

SYNOPSIS
Many detailed studies of the molecular pathogenesis of bladder cancer have been published during the past three decades, identifying important roles for many of the classic cancer pathways in bladder cancer development. Nonetheless, recent large scale mutational and expression analyses in bladder cancer made possible by next generation sequencing and other new techniques have uncovered multiple genes and pathways important for bladder cancer development, many of which were previously unknown. Genes involved in cell cycle control, chromatin regulation, and receptor tyrosine and PI 3-kinase-mTOR signaling pathways are commonly mutated in muscle-invasive bladder cancer, and most cancers have involvement of more than one of these pathways. In addition, expression-based analyses have enabled identification of two distinct types of bladder cancer, basal and luminal, that are similar to those same subsets of breast cancer, and have prognostic and therapeutic significance. These observations are leading to a number of novel therapeutic approaches in bladder cancer, providing optimism for therapeutic progress in the near future.
Keywords: Bladder cancer, urothelial carcinoma, mutations, somatic copy number alterations, cell cycle, epigenetics, molecular subtypes
BACKGROUND
Bladder cancer is a leading cause of morbidity and mortality, with nearly 400,000 new cases and 150,000 deaths worldwide (1). However novel approaches to treatment in the past two decades have been sparse. Since 2006, of 126 approvals granted by the US Food and Drug Administration for hematology/oncology medications, none have been for the treatment of bladder cancer (2), and chemotherapeutic approaches remain rooted in cisplatin-based combinations first introduced thirty years ago. This limited progress has provided major incentive to analyze molecular alterations in bladder cancer in detail in an effort to identify novel treatment approaches.
Bladder cancer genetics and molecular biology have historically provided important general insights into cancer biology, beginning with the discovery of HRAS as the first oncogene in a bladder cancer cell line (3). Since that seminal discovery, multiple genes commonly subject to mutation in bladder cancer have been identified, including TP53 (4), RB1 (5), TSC1 (6), FGFR3 (7), and PIK3CA (8, 9).
Furthermore, comparative genomic hybridization and related techniques were used extensively in bladder cancer, leading to identification of multiple amplified and deleted genes, including PPARG, E2F3, EGFR, CCND1 and MDM2 which are amplified, and CDKN2A and RB1 which are commonly deleted (10–17). These and other molecular alterations involved in bladder cancer are summarized in past reviews (16, 18).
Recently, next generation sequencing (NGS) has enabled large-scale analyses, mainly focused on muscle-invasive bladder cancer, greatly expanding our understanding of this malignancy (19–22). The initial NGS studies were performed by the Beijing Genomics Institute (20, 21), in studies which focused initially on mutation identification (20), and then included both mutation analysis and transcriptome studies (21). More recently The Cancer Genome Atlas (TCGA) project, funded by the National Cancer Institute, has performed a comprehensive analysis of 131 muscle-invasive bladder cancers, including assessment of mutations, copy number changes, expression profiling by RNA-Seq, miRNA analysis, CpG methylation analysis, proteomic analysis of about 150 proteins, and integrated analyses of these data sets.
This review will summarize the current understanding of molecular alterations in bladder cancer, and will focus on the findings of the TCGA project (19) and the Beijing group (20, 21). We will also discuss recent reports providing improved understanding of molecular subtypes of bladder cancer based on expression analyses.
MOLECULAR ALTERATIONS IN BLADDER CANCER
Figure 1 illustrates the major findings of the TCGA study, showing mutation rates and frequencies, gene deletions and amplifications, and changes in expression for genes of interest (19).
Figure 1. The genomic landscape of bladder cancer.

a, Mutation rate and type, histological subtype, smoking status, gender, tumour stage and cluster type. b, Genes with statistically significant levels of mutation and mutation types. c, Deletions and amplifications for genomic regions with statistically significant focal copy number changes. ‘Copy number’ refers to absolute copy number. Note that two amplification peaks (*) contain several genes, any of which could be the target, as opposed to the single gene listed here. d, RNA expression level for selected genes, expressed as fold change from the median value for all samples. Cancers were grouped into three clusters (red, blue and green) using consensus NMF clustering.
From Macmillan Publishers Ltd: Nature. The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014;507:315–322. Copyright 2014; with permission.
From The Cancer Genome Atlas Research Network Nature 507, 315–322 (2014) doi:10.1038/nature12965. Available at: http://www.nature.com/nature/journal/v507/n7492/full/nature12965.html. Accessed Oct 13 2014; with permission.
Mutations in bladder cancer – general findings
The TCGA study identified a relatively high rate of 7.68 mutations per Mb within coding regions, equivalent to 302 exonic mutations per cancer (19). This mutation rate is exceeded among cancers studied in the TCGA project (now > 20) only by lung adenocarcinoma, lung squamous cell carcinoma, and melanoma (23). The mechanism or cause of this high mutation rate in bladder cancer is not known with certainty. Although smoking is associated with mutation rate and spectrum in lung cancer, this was not seen in bladder cancer (19), despite the well-known epidemiologic association between cigarette smoking and bladder cancer. On the other hand, 51% mutations seen in bladder cancer were TCW -> TTW or TGW changes (“TCW mutations” (C > T and C > G mutations at T-C-A/T [TCW] trinucleotides), a class of mutation likely mediated by one of the DNA cytosine deaminases, in the APOBEC gene family (24, 25). In addition, APOBEC3B was expressed at high levels in all bladder cancers examined, suggesting a major role for APOBEC-mediated mutagenesis in the high mutation rate seen in bladder cancer (19).
The Beijing group identified a somewhat lower overall mutation rate, but by statistical analyses identified significant levels of mutation in 37 genes. This included many genes identified previously, as well as multiple chromatin remodeling genes: KDM6A, ARID1A, CREBBP, EP300, KMT2A, NCOR1, CHD6, KMT2C (20, 21). In the TCGA analysis, 32 genes were identified as sustaining mutations at a statistically significant rate (Figure 1b). There was considerable overlap between the genes identified by the Beijing group and those identified in the TCGA analysis, providing significant confirmation, despite the distinctly different populations being studied (Beijing – Chinese; TCGA – mostly American and European origin). Genes sustaining significant levels of mutation can be organized into several classes.
