

Abstract
To contribute solutions for current energy concerns, improvements in the efficiency of C-H bond cleavage chemistry, e.g., selective oxidation of methane to methanol, could minimize losses in natural gas usage or produce feedstocks for fuels. Oxidative C-H activation is also a component of polysaccharide degradation, affording alternative biofuels from abundant biomass. Thus, an understanding of active-site chemistry in copper monooxygenases, those activating strong C-H bonds is briefly reviewed. Then, recent advances in the synthesis-generation and study of various copper-oxygen intermediates are highlighted. Of special interest are cupric-superoxide, Cu-hydroperoxo and Cu-oxy complexes. Such investigations can contribute to an enhanced future application of C-H oxidation or oxygenation processes using air, as concerning societal energy goals.
Introduction
In this article, we highlight certain copper-dioxygen (O2; molecular oxygen) chemistries, providing information or concepts derived from biochemical or synthetic bioinorganic systems. The emphasis is “oxygen activation” with Cu complexes, especially those containing a single copper ion site, and C-H bond oxidative cleavage in the context of reactions mediated by monooxygenases [1,2], cf., R–H + O2 + 2e− + 2H+ → R–OH + H2O, utilizing molecular oxygen as the ‘O’ atom source; the ‘other’ atom derived from O2 is released in the reduced form of water.
With the decades long soaring of energy demands, one envisions that finding efficient ways to improve methods to utilize current energy resources, or to develop new ones is an important societal goal. One of various targets of interest is to enhance the application of energy sources such as natural gas (i.e., methane; CH4). The development of new shale gas fracking technology has enabled the very recent steep growth in natural gas output. The efficient control of overflowed shale gas includes liquefaction of methane. Here, a key reaction would be the activation of the inert methane C–H bond (BDE = 104 kcal/mol-1). High temperature and pressure (800 °C, 40 atm) are required for the industrial partial oxidation of CH4 to produce methanol (CH3OH) [3]. This energy-dense liquid can be a fuel (e.g., in fuel cells or as an additive to blended gasoline) or feedstock. Methanol also provides advantages in its utilization, as there would be minimal loss in transport from a storage site.
Alternative energy resources include biofuels, available from currently plentiful and renewable biomass [4]. Abundant biopolymers such as cellulose, chitin, lignin and starches provide us with potential future sources of primary feedstock in the production of fuels [5]. Hydrolytic chemistry by enzymes or extreme chemical conditions, such as heating with sulfuric acid [6], can lead to useful breakdown products. Recent advances have revealed new classes of enzymes which effect biopolymer breakdown via oxidative chemistry.
In fact, for C–H oxidative cleavage of methane, biopolymers (vide supra), or other substrates, enzymes with copper active sites include lytic polysaccharide monooxygenases (LPMOs), peptidylglycine-α-hydroxylating monooxygenase (PHM), tyrosinases (phenol o-hydroxylases) and particulate methane monoxygenase (pMMO), which possess mononuclear (single Cu), non-coupled dicopper or adjacent dicopper active centers. The understanding of the formation, chemical-physical properties and reactivity of diverse biochemical or synthetic copper-oxygen adducts is of great interest. Here, we highlight the structures, functions and nature of copper centers in LPMOs, PHMs, and pMMOs. Highlighting of more recent efforts to generate synthetic analogs, notably those with single Cu active sites, as possibly relevant to monooxygenases will also be provided. If we can learn the manner in which biological systems efficiently activate C-H bond oxidation under ambient conditions, we have the potential to aid efforts to relieve our energy concerns by developing copper catalysts that can oxidatively activate strong C-H bonds.
Lytic Polysaccharide Monooxygenases (LPMOs)
LPMOs have aroused much attention due to their significant roles in potential biomass conversion via oxidative polysaccharide breakdown [5,7,8]. Several families of LPMOs have been categorized; cellulose-active fungal AA9, chitin-degradable bacterial AA10, and chitin-active fungal AA11 [5,7]. Most recently, other LPMOs are shown to be capable of degrading starch [9,10]. There are now many X-ray crystallographic studies showing that all LPMOs possess a mononuclear copper active site where binding to a chelating (bidentate) histidine amino terminus and a second His via εN imidazole ligation (Figure 1a) [7,11,12]. These three N-donors comprise a roughly T-shaped coordination (especially for reduced CuI) [13], with the chelating histidine referred to as a ‘His-brace’. In oxidized forms, additional waters coordinate and tyrosine or other aromatic amino-acids reside nearby. Subtle differences in the LPMO active-site structures occur. AA9 enzymes possess an εN-methylated His-brace, a highly unusual biochemical feature (Figure 1a). The type of copper-oxygen intermediate formed during catalysis is unknown, but it must be highly reactive in order to cleave the very strong sugar C–H bond (C1 or C4) (also see below).
Figure 1.
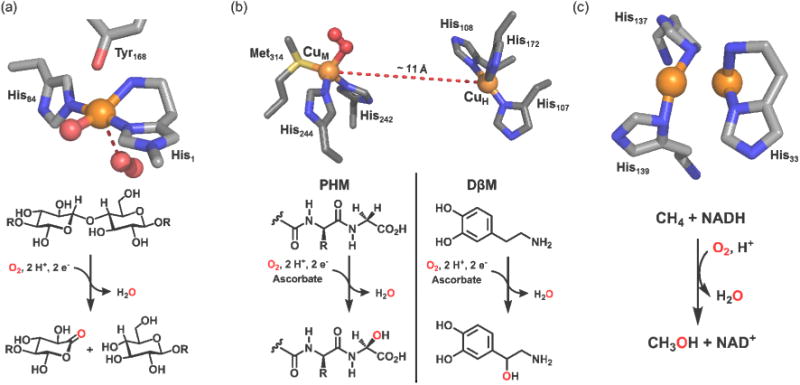
The X-ray crystal structures of copper active sites and roles of the monooxygenases involved in C-H bond activation; (a) LPMO: AA9 (PDB: 4EIR), (b) PHM (PDB: 1SDW), (c) pMMO (PDB: 1YEW).
