

Summary
Mitochondrial diseases and aging are associated with defects in the oxidative phosphorylation machinery (OXPHOS), which are the only complexes composed of proteins encoded by separate genomes. To better understand genome coordination and OXPHOS recovery during mitochondrial dysfunction, we examined ATFS-1, a transcription factor that regulates mitochondria-to-nuclear communication during the mitochondrial UPR, via ChIP-sequencing. Surprisingly, in addition to regulating mitochondrial chaperone, OXPHOS complex assembly factor, and glycolysis genes, ATFS-1 bound directly to OXPHOS gene promoters in both the nuclear and mitochondrial genomes. Interestingly, atfs-1 was required to limit the accumulation of OXPHOS transcripts during mitochondrial stress, which required accumulation of ATFS-1 in the nucleus and mitochondria. Because balanced ATFS-1 accumulation promoted OXPHOS complex assembly and function, our data suggest that ATFS-1 stimulates respiratory recovery by fine-tuning OXPHOS expression to match the capacity of the suboptimal protein-folding environment in stressed mitochondria, while simultaneously increasing proteostasis capacity.
Introduction
Mitochondria generate ATP via chemical reactions mediated by the tricarboxylic acid (TCA) cycle and OXPHOS. The TCA cycle requires eight mitochondrial matrix-localized enzymes while OXPHOS requires the four respiratory chain complexes and the ATP synthase. The OXPHOS complexes are comprised of over 70 proteins that assemble into individual complexes on the mitochondrial inner membrane with the assistance of molecular chaperones and complex-specific assembly factors (Mimaki et al., 2012). Nuclear genes encode most subunits, but 13 are encoded by the mitochondrial genome (mtDNA) (Pagliarini et al., 2008) (12 in C. elegans (Bratic et al., 2010)). Therefore, expression from each genome must be coordinated to promote proper stoichiometry, efficient assembly and prevent the accumulation of deleterious orphaned subunits (Nolden et al., 2005; Rugarli and Langer, 2012).
Mitochondrial dysfunction occurs during aging as well as in a number of diseases, however the mechanisms a cell employs to stabilize defective mitochondria and regenerate functional OXPHOS complexes under such conditions are unclear (Rugarli and Langer, 2012; Vafai and Mootha, 2012). The UPRmt is a mitochondrial-specific stress response mediated by the bZip transcription factor ATFS-1, which is normally efficiently imported into mitochondria and degraded. However, during mitochondrial dysfunction, a percentage of ATFS-1 fails to be imported and accumulates in the cytosol. Because ATFS-1 has a nuclear localization sequence (NLS), it traffics to the nucleus and mediates the induction of protective genes including mitochondrial chaperones and proteases, antioxidant machinery as well as components of the glycolysis pathway (Nargund et al., 2012). These findings suggest that the UPRmt serves to stabilize the mitochondrial protein-folding environment and up-regulate a separate source of ATP production to promote survival and ultimately recover mitochondrial function (Haynes et al., 2013; Houtkooper et al., 2013; Nargund et al., 2012). However, the cellular processes affected by ATFS-1 and UPRmt activation have yet to be fully resolved.
Transcription of nuclear-encoded OXPHOS genes is regulated by multiple transcription factors in mammals including NRF-1, NRF-2, PPARα, the estrogen-related receptors and the transcriptional co-activator PGC-1 (Scarpulla et al., 2012; Schreiber et al., 2004; Wu et al., 1999). The remaining OXPHOS subunits are transcribed from mtDNA via a process that has been extensively characterized in mammalian systems and requires three nuclear-encoded components; the mitochondrial RNA polymerase (POLRMT), a transcription initiation factor mtTFB2/TFB2M, and TFAM which functions as a transcriptional activator as well as a mtDNA packaging protein (Bestwick and Shadel, 2013; Falkenberg et al., 2007).
Once transcribed, the OXPHOS subunits are translated by cytosolic or mitochondrial ribosomes and trafficked to the mitochondrial inner membrane where they assemble into stoichiometric complexes, which presents considerable challenges. For example, the individual OXPHOS components are highly expressed and require multiple assembly factors and chaperones to efficiently complete the assembly process. Orphaned subunits that fail to incorporate into functional complexes can be degraded by either the i-AAA or m-AAA proteases located in the mitochondrial inner membrane facing the intermembrane space and matrix, respectively (Leonhard et al., 2000; Rugarli and Langer, 2012). Interestingly, subunits of both protease complexes are induced by ATFS-1 during stress (Nargund et al., 2012), suggesting complex assembly is especially challenging during mitochondrial dysfunction.
To complement our previous mRNA profiling experiments, we performed chromatin immunoprecipitation coupled with DNA sequencing (ChIP-seq) to determine where in the genome ATFS-1 bound during mitochondrial stress. In addition to mitochondrial protective and glycolysis genes, ATFS-1 interacted with the promoters of TCA cycle and OXPHOS genes suggesting that ATFS-1 regulates multiple metabolic pathways. Interestingly, in addition to interacting with nuclear DNA, ATFS-1 also bound mtDNA. Surprisingly, we found that atfs-1 acts as a negative regulator of TCA cycle and OXPHOS transcript accumulation. And, balanced mitochondrial and nuclear accumulation of ATFS-1 is essential for coordinated OXPHOS gene expression, efficient OXPHOS complex assembly, respiration, and development during mitochondrial dysfunction. Thus, in addition to increasing mitochondrial protein folding and complex assembly capacity, our data suggest that ATFS-1 stimulates OXPHOS recovery by fine-tuning OXPHOS biogenesis rates to match the capacity of the suboptimal mitochondrial protein-folding environment.