Somatic copy number alterations (SCNA) in bladder cancer
SCNAs were also very common in the TCGA data set, with an average of 204 segmental SCNAs and 22 genomic rearrangements per cancer analyzed (19). Statistical methods (GISTIC) were used to identify statistically significant recurrent focal SCNAs, and 27 amplified and 30 deleted regions were identified (Figure 1c). CDKN2A was deleted in nearly half of samples, and other genes that were probable targets of deletion included: KDM6A, RB1, WWOX, PDE4D, FOXQ1, FAM190A, LRP1B, and CREBBP. Many deletions extended over a large genomic region and a single gene target could not be identified. Many genes were focally amplified including: E2F3/SOX4, CCND1, EGFR, PPARG, MDM2, ERBB2, YAP1, CCNE1, MYC, ZNF703, FGFR3, MYCL1, and BCL2L1. Other chromosomal regions of amplification extended over a large genomic region, containing more than a single gene. This included a region on chromosome 1q22-23.2 containing PVRL4, and a region on chromosome 8q22.3 containing YWHAZ. Multiple past reports made similar though more limited findings (14, 26–28).
Cell cycle gene aberrations in bladder cancer
Mutation or deletion of cell cycle genes in cancer has been known for many years, and reported previously in bladder cancer for over 2 decades, e.g. (4, 17, 29, 30). The recent in-depth genomic analyses performed have confirmed and extended our knowledge of cell cycle gene events in bladder cancer. Reviewing these alterations in the order of frequency, TP53 (encodes p53) mutations were found in 49% and 24% of bladder cancers by the TCGA and Beijing groups (19–21). p53 is the ‘guardian of the genome’, and it responds to cellular stress by inducing cell cycle arrest, apoptosis, senescence, and DNA repair. Its loss leads to bypass of these important effects, enhancing further genome damage, and continued proliferation. CDKN2A deletions are also very common in bladder cancer, similar to many other malignancies, with deletions seen in about 50% of samples in both the TCGA and Beijing studies (19–21). Mutations in CDKN2A were also seen in 5% of bladder cancers (19). CDKN2A encodes both p19ARF and p16INK4A proteins, which regulate the p53 and RB pathways, respectively. p16INK4A is a cyclin-dependent kinase (cdk) inhibitor for cdk4 and cdk6, and its loss enhances cell cycle progression.
RB1 was mutated in 10% and deleted in 15% of TCGA tumors (19), with similar findings by the Beijing group (20, 21). The RB protein regulates the cell cycle by binding to the E2F transcription factor (31), and its loss also leads to enhanced cell cycle progression despite DNA damage or other signals.
A novel finding in the TCGA analysis was inactivation of CDKN1A by mutation in 14% and deletion in 6% (19). Another group reported similar findings simultaneously (22). CDKN1A encodes another cyclin dependent kinase inhibitor, p21, distinct from p16INK4A, but its loss similarly pushes the cell toward continued division despite DNA damage or other signals, enhancing cell proliferation and accumulation of additional DNA damage. CDKN1A mutation has not been seen at appreciable frequency in any other cancer studied to date by the TCGA.
Three cell cycle genes were amplified in bladder cancer in these studies: CCND1 (cyclin D1) in 10% – 21% of bladder cancers; CCNE1 (cyclin E1) in 12% – 13%; and MDM2 in 9% – 4% of samples respectively in the TCGA and Beijing analyses (19–21). Both cyclin D1 and cyclin E1 are necessary co-factors for different cdk’s and hence their overexpression enhances cell proliferation. MDM2 encodes an E3 ubiquitin-protein ligase, the enzyme which degrades p53 protein, and hence its overexpression lowers p53 levels, thereby inhibiting p53 function. MDM2 amplification was mutually exclusive of TP53 mutation for the most part in the TCGA analysis (Figure 1) (19).
Overall one or more cell cycle genes were mutated, deleted, or activated by amplification in 93% of the TCGA tumor samples, most often the TP53 and/or CDKN2A genes (Figure 1).
Kinase signaling: receptor tyrosine kinases and the PI3K-mTOR pathway
Given the well-known successes of tyrosine kinase inhibitor therapy for activated kinases in both chronic myelogenous leukemia, driven by BCR-ABL, and lung adenocarcinoma, driven by activating mutations in EGFR, there is major interest in the identification of similar activated tyrosine kinases in all types of cancer. Activating mutations in FGFR3 were first identified in bladder cancer 15 years ago (7), though more commonly seen in superficial than in muscle-invasive cancers. FGFR3 mutations were seen at 11% frequency in the muscle-invasive cancers studied by the TCGA and Beijing groups (19–21). In addition, 3% of cases had FGFR3 amplification, and another 3% had FGFR3-TACC3 gene fusions (19). These latter gene fusions have recently been reported by several groups, and shown to be activating (32, 33). Three members of the ERBB family of receptor tyrosine kinases were also altered at significant frequency in bladder cancer (19). EGFR amplification was seen in 11%, ERBB2 was mutated in 5% and amplified in 7%, and ERBB3 was mutated in 11% and amplified in 2% of bladder cancers. These events in FGFR3, and ERBB family members are all potentially targetable using either tyrosine kinase inhibitors, or antibody-mediated therapies (34, 35). This potential is underscored by a recent report of the benefit of both erlotinib (EGFR tyrosine kinase inhibitor) and cetuximab (monoclonal antibody against EGFR) in some subsets of muscle-invasive bladder cancers (36).