Peptidylglycine-α-Hydroxylating Monooxygenase (PHM) and Dopamine-β-Monooxygenase (DβM)
These Cu monooxygenases mediate strong C-H bond activation; for PHM, a glycine-extended prohormone undergoes α-carbon hydroxylation while DβM catalyzes the benzylic hydroxylation of dopamine to give norepinephrine [14,15]. They possess a dicopper active site which is “noncoupled”; the two copper ions are about 11 Å apart (Figure 1b) [16]. One copper ion (Cuh) receives and passes reducing equivalents from ascorbate to CuM, where O2 and substrate binding occurs. An X-ray crystallographic study on a PHM derivative reveals a dioxygen-derived species at the CuM site, judged to be a CuI/O2 adduct, formally an end-on superoxide (O2•−) ligand-CuII complex (ES; Figure 2) [17]; Dioxygen binding to transition metals involves electron-transfer from metal ion to O2. Other biochemical and computational studies implicate a CuII-O2•− species formed in PHM which effects substrate hydrogen-atom transfer (HAT), eventually leading to product [14,15,18]. With these protein studies and more recent model compound work, fundamental studies on the physical properties and reactivity of mononuclear cupric superoxo species have thus been evoked (vide infra).
Figure 2.
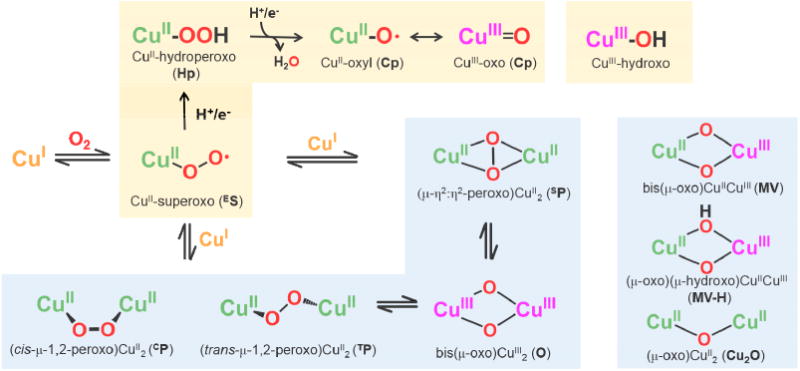
O2-derived copper species proposed as reactive intermediates mediating C-H bond activation experimentally or computationally.
Particulate Methane Monooxygenase (pMMO)
pMMO is found in bacterial methanotrophs, and if enough copper is biologically available, this copper-dependent membrane protein is employed to oxidize methane to methanol in order to acquire a carbon source [19,20]. X-ray crystallographic studies exhibit that pMMO possesses three different metal active sites; dicopper, monocopper, and a zinc center [21]. Biochemical studies implicate an asymmetrical dinuclear copper active site [22-24], although Chan et al. [25] hold the view that an as yet unseen pMMO trinuclear Cu center constitutes the active site. The dicopper site displays a short Cu–Cu distance, where the ligands are provided by two His's for one copper and the bidentate N-terminal histidine (‘His-brace’, as in LPMOs) for the other (Figure 1c).
Closer examination of the dicopper centers in X-ray structures (albeit the structures are not of high-resolution) reveals two very tilted Cu-NHis bonds (with the Cu-N vector reaching far out of the plane of the imidazole ring), not reasonable for inorganic coordination structures. As LPMO structures also having the His-brace are mononuclear in Cu, we (following others [26]) suggest the pMMO active site may possess only one copper, coordinated by the His-brace and one another His residue. Nevertheless, a dicopper center capable of methane or other alkyl C–H oxidations is worthy of studying, and there are many 2:1 Cu/O2 derived entities in synthetic chemistry (vide infra).
Synthetic Cu2/O2 Complexes and C-H Activation
As copper monooxygenases mediating C-H bond activation have different kinds of active sites (vide supra), it is critical to clarify fundamental aspects for the formation, structural/spectroscopic characteristics, and reactivity of the known CuI-O2 derived species, Figure 2.
Upon oxygenation of a reduced copper ion, an initial product is a mononuclear cupric superoxo (CuII-O2•−, ES) species. Over the years, species ES have been difficult to observe because the reactions of ES or “side-on” η2-bound forms (SS) with a second mole-equiv of the starting ligand Cu(I) complex are very fast and the binuclear dicopper complexes (μ-1,2-peroxo)CuII2 (“trans” TP; or a newer “cis” CP form [27]), (μ-η2:η2-peroxo)CuII2 (SP) and bis(μ-oxo)CuIII3 (O) (Figure 2) are stable if handled at cryogenic temperatures [28-32]. The dicopper(III) complex O seems to be most stable among the three, possibly due to the strong CuIII-oxo bonds, but the nature of the chelating ligand employed controls the chemistry and final structure. However, O is well known to attack exogenous substrates, including C–H bonds, in a wide variety of reactions as outlined by Stack, Tolman, Itoh and others [28,33-36].
As described, pMMO is well thought to effect dicopper methane oxygenation chemistry and O has been suggested as an intermediate which may form at the active site. Related species which are probably more reactive toward oxidative (oxygenative; overall O-atom insertion) chemistry involving very strong C–H bonds are bis(μ-oxo)CuIICuIII (MV) or (μ-oxo)(μ-hydroxo)CuIICuIII (MV-H) (Figure 2). MV is suggested by examination of the literature [23-25,37] or from calculations [38]; Yoshizawa's newer computational analysis [39] suggests further activation, i.e., implementation of enhanced reactivity toward CH4 as substrate, could come by starting with O and injecting a proton and an electron from a nearby tyrosine to give MV-H. However, neither MV nor MV-H species has ever been directly synthesized or identified. These remain as important synthetic targets.
Solomon, Schoonheydt and co-workers have shown that a (μ-oxo)CuII2 (Cu2O, Figure 2) attacks methane in Cu-loaded zeolites (Cu-ZSM-5) [40], producing CH3OH at low temperature (∼100 °C) with high selectivity. Some Cu2O synthetic complexes have been reported [41], but they have not yet been shown to undergo interesting C-H bond oxidation chemistry.
An important recent contribution from Stack and coworkers [36] establishes the chemistry of complexes O when ligated by a bidentate ligands where at least one donor is a primary amine (-NH2) (Figure 3). A combination of experimental and computational studies leads to conclusions that might not be expected – the smaller size of the –NH2 ligand (relative to – NR2) allows for tight/strong Cu-N bonding and achievement of Cu high-valency in O, without loss of oxidative power. Also, the small −NH2 ligand also allows a substrate closer approach to the O2-derived copper-oxygen species. This study thus provides possible insight into the presence of the protein terminal His residue as chelating ligand in pMMO and/or LPMOs (Figure 1), enzymes with very strong C–H bond containing substrates.
Figure 3.
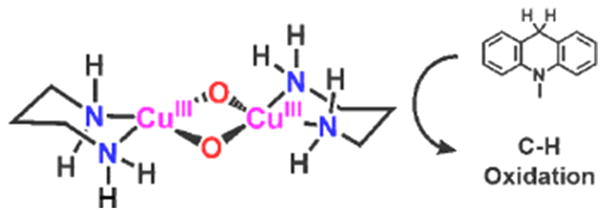
A high-valent dicopper(III) species O ligated by the primary amine, 1,3-propylenediamine, is capable of H-atom abstraction from the C–H bond in 9,10-dihydromethylacridine.
