Roman S., Reginato A.M., Wang H., Cundy T., Glorieux F.H., Lev D., Osteoporosis-Pseudoglioma Syndrome Collaborative Group LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 11.Leupin O., Piters E., Halleux C., Hu S., Kramer I., Morvan F., Bouwmeester T., Schirle M., Bueno-Lozano M., Fuentes F.J. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 2011;286:19489–19500. doi: 10.1074/jbc.M110.190330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boudin E., Steenackers E., de Freitas F., Nielsen T.L., Andersen M., Brixen K., Van Hul W., Piters E. A common LRP4 haplotype is associated with bone mineral density and hip geometry in men-data from the Odense Androgen Study (OAS) Bone. 2013;53:414–420. doi: 10.1016/j.bone.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 13.Kato M., Patel M.S., Levasseur R., Lobov I., Chang B.H., Glass D.A., 2nd, Hartmann C., Li L., Hwang T.H., Brayton C.F. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 2002;157:303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kokubu C., Heinzmann U., Kokubu T., Sakai N., Kubota T., Kawai M., Wahl M.B., Galceran J., Grosschedl R., Ozono K., Imai K. Skeletal defects in ringelschwanz mutant mice reveal that Lrp6 is required for proper somitogenesis and osteogenesis. Development. 2004;131:5469–5480. doi: 10.1242/dev.01405. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y., Li Y.P., Paulson C., Shao J.Z., Zhang X., Wu M., Chen W. Wnt and the Wnt signaling pathway in bone development and disease. Front Biosci (Landmark Ed) 2014;19:379–407. doi: 10.2741/4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao C., Chen Y.G. Dishevelled: The hub of Wnt signaling. Cell. Signal. 2010;22:717–727. doi: 10.1016/j.cellsig.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 17.Bunn K.J., Lai A., Al-Ani A., Farella M., Craw S., Robertson S.P. An osteosclerotic form of Robinow syndrome. Am. J. Med. Genet. A. 2014;164A:2638–2642. doi: 10.1002/ajmg.a.36677. [DOI] [PubMed] [Google Scholar]
- 18.Eijkenboom D.F., Verbist B.M., Cremers C.W., Kunst H.P. Bilateral conductive hearing impairment with hyperostosis of the temporal bone: a new finding in Robinow syndrome. Arch. Otolaryngol. Head Neck Surg. 2012;138:309–312. doi: 10.1001/archoto.2011.1459. [DOI] [PubMed] [Google Scholar]
- 19.Bain M.D., Winter R.M., Burn J. Robinow syndrome without mesomelic ‘brachymelia’: a report of five cases. J. Med. Genet. 1986;23:350–354. doi: 10.1136/jmg.23.4.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Al Kaissi A., Bieganski T., Baranska D., Chehida F.B., Gharbi H., Ghachem M.B., Hendaoui L., Safi H., Kozlowski K. Robinow syndrome: report of two cases and review of the literature. Australas. Radiol. 2007;51:83–86. doi: 10.1111/j.1440-1673.2006.01668.x. [DOI] [PubMed] [Google Scholar]
- 21.Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;11:1–11. doi: 10.1002/0471250953.bi1110s43. 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cingolani P., Platts A., Wang L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cingolani P., Patel V.M., Coon M., Nguyen T., Land S.J., Ruden D.M., Lu X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012;3:35. doi: 10.3389/fgene.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilkins M.R., Gasteiger E., Bairoch A., Sanchez J.C., Williams K.L., Appel R.D., Hochstrasser D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 27.Dillman A.R., Minor P.J., Sternberg P.W. Origin and evolution of dishevelled. G3 (Bethesda). 2013;3:251–262. doi: 10.1534/g3.112.005314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wynshaw-Boris A. Dishevelled: in vivo roles of a multifunctional gene family during development. Curr. Top. Dev. Biol. 2012;101:213–235. doi: 10.1016/B978-0-12-394592-1.00007-7. [DOI] [PubMed] [Google Scholar]
- 29.Lijam N., Paylor R., McDonald M.P., Crawley J.N., Deng C.X., Herrup K., Stevens K.E., Maccaferri G., McBain C.J., Sussman D.J., Wynshaw-Boris A. Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell. 1997;90:895–905. doi: 10.1016/s0092-8674(00)80354-2. [DOI] [PubMed] [Google Scholar]
- 30.Hamblet N.S., Lijam N., Ruiz-Lozano P., Wang J., Yang Y., Luo Z., Mei L., Chien K.R., Sussman D.J., Wynshaw-Boris A. Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development. 2002;129:5827–5838. doi: 10.1242/dev.00164. [DOI] [PubMed] [Google Scholar]