Mutations involving genes in the PI3K-PTEN-AKT-TSC1-TSC2-mTOR signaling pathway have long been recognized in bladder cancer (6, 8, 9). The recent large-scale studies provided further documentation of mutation of both positive and negative regulators in this signaling cascade. PIK3CA was mutated in 15% and amplified in 5%; PTEN was mutated in 3% and deleted in 13%; TSC1 was mutated in 8%; and TSC2 was mutated in 2% in the TCGA analyses, with similar results from the Beijing studies (19–21). All of these mutations lead to potential therapeutic targets, as highlighted by two recent studies showing that bladder cancer patients with TSC1 and MTOR mutations were exceptional responders to treatment with/including everolimus, an mTOR allosteric inhibitor (37, 38).
Other genes and pathways
Two genes involved in the response to oxidative stress were found to be mutated in bladder cancer in the TCGA analysis. NFE2L2 encodes a transcription factor which is induced to mediate the cellular response to oxidative stress, and missense mutations in this gene were identified in 8% of the TCGA samples (19). Mutations in NFE2L2 were previously reported in cancer and shown to promote malignancy (39), although not previously known for bladder cancer. TXNIP encodes thioredoxin interacting protein and is also involved in mediating the response to oxidative stress (40). Mutations were seen in 7% of cancers in the TCGA study (19).
Mutations in genes involved in lipid metabolism were also identified in the TCGA analysis (19). RXRA, which encodes the retinoid X nuclear receptor alpha (41) was mutated in 9% of bladder cancers, with 7 of 12 mutations occurring at the same amino acid in the ligand-binding domain. All seven of these cancers showed increased expression of genes involved in lipid metabolism, consistent downstream effects of RXR activation. PPARG, which encodes the peroxisome proliferator-activated receptor gamma (glitazone receptor), was amplified in 17% of bladder cancers.
STAG2 is another relatively novel gene found to be mutated in bladder cancer by several groups at frequencies from 10 – 20% (19, 21, 42–44). The STAG2 protein is a subunit of cohesin, a protein complex that regulates the separation of sister chromatids during cell division. Conflicting results were seen in terms of a potential association between STAG2 mutation and survival in bladder cancer (21, 42–44).
ERCC2 was observed to be mutated in 12% and 7% of bladder cancers in the TCGA and Beijing analyses (19–21). ERCC2 is a nucleotide excision repair gene that causes xeroderma pigmentosum (45). It appears that these mutations may act in a dominant negative fashion, as 15 of 16 were deleterious missense mutations (19). A recent report described a positive association between ERCC2 mutation and response to cisplatin-based chemotherapy in bladder cancer, consistent with loss of ERCC2 function leading to an Achilles heel-like sensitivity to cisplatin (46).
Epigenetics: chromatin regulation
The role of epigenetic effects on regulation of gene expression, and their alterations in cancer in general have been studied for many years. However, the potential importance of these events in bladder cancer was advanced considerably by the recent in-depth genomic analyses. Mutations in chromatin regulatory genes in bladder cancer were first identified by the Beijing group, and then confirmed and extended by the TCGA analyses (19–21). It is known that there are two main categories of chromatin modification that influence gene expression. The first is methylation at the C position of CG nucleotide sequences, and other less common modifications to the nucleotide sequence itself. The second is histone modifications, which are generated by chromatin regulatory genes. Chromatin regulatory genes are often referred to as ‘writers’, ‘erasers’, and ‘readers’, based on their function in creating covalent modification of histones (methylation, acetylation), removing such modifications (de-methylation, de-acetylation), and in binding to such modifications to influence gene transcription. Genes involved in all three of these functions are commonly mutated in bladder cancer.
Mutations in KDM6A, an ‘eraser’ demethylase acting on histone H3 at lysine 27, are seen in 20–25% of bladder cancers (19–21). MLL2, encoding a histone 3 lysine 4 (H3K4) methyltransferase of the trithorax group, is a ‘writer,’ and is mutated in 27% of bladder cancers (19). ARID1A, encoding a member of the SWI/SNF family, has helicase and ATPase activity, is a ‘reader,’ and is mutated in 25% of bladder cancers (19). EP300, encoding a histone acetylase, is a ‘writer’ which is also a transcription factor, and is mutated in 15% of bladder cancers (19). Many other chromatin regulatory genes are mutated in over 10% of bladder cancer samples, including MLL3, MLL, CREBBP, CHD7 and SRCAP (19). In addition, CREBBP and NCOR1 were deleted in 13% and 25% of bladder cancers, respectively (Figure 1). Despite the likely importance of these mutations in the chromatin regulation of the genome in bladder cancer cells, there was no significant association between any of these gene mutations and expression profile or other features in the TCGA analysis (19).
Viral infection in bladder cancer: CMV, BK polyomavirus, and HPV16
Exploration of infectious etiologies has long been of interest in bladder cancer: a viral etiology for bladder cancer has been considered for some time, and there is a clear epidemiologic relationship to chronic Schistosoma infection. The TCGA analysis identified seven tumors (6%) with viral DNA sequences and five tumors with viral RNA transcripts (19). The tumors with viral transcripts were: 3 with CMV, 1 with BK polyomavirus, and 1 with HPV16. Some of those with viral DNA sequences showed integration of viral DNA into the genome, with one instance of integration into BCL2L1, an apoptosis regulatory gene, suggesting the possibility that insertional mutagenesis might contribute to bladder cancer development in these cases.