Primary CuI/O2 Adducts and Reactivity
As stated above, a CuII-O2•− (ES) species is the initial adduct produced from the internal electron transfer between a single ligand-CuI complex and O2. Then, sequential electron-proton addition from reductants (e.g., ascorbate) and solvent, or H-atom transfer from substrates (see below) to species ES may lead to a CuII-OOH species (Hp). Similar reduction-protonation accompanied by O-O bond homolysis should afford a cupric oxyl (Cp) (CuII-O• ↔ CuIII=O) species (Figure 2); the latter has only been detected in the gas phase [42-44].
Cupric superoxo species are important entities to study, see above [13,17,45]. In fact, previous work has led to X-ray structures (not shown) of one CuII-O2•− (ES) species with tris(tetramethylguanidino)tren [46], along with two structures [47,48] where the superoxo (or peroxo) [48] moiety is ligated in a “side-on” η2-binding fashion (SS), the latter exhibit limited or no exogenous substrate reactivity due to ligand imposed steric effects. The reactivity toward substrates where C–H oxygenation chemistry is achieved has been a critically lacking aspect of this subfield. However, recent newer ligand designs, e.g., with electron-releasing ligand substituents [49], or application of extreme cryogenic conditions (e.g., down to < 125 °C) [50], have enabled generation of new ES complexes which exhibit important reactivity (Figure 4).
Figure 4.
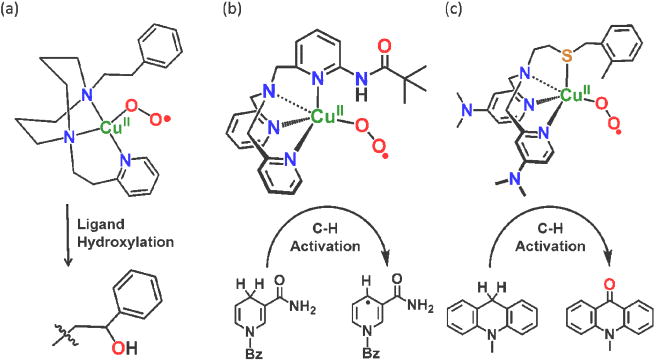
Spectroscopically characterized CuII(O2•−) species: (a) Itoh's ES complex effecting intramolecular benzylic oxygenation (net hydroxylation), (b) Hydrogen-bond stabilized ES complex effecting exogenous (weak) C-H bond HAT chemistry and (c) Thioether-ligated ES complex with enhanced reactivity, here, toward 9,10-dihydromethylacridine. Also, see text.
Itoh and co-workers [51,52] reported an exciting advance starting in 2009; their −80 °C stabilized ES complex with a tridentate alkylamino donor ligand, undergoes intramolecular benzylic C–H oxygenation (Figure 4a). Such reactivity is similar to that of DβM (Figure 1b), thus giving credence to the biochemical [15] and computational studies [18] suggesting that such a CuII-(O2•−) species ES (or SS) is responsible for the enzyme substrate HAT.
Karlin and co-workers [50] have also carried out detailed mechanistic examinations of ES species toward exogenous substrates with weak C-H bonds (Figure 4b). The ligand internal H-bonding group affords enough stability to generate the complex at – 125 °C, yet substrate H-atom abstraction occurs.
Another set of −80 °C stabilized ES complexes possess 4-dimethylamino [49] or 4-methoxy-pyridyl ligand donors [53]. For the latter, an in-depth mechanistic study was carried out employing phenolic substrates: the ES complex effects initial and rate-determining substrate O–H bond HAT, then the phenoxyl radical produced is attacked by a second ES complex, all leading to phenol oxygenation and p-benzoquinone formation [53].
Kim and co-workers recently employed a new thioether sulfur ligated (N3S) complex (Figure 4c) [54]. It exhibits enhanced reactivity compared to N4 ligated ES analogs. This work is a breakthrough in synthetic biomimetic research in that the complex mimics the CuM center of PHM (Figure 1b) and such studies should lead to a better understanding of the role of the PHM active site methionine ligand.
Mononuclear CuII-OOH (Hp) systems have been also generated and studied, again usually at low temperatures (−80 °C). They may derive from (i) addition of simple copper(II) complex precursors plus H2O2 in the presence of base [55,56], perhaps referred to as a shunt pathway (i.e., not derived from dioxygen chemistry), (ii) HAT from e-/H+ donors (e.g., phenols) to ES, or (iii) addition of 1.5 equiv H2O2 to a fully reduced CuI complex, possibly initially a Fenton chemistry type reaction [55]. In our hands, well-characterized Hp complexes may effect oxidative N-dealkylation chemistry (e.g., R2-NCH2R′ →→ R2NH + R′C(O)H), that analogous to PHM [57]. However, we found a recent case where the chemistry appears to proceed through Cu–O homolytic cleavage of CuII– OOH and subsequent site-specific Fenton chemistry derived from the Cu(I) thus produced reacting with the excess H2O2 present [58]. In general, CuII-OOH (Hp) appear experimentally [55,59] and computationally [60] to not be effective oxidants toward C–H HAT.
As mentioned, there is yet no direct evidence or characterization of a CuII-oxyl (Cp) complex, in solution chemistry. However, in one study, Itoh and co-workers [61] provided compelling evidence for the generation of Cp, produced from O-O homolysis of a CuII-OOR complex, leading to a substrate C-H oxygenation. In a DFT computational study, Beckham and co-workers [62] suggest that net C–H hydroxylation in fungal LPMOs may occur by generation of Cp (of course starting from CuI-O2 chemistry). In relevant studies, Tolman and co-workers [63] have been able to study a high-valent CuIII-OH species (Figure 2) (perhaps a protonated CuII-oxyl) which effects dihydroanthracene rate-limiting HAT oxidative chemistry.
Aqueous O2-Reduction, CuI/O2 Oxidase or Monooxygenase Chemistries
Aqueous reduction and protonation of molecular oxygen involve sequential electron-proton addition, as shown in Figure 5a; many relevant (de)protonation equilibria, not shown (e.g., HO2/O2•−; H2O2/HO2−), are also involved. Note that as O2 becomes further reduced, the products become stronger oxidants, such as superoxide, hydrogen peroxide (H2O2 + 2e− + 2H+ → 2H2O; E°= +1.35 V (pH = 7) vs. NHE; not shown), and hydroxyl radical (Figure 5a). In (bio)chemical systems, reduced iron or copper complexes ‘reductively activate’ O2 forming CuI/O2 derived species which themselves can continue to be reduced and protonated (Figure 5b) and/or they attack R–H substrates leading R-OH products (Figure 5c,d,e). Ground-state triplet dioxygen is essentially inert to direct reaction with organics (as ground state singlets) [64]. In all cases, the overall reactions are four-electron four-proton processes (Figure 5).