- 31.Etheridge S.L., Ray S., Li S., Hamblet N.S., Lijam N., Tsang M., Greer J., Kardos N., Wang J., Sussman D.J. Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 2008;4:e1000259. doi: 10.1371/journal.pgen.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.González-Sancho J.M., Greer Y.E., Abrahams C.L., Takigawa Y., Baljinnyam B., Lee K.H., Lee K.S., Rubin J.S., Brown A.M. Functional consequences of Wnt-induced dishevelled 2 phosphorylation in canonical and noncanonical Wnt signaling. J. Biol. Chem. 2013;288:9428–9437. doi: 10.1074/jbc.M112.448480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyazaki K., Fujita T., Ozaki T., Kato C., Kurose Y., Sakamoto M., Kato S., Goto T., Itoyama Y., Aoki M., Nakagawara A. NEDL1, a novel ubiquitin-protein isopeptide ligase for dishevelled-1, targets mutant superoxide dismutase-1. J. Biol. Chem. 2004;279:11327–11335. doi: 10.1074/jbc.M312389200. [DOI] [PubMed] [Google Scholar]
- 34.Wei W., Li M., Wang J., Nie F., Li L. The E3 ubiquitin ligase ITCH negatively regulates canonical Wnt signaling by targeting dishevelled protein. Mol. Cell. Biol. 2012;32:3903–3912. doi: 10.1128/MCB.00251-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding Y., Zhang Y., Xu C., Tao Q.H., Chen Y.G. HECT domain-containing E3 ubiquitin ligase NEDD4L negatively regulates Wnt signaling by targeting dishevelled for proteasomal degradation. J. Biol. Chem. 2013;288:8289–8298. doi: 10.1074/jbc.M112.433185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jung H., Kim H.J., Lee S.K., Kim R., Kopachik W., Han J.K., Jho E.H. Negative feedback regulation of Wnt signaling by Gbetagamma-mediated reduction of Dishevelled. Exp. Mol. Med. 2009;41:695–706. doi: 10.3858/emm.2009.41.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung H., Kim B.G., Han W.H., Lee J.H., Cho J.Y., Park W.S., Maurice M.M., Han J.K., Lee M.J., Finley D., Jho E.H. Deubiquitination of Dishevelled by Usp14 is required for Wnt signaling. Oncogenesis. 2013;2:e64. doi: 10.1038/oncsis.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goto T., Sato A., Shimizu M., Adachi S., Satoh K., Iemura S., Natsume T., Shibuya H. IQGAP1 functions as a modulator of dishevelled nuclear localization in Wnt signaling. PLoS ONE. 2013;8:e60865. doi: 10.1371/journal.pone.0060865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tauriello D.V., Jordens I., Kirchner K., Slootstra J.W., Kruitwagen T., Bouwman B.A., Noutsou M., Rüdiger S.G., Schwamborn K., Schambony A., Maurice M.M. Wnt/β-catenin signaling requires interaction of the Dishevelled DEP domain and C terminus with a discontinuous motif in Frizzled. Proc. Natl. Acad. Sci. USA. 2012;109:E812–E820. doi: 10.1073/pnas.1114802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M., Carter N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X., Zhang Y., Kang H., Liu W., Liu P., Zhang J., Harris S.E., Wu D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 2005;280:19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 42.Korinek V., Barker N., Willert K., Molenaar M., Roose J., Wagenaar G., Markman M., Lamers W., Destree O., Clevers H. Two members of the Tcf family implicated in Wnt/beta-catenin signaling during embryogenesis in the mouse. Mol. Cell. Biol. 1998;18:1248–1256. doi: 10.1128/mcb.18.3.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwarz-Romond T., Merrifield C., Nichols B.J., Bienz M. The Wnt signalling effector Dishevelled forms dynamic protein assemblies rather than stable associations with cytoplasmic vesicles. J. Cell Sci. 2005;118:5269–5277. doi: 10.1242/jcs.02646. [DOI] [PubMed] [Google Scholar]
- 44.Schwarz-Romond T., Fiedler M., Shibata N., Butler P.J., Kikuchi A., Higuchi Y., Bienz M. The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat. Struct. Mol. Biol. 2007;14:484–492. doi: 10.1038/nsmb1247. [DOI] [PubMed] [Google Scholar]
- 45.Wang J.Y., Chen F., Fu X.Q., Ding C.S., Zhou L., Zhang X.H., Luo Z.G. Caspase-3 cleavage of dishevelled induces elimination of postsynaptic structures. Dev. Cell. 2014;28:670–684. doi: 10.1016/j.devcel.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 46.Tree D.R., Shulman J.M., Rousset R., Scott M.P., Gubb D., Axelrod J.D. Prickle mediates feedback amplification to generate asymmetric planar cell polarity signaling. Cell. 2002;109:371–381. doi: 10.1016/s0092-8674(02)00715-8. [DOI] [PubMed] [Google Scholar]
- 47.Liu C., Lin C., Gao C., May-Simera H., Swaroop A., Li T. Null and hypomorph Prickle1 alleles in mice phenocopy human Robinow syndrome and disrupt signaling downstream of Wnt5a. Biol. Open. 2014;3:861–870. doi: 10.1242/bio.20148375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernatik O., Ganji R.S., Dijksterhuis J.P., Konik P., Cervenka I., Polonio T., Krejci P., Schulte G., Bryja V. Sequential activation and inactivation of Dishevelled in the Wnt/beta-catenin pathway by casein kinases. J. Biol. Chem. 2011;286:10396–10410. doi: 10.1074/jbc.M110.169870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.