Molecular subtypes of bladder cancer
There is considerable heterogeneity in bladder cancer, both in terms of natural history and response to chemotherapy. Many efforts have been made to define molecular subsets of bladder cancer based upon mutation profile and/or expression features to attempt to provide prognostic information, as well as guidance in selection of chemotherapy. For example, Takata et al. analyzed gene expression profiles of biopsy materials from 27 invasive bladder cancers by microarray to identify 14 “predictive” genes whose expression levels were most correlated with response to methotrexate, vinblastine, doxorubicin, and cisplatin (M-VAC) chemotherapy (47). Ongoing studies are attempting to validate this expression profile prospectively. In the TCGA analysis, unsupervised clustering by non-negative matrix factorization of mutations and focal SCNAs identified three groups (Fig. 1a) (19). Group A (red), labeled ‘focally amplified’, was highly enriched in focal SCNAs in several genes, as well as mutations in MLL2. Group B (blue), labeled ‘papillary CDKN2A-deficient FGFR3 mutant’, was enriched in papillary histology, and the majority had loss of CDKN2A, and one or more alterations in FGFR3. Group C (green), labeled ‘TP53/cell-cycle-mutant’, had TP53 mutations in nearly all samples, and enrichment for RB1 mutations, and amplifications of E2F3 and CCNE1 (Fig. 1).
The TCGA analysis also used RNA-Seq based expression profiling to identify four distinct mRNA expression clusters (Figure 2A) (19). Cluster I (‘papillary-like’) was enriched in cancers with papillary morphology, FGFR3 mutations, FGFR3 copy number gain, elevated FGFR3 expression, and FGFR3–TACC3 gene fusions. Cluster II was similar to Cluster I in several respects but did not have an association with papillary morphology or FGFR3 events. Clusters I and II both expressed high HER2 (ERBB2) levels and had an elevated estrogen receptor beta (ESR2) signaling signature, suggesting a relationship to HER2-positive breast cancers. Clusters I and II also had features similar to those of luminal A breast cancer, with high expression of GATA3 and FOXA1. I and II also expressed uroplakins (markers of urothelial differentiation), E-cadherin, and members of the miR-200 family of miRNAs (which target multiple regulators of epithelial–mesenchymal transition), suggesting that these cancers had urothelial differentiation to some extent. In contrast, cluster III (‘basal/squamous-like’) was similar in some respects to both basal-like breast cancer, and squamous cell cancers of the head and neck and lung with expression of epithelial lineage genes, including several keratin genes. Some also showed some degree of variant squamous histology on pathology review.
Figure 2. Molecular subtypes of bladder cancer defined by expression profiling in 3 studies.


A. Expression clustering by the TCGA. a, Papillary histology, FGFR3 alterations, FGFR3 expression and reduced FGFR3-related miRNA expression are enriched in cluster I. b, Expression of epithelial lineage genes and stem/progenitor cytokeratins are generally high in cluster III, some of which show variant squamous histology. c, Luminal breast and urothelial differentiation factors are enriched in clusters I and II. d, ERBB2 mutation and oestrogen receptor beta (ESR2) expression are enriched in clusters I and II.
B. Expression clustering and survival by Choi et al.
a. Array-based RNA expression profiling and hierarchical cluster analysis of a cohort of 73 muscle-invasive bladder cancers. RAS, TP53, RB1, and FGFR3 mutations are indicated below the dendrogram. Black, mutation; white, wildtype; gray, data not available. Right: Kaplan-Meier overall survival (p=0.098) and disease-specific survival (p=0.028) in the three tumor subtypes.
b. Expression of basal and luminal markers in the three subtypes. The heat maps depict relative expression of basal (left) and luminal (right) biomarkers. GSEA analyses (not shown) were used to confirm that basal and luminal markers were enriched in the subtypes.
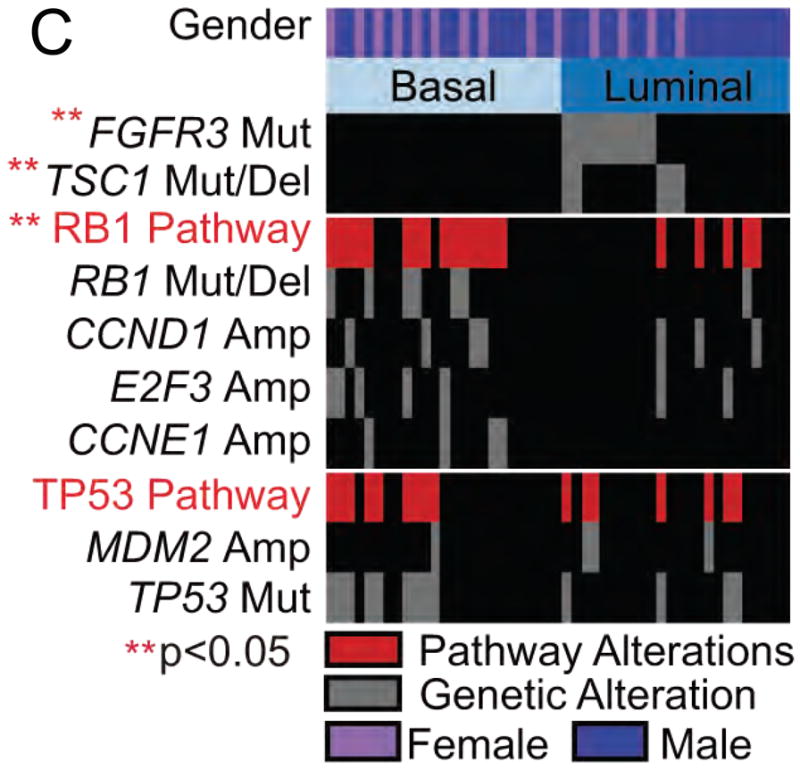
C. Associations between mutations and basal subtypes of bladder cancer by Damrauer et al. FGFR3 and TSC1 alterations were significantly enriched in luminal bladder cancer, whereas alterations of the RB1 pathway were enriched in basal-like bladder cancer. TP53 alterations were distributed evenly in the two subtypes.
Fig 2A: From The Cancer Genome Atlas Research Network Nature 507, 315–322 (2014) doi:10.1038/nature12965. Available at: http://www.nature.com/nature/journal/v507/n7492/full/nature12965.html. Accessed Oct 13 2014; with permission.
Fig 2B: From Choi W, Porten S, Kim S, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014 25:152–65; with permission.