Figure 5.
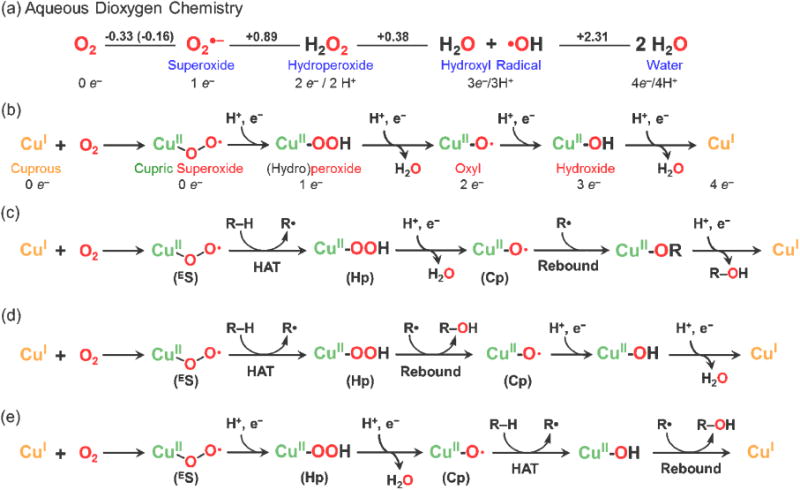
(a) Aqueous four-electron four-proton O2-reduction to water; reduction potentials (volts, pH 7). (b) Copper oxidase reactivity and intermediates relevant for single copper site chemistry. (c),(d),(e) Single-copper site stepwise monooxygenase chemistry. As must be the case, overall four-electron reduction chemistry occurs, where two derive from a substrate R–H bond; the other two electrons come from donors such as ascorbic acid. All O2-derived species, superoxo (ES), (hydro)peroxo (Hp) or cupryl (Cp), must be generated (in some form). The variations in chemistry, (c) vs (d) vs (e), are that for each, initial HAT chemistry with the substrate (R–H → R•) is effected by a different copper-oxygen intermediate, (ES), (Hp) or (Cp). Still, other pathways or mechanisms are possible. Also, see text.
Oxidase enzymes reduce O2 to water (or H2O2 in some cases) where the electrons come from reductants (e.g., cytochrome c in cytochrome c oxidase (CcO)) or from substrates which undergo dehydrogenations (providing protons + electrons; e.g., in ascorbate, diphenols) or multicopper oxidase enzymes such as laccase) [2]. Copper oxidases can be considered or compared to catalysts which mediate cathodic reactions in fuel cells [65], further making an understanding of their chemistry of great relevance in the field of energy. Figure 5b depicts single-Cu site oxidase chemistry, with the point being that here we find O2-derived copper-oxygen species which must form, i.e., they are critical to the whole process of capturing the energy stored in dioxygen when reduced to water, that being +0.82 V at pH = 7 [2].
However, what is critically lacking is our knowledge of the details depicted in Figure 5b. Future research is required to elucidate fundamental information such as reduction potentials (E° or E°′(pH = 7)), e.g., for CuII-O2•− (ES or SS) species, and their reduction to CuII-peroxo complexes. Alternatively, stepwise or proton-coupled reduction to CuII-OOH (Hp) may be preferred or required; aqueous superoxide anion (O2•−) cannot be reduced without the presence of a proton [64].
Thus, we also need to learn about the basicity of the superoxo complex O-atoms, since a proton is normally required to effect an electron-transfer reduction, but what acid strength (pKa) is needed? The same kind of information, reduction potentials and O-atom basicities, is needed for each subsequent step, CuII-OOH (Hp) + H+/e− → CuII-O• (Cp) + H2O, and Cp + H+/e− → CuII-OH (Figure 5b). The former step is the critically important reductive cleavage reaction, that important for oxidase (O2-reduction to water) and for substrate oxygenation (e.g., Figure 5c,d,e). Such chemistries are thus widely applicable to heme, non-heme iron, copper biochemistries and metal-mediated stoichiometric or catalytic substrate oxidation-oxygenation chemistries, thus important for practical and/or industrial situations.
In reality, the amount of basic/fundamental information needed is in fact vastly multiplied from what is discussed and illustrated here; (i) the environment around the metal (the ligand and second sphere) very much influences or even controls factors such as reduction potentials and M-(H)O2n− basicity, and (ii) all these chemistries need to be evaluated in more complex systems where 2 or 3 proximate copper ions constitute the catalyst center, as occurs in multi-copper oxidases or in CcO (with heme-Cu dimetal active site) [1,2]. (iii) Simultaneous multi-electron (i.e., 2e−) processes may occur [1], so different with what is represented in Figure 5. Thus, in practice, nature does not utilize mononuclear Cu centers to effect oxidase chemistry; however, an understanding of the Figure 5b chemistry is a critical start.
Figure 5c, d, and e outline pathways in which a monooxygenase may function, as possibly occurs at the PHM CuM active site. See earlier works in which such comparisons of pathways have been presented [66,67]. But which entity, ES, SS, Hp or Cp is the species that initially attacks the substrate to effect HAT from R–H, giving R•? Pathways c and d involve ES or SS substrate HAT, but they differ in that either ‘rebound’ of the O-atom to R• occurs at different stages. Klinman and co-workers prefer pathway c, but calculations from Chen & Solomon suggest pathway d [67]. Other mechanisms have also been suggested [67,68].
If, as strongly suggested, an ES or SS copper-superoxo intermediate effects substrate HAT in PHM, than this dramatically differs from the situation for cytochrome P-450 monooxygenase, the ‘classic’ system known for C–H oxygenation [69,70]. There, protons and electrons are stepwise added the ferrous-heme O2-adduct, to form an ironIV-porphrin-π-cation-radical (≡ FeV), that derived from heterolytic cleavage of an FeIII-OOH intermediate. Closest to that by analogy would be that illustrated by pathway Figure 5e. But there, homolytic O-O reductive cleavage occurs to give a CuII-oxyl species (Cp). If a proton plus protein (or ligand)-derived electron could be further provided, heterolysis could occur, giving a (X•+)CuII-oxyl intermediate (≡ CuIV=O formally) plus H2O [68].