Fig 2C: From Damrauer JS, Hoadley KA, Chism DD, Fan C, Tiganelli CJ, Wobker SE, Yeh JJ, Milowsky MI, Iyer G, Parker JS, Kim WY. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A 2014 111:3110–5; with permission.
Similar but more extensive findings were made simultaneously by two other groups (48, 49). Unsupervised clustering using array-based RNA expression data identified three robust clusters, termed basal, luminal and p53-like in one of these studies (Figure 2B) (48). Samples in the basal cluster expressed high levels of keratins 5, 6, and 14, similar to Cluster III from the TCGA analysis. This cluster was labeled basal since the authors recognized that this expression pattern was similar to that of the basal layer of the normal urothelium, which is least differentiated and does not express the usual urothelial marker genes. Several other expression features were common to these two independently derived clusters by the TCGA and Choi et al. Choi et al. also showed that basal bladder cancers had the poorest prognosis, with the shortest disease-specific survival (48). The luminal cluster was similar to clusters I and II identified in the TCGA analysis, with expression of luminal breast cancer markers, as noted above. The last cluster, p53-like, was similar to the luminal cluster in terms of luminal breast marker expression, but also had an activated wild-type p53 gene signature. Choi et al. did extensive replication analyses to validate their clustering analysis, including reanalysis of a large data set in which clustering had identified clusters of a similar nature, though named somewhat differently (50, 51). Pathway analysis led to identification of Stat-3, NFkB, Hif-1, and p63 as probable transcriptional drivers of basal gene expression (48); and correspondingly PPARγ and the estrogen receptor (ER) as drivers of the luminal gene expression pattern. shRNA knockdown of p63 in a basal-like bladder cancer cell line, UM-UC14, showed decreased basal gene expression, and increased PPARγ pathway expression. Treatment with rosiglitazone, a PPARγ activator, also reduced basal gene expression, and enhanced luminal cluster gene expression, providing evidence that p63 and PPARγ antagonize each other. Choi et al. also noted that patients with bladder cancers in the p53-like expression cluster showed a poor response to cisplatin-based chemotherapy, finding this to be consistent among several different treatment groups they were able to collect (48). Notably, the p53-like expression cluster, defined by expression of a p53 gene set, did not correlate with mutation of TP53. There were tumors in the p53-like cluster that had TP53 mutations, as well as tumors that were wild-type for TP53 but did not segregate to this cluster. Gene expression pattern could be assayed using DAZL technology on formalin-fixed paraffin-embedded samples, enabling wide adoption of this method of bladder cancer expression clustering (48).
A similar clustering of bladder cancer into basal and luminal subtypes was made independently by the third group, also using multiple data sets (49). In addition, these investigators found that there was a significant association between mutation and expression subtype. FGFR3 and TSC1 mutations were significantly enriched in the luminal subtype, whereas RB1 pathway alterations including RB1 mutation/deletion, CCND1 amplification, SOX4/E2F3 amplification, and CCNE1 amplification were significantly enriched in basal-like bladder cancer (Figure 2C) (49).
SUMMARY
Great progress has been made in deciphering in considerable detail the molecular events that occur in muscle-invasive bladder cancer (19–22). Mutations are common in multiple signaling pathways in bladder cancer, and typically cancers have various alterations in different pathways that likely all contribute to cancer development. Cell cycle gene alterations are very common in bladder cancer, similar to many other adult malignancies, but therapeutic development in this sphere has not progressed. In contrast, multiple alterations in both receptor tyrosine kinases and the PI3K-mTOR pathway occur commonly in bladder cancer, and there is significant hope that many of these can be targeted effectively with specific kinase inhibitors either in use currently or under development. Chromatin regulatory gene mutations are especially common in bladder cancer, and there is hope for therapeutic development directed at these mutations.
Recent comprehensive expression profiling studies have been convergent in identifying two major types of bladder cancer, basal and luminal, with some significant similarities to the same subtypes in breast cancer (19, 48, 49). Moreover, these subtypes have different prognoses and natural histories, and response to conventional chemotherapy. Thus, there is hope that this new understanding will also translate to improved care for the bladder cancer patient.
KEY POINTS.
There is a high rate of both mutations and genomic amplifications and deletions in muscle-invasive bladder cancer.
The ERBB family sustains mutation and copy number changes at a cumulative high frequency in bladder cancer, including EGFR, ERBB2, and ERBB3, which are potentially targetable using small molecule kinase inhibitors or antibody therapeutics.
FGFR3 is commonly activated in bladder cancer, through either mutation, gene fusion events, or elevated expression, and is another potential therapeutic target.
The PI-3-kinase-mTOR signaling cascade also sustains many mutations in bladder cancer, including PI3KCA, PTEN, and TSC1, and all of these are also potential therapeutic targets.
Chromatin regulatory gene mutations occur at high frequency in bladder cancer, higher than any other epithelial cancer, and often there is more than one per cancer. Although this pathway has historically not been druggable, there is some optimism that agents may be developed, and would therefore present therapeutic opportunity.
Cell cycle gene mutations, amplifications (cyclins), and deletions (cdk inhibitors) are very common in bladder cancer.
Viral infection appears to contribute to bladder cancer development in 5–10% of cancers.
The main RNA expression subtypes of bladder cancer are basal and luminal, similar to breast cancer, and confer both prognostic and therapeutic significance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global Cancer Statistics. Ca-a Cancer Journal for Clinicians. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.United States Food and Drug Administration. Hematology/Oncology (Cancer) Approvals & Safety Notifications. 2014 Aug 24; Available from: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm279174.htm.