Figure 5e, itself, represents the situation where CuI/O2 chemistry proceeds to a CuII–O• (Cp) species, the last reactive intermediate possible following reduction-protonation of O2 at a mononuclear Cu site. As mentioned, based on one recent computational study, it is pathway 5e and a Cp reactive intermediate which is relevant at polysaccharide monooxygenation at the active site of LPMOs [62].
Conclusion and Future Perspectives
The chemistry represented by Figure 5, with all the possible CuI/O2 derived reduced and/or protonated intermediates, is for coordination chemists or synthetic bioinorganic chemists, a vast field of research targets for study. The relevance to energy, including savings and increasing efficiency, is clear; the understanding of copper ion complexes as biochemical or synthetic entities able to catalyze oxidation reactions using readily available molecular oxygen is critically important. As described, precedence exists for copper mediated oxidation, i.e., C–H activation and net hydroxylation, of very tough substrates such as methane or polysaccharides. The understanding of such chemistries may also be applicable to the design of copper ion based fuel-cell catalysts. Elucidation of underlying principles and details of reduction, pronation, the timing of these preceding events, Cu-oxy species' (i.e., ES, SS, Hp, Cp, or even multicopper containing analogs) structure, spectroscopy and electronic-structure (bonding) and determination of reaction mechanisms is an important goal. Determination of reduction potentials for CuI/O2 derived reduced species (e.g., ES, SS, TP, SP, CP, O, Hp and Cp), and comparison in an absolute or relative sense to those of free O2-reduced species, is also of fundamental importance.
Some specific conclusions or perspectives are:
1. Energy – copper enzymes
Looking to the chemistry involved at copper enzyme active sites can lead to new insights and thus strategies to save energy via the development of new reagents or catalysts for (i) selective and efficient substrate oxidations-oxygenations, and (ii) fuel cell cathodic O2-reduction.New chemistries may in the future substitute for the activities of LPMOs, PHM, and pMMO. In addition to protein studies, establishing small molecule model systems and investigating their fundamental properties will be important. Which Cu-O2 intermediate is effective to carry out what chemistry?
2. Mixed-valent dicopper complexes
Although the dicopper complexes, TP, SP and O are well studied, further insights are still needed. However, it is especially important to target the generation and investigations of the chemistry of mixed-valent species like MV or MV-H. Also, can different sorts Cu2O species be discovered that will enable the oxidative attack of strong C-H bonds?
3. Cu-oxyl (Cp) species
This is surely a critical target for study, not only in how it forms (from CuII-OOH (HP)) but also as to what is its solution substrate reactivity profile. It is expected to be the most reactive, “hottest”, CuI-O2 derived species, and a Cp species is most likely important in LPMOs, or possibly in pMMO.
4. His-braced monocopper complex
As the His-brace copper ion ligation is critical in LPMOs and pMMO, elucidation of its effect on CuI/O2 chemistry is needed. Perhaps the recent study by Stack and co-workers [36] has already provided insight.
5. CuII-O2•− Complexes
Very recent breakthroughs in the generation and reactivity of such species indicate that this sub-field can be further explored. The relationship between copper-ligation and ES or SS reactivity requires elucidation. An intriguing point for consideration of monocopper O2-chemistry is that a new computational study [71] suggests that the reactive species in PHM is a [CuII-O2•−(H)]2+ entity, i.e., with a protonated superoxide. HO2 (aq) is a much stronger HAT reagent than is O2•− anion [64]. Even with the Lewis acidic Cu(II) ion present and ligated, perhaps [CuII-O2•−(H)]2+, as suggested [71], is required to effect HAT chemistry producing R• + CuII-OOH plus release of that “extra” proton.
6. Thioether-ligated CuII-O2•− species
In addition to the recently developed tetradentate sulfur-ligated N3S-ES complex, new reactive overall tridentate N2S-ES are targets for synthetic studies, in relationship to the active site in PHM.
7. CuII-OOH (Hp) reductive O–O cleavage
Detailed mechanistic investigation of this reaction, i.e., elucidating insights (into pKa of added acids, site of protonation, etc.) is not only critical to the field of CuI/O2 chemistry, but also for all fields and aspects of metal-O2 (bio)chemical reactivity. Such studies can of course also provide insights to the formation and chemistry of the cupryl CuII-O• (Cp) species, see above.
The field of biochemical and synthetic bioinorganic O2-activation by copper ion is exciting. While numerous insights have been obtained in the last 30 years, many challenges for both fundamental and practical chemistry applications (including to ‘energy’) remain.
Copper-oxygen complexes can effect difficult C–H oxidations in copper monooxygenases.
The advancement of Cu-oxygen reactivity can contribute to societal energy concerns.
Differing types of CuO2 and Cu2O2 complexes have been synthesized.
Copper-superoxo, Cu-hydroperoxo, and cupryl species are key reactive intermediates.
Copper-monooxygenase substrate likely proceed by varying mechanisms.