- 3.Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature. 1982;297:474–8. doi: 10.1038/297474a0. [DOI] [PubMed] [Google Scholar]
- 4.Esrig D, Spruck CH, Nichols PW, Chaiwun B, Steven K, Groshen S, Chen SC, Skinner DG, Jones PA, Cote RJ. P53 Nuclear-Protein Accumulation Correlates With Mutations In The P53 Gene, Tumor Grade, And Stage In Bladder-Cancer. American Journal of Pathology. 1993;143:1389–1397. [PMC free article] [PubMed] [Google Scholar]
- 5.Cote RJ, Dunn MD, Chatterjee SJ, Stein JP, Shi SR, Tran QC, Hu SX, Xu HJ, Groshen S, Taylor CR, Skinner DG, Benedict WF. Elevated and absent pRb expression is associated with bladder cancer progression and has cooperative effects with p53. Cancer Research. 1998;58:1090–1094. [PubMed] [Google Scholar]
- 6.Hornigold N, Devlin J, Davies AM, Aveyard JS, Habuchi T, Knowles MA. Mutation of the 9q34 gene TSC1 in sporadic bladder cancer. Oncogene. 1999;18:2657–61. doi: 10.1038/sj.onc.1202854. [DOI] [PubMed] [Google Scholar]
- 7.Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, Chopin D, Thiery JP, Radvanyi F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Knowles E, Hernandez S, Malats N, Kogevinas M, Lloreta J, Carrato A, Tardon A, Serra C, Real FX. PIK3CA mutations are an early genetic alteration associated with FGFR3 mutations in superficial papillary bladder tumors. Cancer Res. 2006;66:7401–4. doi: 10.1158/0008-5472.CAN-06-1182. [DOI] [PubMed] [Google Scholar]
- 9.Platt FM, Hurst CD, Taylor CF, Gregory WM, Harnden P, Knowles MA. Spectrum of Phosphatidylinositol 3-Kinase Pathway Gene Alterations in Bladder Cancer. Clinical Cancer Research. 2009;15:6008–6017. doi: 10.1158/1078-0432.CCR-09-0898. [DOI] [PubMed] [Google Scholar]
- 10.Blaveri E, Brewer JL, Roydasgupta R, Fridlyand J, DeVries S, Koppie T, Pejavar S, Mehta K, Carroll P, Simko JP, Waldman FM. Bladder cancer stage and outcome by array-based comparative genomic hybridization. Clin Cancer Res. 2005;11:7012–22. doi: 10.1158/1078-0432.CCR-05-0177. [DOI] [PubMed] [Google Scholar]
- 11.Simon R, Burger H, Brinkschmidt C, Bocker W, Hertle L, Terpe HJ. Chromosomal aberrations associated with invasion in papillary superficial bladder cancer. J Pathol. 1998;185:345–51. doi: 10.1002/(SICI)1096-9896(199808)185:4<345::AID-PATH109>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 12.Richter J, Jiang F, Gorog JP, Sartorius G, Egenter C, Gasser TC, Moch H, Mihatsch MJ, Sauter G. Marked genetic differences between stage pTa and stage pT1 papillary bladder cancer detected by comparative genomic hybridization. Cancer Res. 1997;57:2860–4. [PubMed] [Google Scholar]
- 13.Voorter C, Joos S, Bringuier PP, Vallinga M, Poddighe P, Schalken J, du Manoir S, Ramaekers F, Lichter P, Hopman A. Detection of chromosomal imbalances in transitional cell carcinoma of the bladder by comparative genomic hybridization. Am J Pathol. 1995;146:1341–54. [PMC free article] [PubMed] [Google Scholar]
- 14.Kallioniemi A, Kallioniemi OP, Citro G, Sauter G, DeVries S, Kerschmann R, Caroll P, Waldman F. Identification of gains and losses of DNA sequences in primary bladder cancer by comparative genomic hybridization. Genes Chromosomes Cancer. 1995;12:213–9. doi: 10.1002/gcc.2870120309. [DOI] [PubMed] [Google Scholar]
- 15.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, Futreal PA. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–50. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goebell PJ, Knowles MA. Bladder cancer or bladder cancers? Genetically distinct malignant conditions of the urothelium. Urol Oncol. 2010;28:409–28. doi: 10.1016/j.urolonc.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Williamson MP, Elder PA, Shaw ME, Devlin J, Knowles MA. p16 (CDKN2) is a major deletion target at 9p21 in bladder cancer. Hum Mol Genet. 1995;4:1569–77. doi: 10.1093/hmg/4.9.1569. [DOI] [PubMed] [Google Scholar]
- 18.Knowles MA. Molecular pathogenesis of bladder cancer. International Journal of Clinical Oncology. 2008;13:287–297. doi: 10.1007/s10147-008-0812-0. [DOI] [PubMed] [Google Scholar]
- 19.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–22. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, He M, Li Z, Sun X, Jia W, Chen J, Yang S, Zhou F, Zhao X, Wan S, Ye R, Liang C, Liu Z, Huang P, Liu C, Jiang H, Wang Y, Zheng H, Sun L, Liu X, Jiang Z, Feng D, Chen J, Wu S, Zou J, Zhang Z, Yang R, Zhao J, Xu C, Yin W, Guan Z, Ye J, Zhang H, Li J, Kristiansen K, Nickerson ML, Theodorescu D, Li Y, Zhang X, Li S, Wang J, Yang H, Wang J, Cai Z. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–8. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo GW, Sun XJ, Chen C, Wu S, Huang PD, Li ZS, Dean M, Huang Y, Jia WL, Zhou Q, Tang AF, Yang ZQ, Li XX, Song PF, Zhao XK, Ye R, Zhang SQ, Lin Z, Qi MF, Wan SQ, Xie LF, Fan F, Nickerson ML, Zou XJ, Hu XD, Xing L, Lv ZJ, Mei HB, Gao SJ, Liang CZ, Gao ZB, Lu JX, Yu Y, Liu CX, Li L, Fang XD, Jiang ZM, Yang J, Li CL, Zhao X, Chen J, Zhang F, Lai YQ, Lin ZG, Zhou FJ, Chen H, Chan HC, Tsang S, Theodorescu D, Li YR, Zhang XQ, Wang J, Yang HM, Gui YT, Wang J, Cai ZM. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nature Genetics. 2013;45:1459–U213. doi: 10.1038/ng.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cazier JB, Rao SR, McLean CM, Walker AL, Wright BJ, Jaeger EE, Kartsonaki C, Marsden L, Yau C, Camps C, Kaisaki P, Oxford-Illumina WGSC, Taylor J, Catto JW, Tomlinson IP, Kiltie AE, Hamdy FC. Whole-genome sequencing of bladder cancers reveals somatic CDKN1A mutations and clinicopathological associations with mutation burden. Nat Commun. 2014;5:3756. doi: 10.1038/ncomms4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kandoth C, McLellan MD, Vandin F, Ye K, Niu BF, Lu C, Xie MC, Zhang QY, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, Chen L, Leroy C, Ramakrishna M, Rance R, Lau KW, Mudie LJ, Varela I, McBride DJ, Bignell GR, Cooke SL, Shlien A, Gamble J, Whitmore I, Maddison M, Tarpey PS, Davies HR, Papaemmanuil E, Stephens PJ, McLaren S, Butler AP, Teague JW, Jonsson G, Garber JE, Silver D, Miron P, Fatima A, Boyault S, Langerod A, Tutt A, Martens JW, Aparicio SA, Borg A, Salomon AV, Thomas G, Borresen-Dale AL, Richardson AL, Neuberger MS, Futreal PA, Campbell PJ, Stratton MR Breast Cancer Working Group of the International Cancer Genome C. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–93. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell. 2012;46:424–35. doi: 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hurst CD, Fiegler H, Carr P, Williams S, Carter NP, Knowles MA. High-resolution analysis of genomic copy number alterations in bladder cancer by microarray-based comparative genomic hybridization. Oncogene. 2004;23:2250–63. doi: 10.1038/sj.onc.1207260. [DOI] [PubMed] [Google Scholar]
- 27.Richter J, Beffa L, Wagner U, Schraml P, Gasser TC, Moch H, Mihatsch MJ, Sauter G. Patterns of chromosomal imbalances in advanced urinary bladder cancer detected by comparative genomic hybridization. Am J Pathol. 1998;153:1615–21. doi: 10.1016/S0002-9440(10)65750-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoglund M, Sall T, Heim S, Mitelman F, Mandahl N, Fadl-Elmula I. Identification of cytogenetic subgroups and karyotypic pathways in transitional cell carcinoma. Cancer Res. 2001;61:8241–6. [PubMed] [Google Scholar]
- 29.Sidransky D, Von Eschenbach A, Tsai YC, Jones P, Summerhayes I, Marshall F, Paul M, Green P, Hamilton SR, Frost P, et al. Identification of p53 gene mutations in bladder cancers and urine samples. Science. 1991;252:706–9. doi: 10.1126/science.2024123. [DOI] [PubMed] [Google Scholar]
- 30.Xu HJ, Cairns P, Hu SX, Knowles MA, Benedict WF. Loss of RB protein expression in primary bladder cancer correlates with loss of heterozygosity at the RB locus and tumor progression. Int J Cancer. 1993;53:781–4. doi: 10.1002/ijc.2910530513. [DOI] [PubMed] [Google Scholar]
- 31.Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- 32.Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S, Qiu KL, Gao ZB, Ceccarelli M, Riccardi R, Brat DJ, Guha A, Aldape K, Golfinos JG, Zagzag D, Mikkelsen T, Finocchiaro G, Lasorella A, Rabadan R, Iavarone A. Transforming Fusions of FGFR and TACC Genes in Human Glioblastoma. Science. 2012;337:1231–1235. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams SV, Hurst CD, Knowles MA. Oncogenic FGFR3 gene fusions in bladder cancer. Human Molecular Genetics. 2013;22:795–803. doi: 10.1093/hmg/dds486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greulich H, Kaplan B, Mertins P, Chen TH, Tanaka KE, Yun CH, Zhang XH, Lee SH, Cho JH, Ambrogio L, Liao R, Imielinski M, Banerji S, Berger AH, Lawrence MS, Zhang JH, Pho NH, Walker SR, Winckler W, Getz G, Frank D, Hahn WC, Eck MJ, Mani DR, Jaffe JD, Carr SA, Wong KK, Meyerson M. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14476–14481. doi: 10.1073/pnas.1203201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA, Janakiraman V, Scholz RP, Bowman KK, Lorenzo M, Li H, Wu JS, Yuan WL, Peters BA, Kan ZY, Stinson J, Mak M, Modrusan Z, Eigenbrot C, Firestein R, Stern HM, Rajalingam K, Schaefer G, Merchant MA, Sliwkowski MX, de Sauvage FJ, Seshagiri S. Oncogenic ERBB3 Mutations in Human Cancers. Cancer Cell. 2014;25:543–544. doi: 10.1016/j.ccr.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 36.Rebouissou S, Bernard-Pierrot I, de Reynies A, Lepage ML, Krucker C, Chapeaublanc E, Herault A, Kamoun A, Caillault A, Letouze E, Elarouci N, Neuzillet Y, Denoux Y, Molinie V, Vordos D, Laplanche A, Maille P, Soyeux P, Ofualuka K, Reyal F, Biton A, Sibony M, Paoletti X, Southgate J, Benhamou S, Lebret T, Allory Y, Radvanyi F. EGFR as a potential therapeutic target for a subset of muscle-invasive bladder cancers presenting a basal-like phenotype. Sci Transl Med. 2014;6:244ra91. doi: 10.1126/scitranslmed.3008970. [DOI] [PubMed] [Google Scholar]