Acknowledgments
We are thankful to the National Institutes of Health (GM 28962, to K.D.K.) for continued research support in bioinorganic copper chemistry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1**.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Copper Active Sites in Biology. Chem Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. A recent authoritative and comprehensive review of copper in biology and associated copper ion chemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peterson RL, Kim S, Karlin KD. 3.07 - Copper Enzymes. In: Reedijk J, Poeppelmeier K, editors. Comprehensive Inorganic Chemistry II. Second. Vol. 3 Amsterdam: Oxford: Elsevier; 2013. pp. 149–177. http://dx.doi.org/110.1016/B1978-1010-1008-097774-097774.000309-097770. [Google Scholar]
- 3.Casey PS, McAllister T, Foger K. Selective Oxidation of Methane to Methanol at High Pressures. Industrial & Engineering Chemistry Research. 1994;33:1120–1125. [Google Scholar]
- 4.Chang MCY. Harnessing energy from plant biomass. Curr Opin Chem Biol. 2007;11:677–684. doi: 10.1016/j.cbpa.2007.08.039. [DOI] [PubMed] [Google Scholar]
- 5**.Beeson WT, Vu VV, Span EA, Phillips CM, Marletta MA. Cellulose Degradation by Polysaccharide Monooxygenases. Annual Review of Biochemistry. 2015 doi: 10.1146/annurev-biochem-060614-034439. In this latest review, the authors cover the current understanding of Cu-dependent LPMOs, including aspects of discovery, structure and function, and fuels from biomass. [DOI] [PubMed] [Google Scholar]
- 6.Nagasawa K, Tohira Y, Inoue Y, Tanoura N. Reaction between carbohydrates and sulfuric acid: Part I. Depolymerization and sulfation of polysaccharides by sulfuric acid. Carbohydrate Research. 1971;18:95–102. [Google Scholar]
- 7*.Hemsworth GR, Henrissat B, Davies GJ, Walton PH. Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat Chem Biol. 2014;10:122–126. doi: 10.1038/nchembio.1417. The authors report the discovery of new family of LPMOs, AA11. They show that the structure of the enzyme shares some of the features of the previous two classes of LPMOs, but they are distinctive in details and in their EPR spectroscopy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilmot CM. Polysaccharide Monoxygenases: Giving a Boost to Biofuel Production. Structure. 2012;20:938–940. doi: 10.1016/j.str.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Vu VV, Beeson WT, Span EA, Farquhar ER, Marletta MA. A family of starch-active polysaccharide monooxygenases. Proc Natl Acad Sci. 2014;111:13822–13827. doi: 10.1073/pnas.1408090111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo Leggio L, Simmons TJ, Poulsen JCN, Frandsen KEH, Hemsworth GR, Stringer MA, von Freiesleben P, Tovborg M, Johansen KS, De Maria L, et al. Structure and boosting activity of a starch-degrading lytic polysaccharide monooxygenase. Nat Commun. 2015;6 doi: 10.1038/ncomms6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gudmundsson M, Kim S, Wu M, Ishida T, Momeni MH, Vaaje-Kolstad G, Lundberg D, Royant A, Ståhlberg J, Eijsink VGH, et al. Structural and Electronic Snapshots during the Transition from a Cu(II) to Cu(I) Metal Center of a Lytic Polysaccharide Monooxygenase by X-ray Photoreduction. J Biol Chem. 2014;289:18782–18792. doi: 10.1074/jbc.M114.563494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Beeson WT, IV, Phillips CM, Marletta MA, Cate JHD. Structural Basis for Substrate Targeting and Catalysis by Fungal Polysaccharide Monooxygenases. Structure. 2012;20:1051–1061. doi: 10.1016/j.str.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13**.Kjaergaard CH, Qayyum MF, Wong SD, Xu F, Hemsworth GR, Walton DJ, Young NA, Davies GJ, Walton PH, Johansen KS, et al. Spectroscopic and computational insight into the activation of O2 by the mononuclear Cu center in polysaccharide monooxygenases. Proc Natl Acad Sci. 2014:8797–8802. doi: 10.1073/pnas.1408115111. To contribute to an understanding of LPMO reactivity, the authors determined active site solution structural features. With experiments in combination with computations, they demonstrate how the reduced copper(I) active site is capable of O2-activation via initial Cu(II)-superoxide species formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klinman JP. Mechanisms Whereby Mononuclear Copper Proteins Functionalize Organic Substrates. Chem Rev. 1996;96:2541–2561. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- 15.Klinman JP. The Copper-Enzyme Family of Dopamine beta-Monooxygenase and Peptidylglycine alpha-Hydroxylating Monooxygenase: Resolving the Chemical Pathway for Substrate Hydroxylation. J Biol Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 16.Prigge ST, Kolhekar A, Eipper BA, Mains RE, Amzel M. Amidation of Bioactive Peptides: The Structure Peptidylglycine a-Hydroxylating Monooxygenase. Science. 1997;278:1300–1305. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 17.Prigge ST, Eipper B, Mains R, Amzel LM. Dioxygen Binds End-On to Mononuclear Copper in a Precatalytic Enzyme Complex. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 18.Chen P, Solomon EI. Oxygen Activation by the Non-Coupled Binuclear Copper Site in Peptidylglycine a-Hydroxylating Monooxygenase. Reaction Mechanism and Role of Non-Coupled Nature of the Active Sites. J Am Chem Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 19.Tinberg CE, Lippard SJ. Dioxygen Activation in Soluble Methane Monooxygenase. Acc Chem Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Culpepper MA, Rosenzweig AC. Architecture and active site of particulate methane monooxygenase. Critical Reviews in Biochemistry and Molecular Biology. 2012;47:483–492. doi: 10.3109/10409238.2012.697865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieberman RL, Rosenzweig AC. Crystal structure of a membrane-bound metalloenzyme that catalyses the biological oxidation of methane. Nature. 2005;434:177–182. doi: 10.1038/nature03311. [DOI] [PubMed] [Google Scholar]
- 22.Himes RA, Karlin KD. Copper-dioxygen complex mediated C-H bond oxygenation: relevance for particulate methane monooxygenase (pMMO) Curr Opin Chem Biol. 2009;13:119–131. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Himes R, Barnese K, Karlin K. One is Lonely and Three is a Crowd: Two Coppers Are for Methane Oxidation. Angew Chem Intl Ed. 2010;49:6714–6716. doi: 10.1002/anie.201003403. [DOI] [PubMed] [Google Scholar]
- 24*.Culpepper MA, Cutsail GE, Iii, Gunderson WA, Hoffman BM, Rosenzweig AC. Identification of the Valence and Coordination Environment of the Particulate Methane Monooxygenase Copper Centers by Advanced EPR Characterization. J Am Chem Soc. 2014;136:11767–11775. doi: 10.1021/ja5053126. The authors identify coordination environment aspects of the Cu center in pMMO by employing ENDOR-ESEEM spectroscopic measurements, revealing a dicopper center in the form of a valence localized Cu(I)Cu(II) pair. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan SI, Yu SSF. Controlled Oxidation of Hydrocarbons by the Membrane-Bound Methane Monooxygenase: The Case for a Tricopper Cluster. Accounts Chem Res. 2008;41:969–979. doi: 10.1021/ar700277n. [DOI] [PubMed] [Google Scholar]
- 26.Quinlan RJ, Sweeney MD, Lo Leggio L, Otten H, Poulsen JCN, Johansen KS, Krogh KBRM, Jørgensen CI, Tovborg M, Anthonsen A, et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci. 2011;108:15079–15084. doi: 10.1073/pnas.1105776108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*.Dalle KE, Gruene T, Dechert S, Demeshko S, Meyer F. Weakly Coupled Biologically Relevant CuII2(μ-η1:η1-O2) cis-Peroxo Adduct that Binds Side-On to Additional Metal Ions. J Am Chem Soc. 2014;136:7428–7434. doi: 10.1021/ja5025047. Meyer and coworkers have generated and characterized, for the first time, a cis-peroxo-dicopper complex (CP) [DOI] [PubMed] [Google Scholar]
- 28.Que L, Jr, Tolman WB. Bis(m-oxo)dimetal “diamond” cores in copper and iron complexes relevant to biocatalysis. Angew Chem Int Ed. 2002;41:1114–1137. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 29.Mirica LM, Ottenwaelder X, Stack TDP. Structure and Spectroscopy of Copper-Dioxygen Complexes. Chem Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 30.Lucas HR, Li L, Sarjeant AAN, Vance MA, Solomon EI, Karlin KD. Toluene and Ethylbenzene Aliphatic C–H Bond Oxidations Initiated by a Dicopper(II)-mu-1,2-Peroxo Complex. J Am Chem Soc. 2009;131:3230–3245. doi: 10.1021/ja807081d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Würtele C, Sander O, Lutz V, Waitz T, Tuczek F, Schindler S. Aliphatic C-H Bond Oxidation of Toluene Using Copper Peroxo Complexes That Are Stable at Room Temperature. J Am Chem Soc. 2009;131:7544–7545. doi: 10.1021/ja902327s. [DOI] [PubMed] [Google Scholar]
- 32.Matsumoto T, Ohkubo K, Honda K, Yazawa A, Furutachi H, Fujinami S, Fukuzumi S, Suzuki M. Aliphatic C–H Bond Activation Initiated by a (mu-h2:h2-Peroxo)dicopper(II) Complex in Comparison with Cumylperoxyl Radical. J Am Chem Soc. 2009;131:9258–9267. doi: 10.1021/ja809822c. [DOI] [PubMed] [Google Scholar]
- 33.Itoh S, Nakao H, Berreau LM, Kondo T, Komatsu M, Fukuzumi S. Mechanistic studies of aliphatic ligand hydroxylation of a copper complex by dioxygen: A model reaction for copper monooxygenases. J Am Chem Soc. 1998;120:2890–2899. [Google Scholar]
- 34.Shearer J, Zhang CX, Zakharov LN, Rheingold AL, Karlin KD. Substrate oxidation by copper-dioxygen adducts: Mechanistic considerations. J Am Chem Soc. 2005;127:5469–5483. doi: 10.1021/ja045191a. [DOI] [PubMed] [Google Scholar]
- 35.Maiti D, Woertink JS, Narducci Sarjeant AA, Solomon EI, Karlin KD. Copper Dioxygen Adducts: Formation of Bis(mu-oxo)dicopper(III) versus (mu-1,2)Peroxodicopper(II) Complexes with Small Changes in One Pyridyl-Ligand Substituent. Inorg Chem. 2008;47:3787–3800. doi: 10.1021/ic702437c. [DOI] [PubMed] [Google Scholar]
- 36*.Citek C, Lin BL, Phelps TE, Wasinger EC, Stack TDP. Primary Amine Stabilization of a Dicopper(III) Bis(μ-oxo) Species: Modeling the Ligation in pMMO. J Am Chem Soc. 2014;136:14405–14408. doi: 10.1021/ja508630d. Stack and coworkers show that a -NH2 ligand, perhaps as part of ahistidine brace, can be effective in achieving the Cu(III) valency, also allowing for close approach of a substrate. [DOI] [PubMed] [Google Scholar]
- 37.Himes RA, Karlin KD. A new copper-oxo player in methane oxidation. Proc Natl Acad Sci USA. 2009;106:18877–18878. doi: 10.1073/pnas.0911413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiota Y, Yoshizawa K. Comparison of the Reactivity of Bis(mu-oxo)CuIICuIII and CuIIICuIII Species to Methane. Inorg Chem. 2009;48:838–845. doi: 10.1021/ic8003933. [DOI] [PubMed] [Google Scholar]
- 39.Shiota Y, Juhász G, Yoshizawa K. Role of Tyrosine Residue in Methane Activation at the Dicopper Site of Particulate Methane Monooxygenase: A Density Functional Theory Study. Inorg Chem. 2013;52:7907–7917. doi: 10.1021/ic400417d. [DOI] [PubMed] [Google Scholar]
- 40.Woertink JS, Smeets PJ, Groothaert MH, Vance MA, Sels BF, Schoonheydt RA, Solomon EI. A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol. Proc Natl Acad Sci USA. 2009;106:18908–18913. doi: 10.1073/pnas.0910461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haack P, Limberg C. Molecular CuII-O-CuII Complexes: Still Waters Run Deep. 2014;53:4293. doi: 10.1002/anie.201309505. [DOI] [PubMed] [Google Scholar]
- 42.Schröder D, Holthausen MC, Schwarz H. Radicallike activation of Alkanes by the ligated copper oxide cation (Phenanthroline)CuO+ J Phys Chem B. 2004;108:14407–14416. [Google Scholar]
- 43.Decker A, Solomon EI. Dioxygen activation by copper, heme and non-heme iron enzymes: comparison of electronic structures and reactivities. Curr Opin Chem Biol. 2005;9:152–163. doi: 10.1016/j.cbpa.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 44.Dietl N, Schlangen M, Schwarz H. Thermal Hydrogen-Atom Transfer from Methane: The Role of Radicals and Spin States in Oxo-Cluster Chemistry. Angew Chem Intl Ed. 2012;51:5544–5555. doi: 10.1002/anie.201108363. [DOI] [PubMed] [Google Scholar]
- 45.Itoh S. Mononuclear copper active-oxygen complexes. Curr Opin Chem Biol. 2006;10:115–122. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 46.Würtele C, Heinemann FW, Schindler S. Stabilization of a copper peroxido complex with a new binucleating ligand. J Coord Chem. 2010;63:2629–2641. [Google Scholar]
- 47.Fujisawa K, Tanaka M, Morooka Y, Kitajima N. A Monomeric Side-On Superoxocopper(II) Complex - Cu(O2)(Hb(3-tbu-5-iPrpz)3) J Am Chem Soc. 1994;116:12079–12080. [Google Scholar]