- 37.Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, Viale A, Heguy A, Peng LK, Chan TA, Bochner B, Bajorin DF, Berger MF, Taylor BS, Solit DB. Genome Sequencing Identifies a Basis for Everolimus Sensitivity. Science. 2012;338:221–221. doi: 10.1126/science.1226344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagle N, Grabiner BC, Van Allen EM, Hodis E, Jacobus S, Supko JG, Stewart M, Choueiri TK, Gandhi L, Cleary JM, Elfiky AA, Taplin ME, Stack EC, Signoretti S, Loda M, Shapiro GI, Sabatini DM, Lander ES, Gabriel SB, Kantoff PW, Garraway LA, Rosenberg JE. Activating mTOR Mutations in a Patient with an Extraordinary Response on a Phase I Trial of Everolimus and Pazopanib. Cancer Discovery. 2014;4:546–553. doi: 10.1158/2159-8290.CD-13-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, Hirohashi S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy (vol 105, 13568, 2008) Proceedings of the National Academy of Sciences of the United States of America. 2009;106:10392–10393. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou JBA, Yu Q, Chng WJ. TXNIP (VDUP-1, TBP-2): A major redox regulator commonly suppressed in cancer by epigenetic mechanisms. International Journal of Biochemistry & Cell Biology. 2011;43:1668–1673. doi: 10.1016/j.biocel.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 41.Tontonoz P, Graves RA, Budavari AI, Erdjumentbromage H, Lui M, Hu E, Tempst P, Spiegelman BM. Adipocyte-Specific Transcription Factor-ARF6 Is A Heterodimeric Complex Of 2 Nuclear Hormone Receptors, PPAR-gamma And RXR-alpha. Nucleic Acids Research. 1994;22:5628–5634. doi: 10.1093/nar/22.25.5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor CF, Platt FM, Hurst CD, Thygesen HH, Knowles MA. Frequent inactivating mutations of STAG2 in bladder cancer are associated with low tumour grade and stage and inversely related to chromosomal copy number changes. Human Molecular Genetics. 2014;23:1964–1974. doi: 10.1093/hmg/ddt589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balbas-Martinez C, Sagrera A, Carrillo-de-Santa-Pau E, Earl J, Marquez M, Vazquez M, Lapi E, Castro-Giner F, Beltran S, Bayes M, Carrato A, Cigudosa JC, Dominguez O, Gut M, Herranz J, Juanpere N, Kogevinas M, Langa X, Lopez-Knowles E, Lorente JA, Lloreta J, Pisano DG, Richart L, Rico D, Salgado RN, Tardon A, Chanock S, Heath S, Valencia A, Losada A, Gut I, Malats N, Real FX. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nature Genetics. 2013;45:1464–U221. doi: 10.1038/ng.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solomon DA, Kim JS, Bondaruk J, Shariat SF, Wang ZF, Elkahloun AG, Ozawa T, Gerard J, Zhuang DZ, Zhang SZ, Navai N, Siefker-Radtke A, Phillips JJ, Robinson BD, Rubin MA, Volkmer B, Hautmann R, Kufer R, Hogendoorn PCW, Netto G, Theodorescu D, James CD, Czerniak B, Miettinen M, Waldman T. Frequent truncating mutations of STAG2 in bladder cancer. Nature Genetics. 2013;45:1428–U171. doi: 10.1038/ng.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lehmann AR. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes & Development. 2001;15:15–23. doi: 10.1101/gad.859501. [DOI] [PubMed] [Google Scholar]
- 46.Van Allen EM, Mouw KW, Kim P, Iyer G, Wagle N, Al-Ahmadie H, Zhu C, Ostrovnaya I, Kryukov GV, O’Connor KW, Sfakianos JP, Garcia-Grossman I, Kim J, Guancial EA, Bambury R, Bahl S, Gupta N, Farlow D, Qu A, Signoretti S, Barletta JA, Reuter V, Boehm J, Lawrence M, Getz G, Kantoff P, Bochner BH, Choueiri TK, Bajorin DF, Solit DB, Gabriel S, D’Andrea A, Garraway LA, Rosenberg JE. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014 doi: 10.1158/2159-8290.CD-14-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takata R, Katagiri T, Kanehira M, Tsunoda T, Shuin T, Miki T, Namiki M, Kohri K, Matsushita Y, Fujioka T, Nakamura Y. Predicting response to methotrexate, vinblastine, doxorubicin, and cisplatin neoadjuvant chemotherapy for bladder cancers through genome-wide gene expression profiling. Clin Cancer Res. 2005;11:2625–36. doi: 10.1158/1078-0432.CCR-04-1988. [DOI] [PubMed] [Google Scholar]
- 48.Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee IL, Melquist J, Bondaruk J, Majewski T, Zhang S, Pretzsch S, Baggerly K, Siefker-Radtke A, Czerniak B, Dinney CP, McConkey DJ. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25:152–65. doi: 10.1016/j.ccr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Damrauer JS, Hoadley KA, Chism DD, Fan C, Tiganelli CJ, Wobker SE, Yeh JJ, Milowsky MI, Iyer G, Parker JS, Kim WY. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A. 2014;111:3110–5. doi: 10.1073/pnas.1318376111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sjodahl G, Lauss M, Lovgren K, Chebil G, Gudjonsson S, Veerla S, Patschan O, Aine M, Ferno M, Ringner M, Mansson W, Liedberg F, Lindgren D, Hoglund M. A Molecular Taxonomy for Urothelial Carcinoma. Clinical Cancer Research. 2012;18:3377–3386. doi: 10.1158/1078-0432.CCR-12-0077-T. [DOI] [PubMed] [Google Scholar]
- 51.Sjodahl G, Lovgren K, Lauss M, Patschan O, Gudjonsson S, Chebil G, Aine M, Eriksson P, Mansson W, Lindgren D, Ferno M, Liedberg F, Hoglund M. Toward a molecular pathologic classification of urothelial carcinoma. Am J Pathol. 2013;183:681–91. doi: 10.1016/j.ajpath.2013.05.013. [DOI] [PubMed] [Google Scholar]