- 48.Aboelella NW, Lewis EA, Reynolds AM, Brennessel WW, Cramer CJ, Tolman WB. Snapshots of Dioxygen Activation by Copper: The Structure of a 1:1 Cu/O2 Adduct and Its Use in Syntheses of Asymmetric Bis(mu-oxo) Complexes. J Am Chem Soc. 2002;124:10660–10661. doi: 10.1021/ja027164v. [DOI] [PubMed] [Google Scholar]
- 49.Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD. A 1:1 Copper-Dioxygen Adduct is an End-on Bound Superoxo Copper(II) Complex which Undergoes Oxygenation Reactions with Phenols. J Am Chem Soc. 2007;129:264–265. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]
- 50.Peterson RL, Himes RA, Kotani H, Suenobu T, Tian L, Siegler MA, Solomon EI, Fukuzumi S, Karlin KD. Cupric Superoxo-Mediated Intermolecular C–H Activation Chemistry. J Am Chem Soc. 2011;133:1702–1705. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kunishita A, Kubo M, Sugimoto H, Ogura T, Sato K, Takui T, Itoh S. Mononuclear Copper(II)–Superoxo Complexes that Mimic the Structure and Reactivity of the Active Centers of PHM and DbM. J Am Chem Soc. 2009;131:2788–2789. doi: 10.1021/ja809464e. [DOI] [PubMed] [Google Scholar]
- 52.Kunishita A, Ertem MZ, Okubo Y, Tano T, Sugimoto H, Ohkubo K, Fujieda N, Fukuzumi S, Cramer CJ, Itoh S. Active Site Models for the CuA Site of Peptidylglycine alpha-Hydroxylating Monooxygenase and Dopamine beta-Monooxygenase. Inorg Chem. 2012;51:9465–9480. doi: 10.1021/ic301272h. [DOI] [PubMed] [Google Scholar]
- 53.Lee JY, Peterson RL, Ohkubo K, Garcia-Bosch I, Himes RA, Woertink J, Moore CD, Solomon EI, Fukuzumi S, Karlin KD. Mechanistic Insights into the Oxidation of Substituted Phenols via Hydrogen Atom Abstraction by a Cupric– Superoxo Complex. J Am Chem Soc. 2014;136:9925–9937. doi: 10.1021/ja503105b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim S, Lee JY, Cowley RE, Ginsbach JW, Siegler MA, Solomon EI, Karlin KD. A N3S(thioether)-Ligated CuII-Superoxo with Enhanced Reactivity. J Am Chem Soc. doi: 10.1021/ja511504n. just accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim S, Saracini C, Siegler MA, Drichko N, Karlin KD. Coordination Chemistry and Reactivity of a Cupric Hydroperoxide Species Featuring a Proximal H-Bonding Substituent. Inorg Chem. 2012;51:12603–12605. doi: 10.1021/ic302071e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kunishita A, Kubo M, Ishimaru H, Ogura T, Sugimoto H, Itoh S. H2O2-Reactivity of Copper(II) Complexes Supported by Tris[(pyridin-2-yl)methyl]amine Ligands with 6-Phenyl Substituents. Inorg Chem. 2008;47:12032–12039. doi: 10.1021/ic801568g. [DOI] [PubMed] [Google Scholar]
- 57.Maiti D, Narducci Sarjeant AA, Karlin KD. Copper–Hydroperoxo-Mediated N-Debenzylation Chemistry Mimicking Aspects of Copper Monooxygenases. Inorg Chem. 2008;47:8736–8747. doi: 10.1021/ic800617m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim S, Ginsbach JW, Lee JY, Peterson RL, Liu JJ, Siegler MA, Sarjeant AA, Solomon EI, Karlin KD. Amine Oxidative N-Dealkylation via Cupric Hydroperoxide Cu–OOH Homolytic Cleavage Followed by Site-Specific Fenton Chemistry. doi: 10.1021/ja508371q. just accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi YJ, Cho KB, Kubo M, Ogura T, Karlin KD, Cho J, Nam W. Spectroscopic and computational characterization of CuII-OOR (R = H or cumyl) complexes bearing a Me6-tren ligand. Dalton T. 2011;40:2234–2241. doi: 10.1039/c0dt01036g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen P, Fujisawa K, Solomon EI. Spectroscopic and Theoretical Studies of Mononuclear Copper(II) Alkyl- and Hydroperoxo Complexes: Electronic Structure Contributions to Reactivity. J Am Chem Soc. 2000;122:10177–10193. [Google Scholar]
- 61.Kunishita A, Ishimaru H, Nakashima S, Ogura T, Itoh S. Reactivity of Mononuclear Alkylperoxo Copper(II) Complex. O-O Bond Cleavage and C-H Bond Activation. J Am Chem Soc. 2008;130:4244–4245. doi: 10.1021/ja800443s. [DOI] [PubMed] [Google Scholar]
- 62*.Kim S, Ståhlberg J, Sandgren M, Paton RS, Beckham GT. Quantum mechanical calculations suggest that lytic polysaccharide monooxygenases use a copper-oxyl, oxygen-rebound mechanism. Proc Natl Acad Sci. 2014;111:149–154. doi: 10.1073/pnas.1316609111. Quantum mechanical calculations are utilized to predict that a Cu-oxyl species is the likely copper-oxygen intermediate responsible for polysaccharide degradation in LPMOs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63**.Dhar D, Tolman WB. Hydrogen Atom Abstraction from Hydrocarbons by a Copper(III)-Hydroxide Complex. J Am Chem Soc. 2015;137:1322–1329. doi: 10.1021/ja512014z. First reported in 2014, Dhar and Tolman provide details of the HAT chemistry observed for this complex, which may be considered as a protonated copper(II)-oxyl species. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Warren JJ, Tronic TA, Mayer JM. Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thorseth MA, Tornow CE, Tse ECM, Gewirth AA. Cu complexes that catalyze the oxygen reduction reaction. Coord Chem Rev. 2013;257:130–139. [Google Scholar]
- 66.Bollinger JJM, Krebs C. Enzymatic C-H activation by metal-superoxo intermediates. Curr Opin Chem Biol. 2007;11:151–158. doi: 10.1016/j.cbpa.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 67.de la Lande A, Parisel O, Gérard H, Moliner V, Reinaud O. Theoretical Exploration of the Oxidative Properties of a [(trenMe1)CuO2]+ Adduct Relevant to Copper Monooxygenase Enzymes: Insights into Competitive Dehydrogenation versus Hydroxylation Reaction Pathways. Chem – Eur J. 2008;14:6465–6473. doi: 10.1002/chem.200701595. [DOI] [PubMed] [Google Scholar]
- 68.Crespo A, Marti MA, Roitberg AE, Amzel LM, Estrin DA. The Catalytic Mechanism of Peptidylglycine alpha-Hydroxylating Monooxygenase Investigated by Computer Simulation. J Am Chem Soc. 2006;128:12817–12828. doi: 10.1021/ja062876x. [DOI] [PubMed] [Google Scholar]
- 69.Groves JT. Enzymatic C-H bond activation: Using push to get pull. Nat Chem. 2014;6:89–91. doi: 10.1038/nchem.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McQuarters AB, Wolf MW, Hunt AP, Lehnert N. 1958–2014: After 56 Years of Research, Cytochrome P450 Reactivity Is Finally Explained. Angew Chem Intl Ed. 2014;53:4750–4752. doi: 10.1002/anie.201402404. [DOI] [PubMed] [Google Scholar]
- 71.Abad E, Rommel JB, Kaestner J. Reaction Mechanism of the Bicopper Enzyme Peptidylglycine alpha-Hydroxylating Monooxygenase. J Biol Chem. 2014;289:13726–13738. doi: 10.1074/jbc.M114.558494. [DOI] [PMC free article] [PubMed] [Google Scholar]