Results
ATFS-1 interacts with the promoters of mitochondrial protective genes induced during mitochondrial stress
Our previous gene expression profiling studies identified 381 genes that were induced during mitochondrial stress dependent on atfs-1 (Nargund et al., 2012). To determine where in the genome ATFS-1 bound and potentially which genes were regulated by ATFS-1, we performed ChIP-seq. Mitochondrial stress was induced by RNAi knockdown of spg-7, a component of the m-AAA protease required for mitochondrial ribosome biogenesis and OXPHOS component quality control (Leonhard et al., 2000; Rugarli and Langer, 2012). Of the 381 genes transcriptionally induced by ATFS-1 during mitochondrial stress (Nargund et al., 2012), ATFS-1 interacted with 70 of the corresponding gene promoters (Figure 1A, Tables S1-S2), which included genes such as the mitochondrial chaperones mthsp-70 (hsp-6) (Figure 1B), dnj-10 (Figure S1A) and hsp-60 (Figure 2), the i-AAA protease ymel-1 (Figure 1C), and the glycolysis component gpd-2 (Figure 1D). ATFS-1 was also enriched at the promoters of many other genes required for mitochondrial function including the mitochondrial fission component drp-1 (Figure S1B) and components of the mitochondrial protein import machinery including timm-17 and timm-23 (Figures S1C-D). Of note, ATFS-1 did not bind or activate the promoter of the endoplasmic reticulum (ER) localized chaperone hsp-4 (Figures 1E and 1M) consistent with ATFS-1 regulating a mitochondrial-protective response (Nargund et al., 2012).
Fig. 1. ATFS-1 interacts with the promoters of mitochondrial protective genes induced during mitochondrial stress.

A. Venn diagram illustrating the genes induced in an atfs-1-dependent manner during mitochondrial stress (Nargund et al., 2012), and the gene promoters that ATFS-1 bound during mitochondrial stress identified by ChIP-seq, and the overlap.
B-E. ChIP-seq profiles of the mthsp-70, ymel-1, gpd-2 and hsp-4 promoters in wild-type worms raised on spg-7(RNAi), using ATFS-1 antibody (upper panel) or no antibody (lower panel). The y-axis is the number of sequence reads and the x-axis is approximately 2.5 kilobases with the start codon marked with an arrowhead.
F-I. ChIP of the promoters in Figures 1B-E in the presence or absence of ATFS-1 antibody from wild-type worms raised on control or spg-7(RNAi) as measured by qPCR (N = 3, ± SD, p* (student t-test) < 0.05).
J-M. Expression levels of mthsp-70, ymel-1, gpd-2 and hsp-4 mRNA in wild-type or atfs-1(tm4525) worms raised on control or spg-7(RNAi) determined by qRT-PCR (N = 3, ± SD, p* (student t-test) < 0.05) (right panels).
See also Figure S1 and Tables S1 and S2.
Fig. 2. ATFS-1 binds to the UPRmtE and induces mitochondrial chaperone transcription during stress.
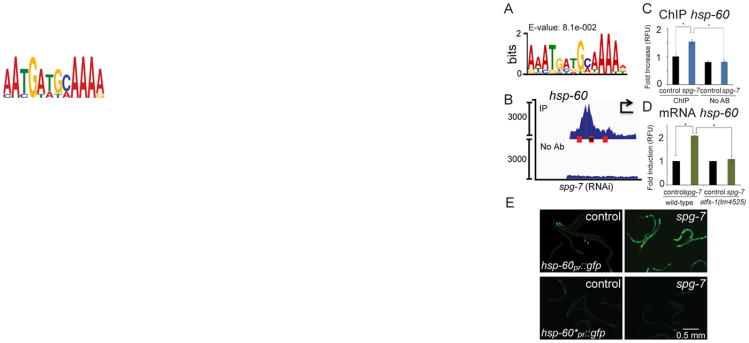
A. The UPRmtE consensus sequence.
B. ChIP-seq profile of the hsp-60 promoter in wild-type worms raised on spg-7(RNAi) using ATFS-1 antibody or no antibody. The three UPRmtEs within the promoter are marked with red boxes, and the element mutated in Figure 2E is marked with a black dot. The y-axis is the number of sequence reads and the x-axis is approximately 2.5 kilobases with the start codon marked with an arrowhead. Of note, the majority of the reads come from the multi-copy hsp-60pr∷gfp transgene likely accounting for the high number of reads and the sharp profile borders.
C. ChIP of the hsp-60 promoter in the presence or absence of ATFS-1 antibody from wild-type worms raised on control or spg-7(RNAi) as measured by qPCR (N = 3, ± SD, p* (student t-test) < 0.05).
D. Expression levels of hsp-60 mRNA in wild-type or atfs-1(tm4525) worms raised on control or spg-7(RNAi) determined by qRT-PCR (N = 3, ± SD, p* (student t-test) < 0.05).
E. Photomicrographs of hsp-60pr∷gfp (top panels) or hsp-60pr∷gfp lacking a UPRmtE (*) (bottom panels) transgenic worms raised on control or spg-7(RNAi). Scale bar, 0.5 mm.
See also Figure S2.
To validate ATFS-1 binding, ChIP followed by qPCR was performed to determine if ATFS-1 preferentially bound these promoters specifically during stress (Figures 1F-I). Consistent with ATFS-1 accumulating in the nucleus during mitochondrial stress (Nargund et al., 2012), ATFS-1 was enriched at the mthsp-70, ymel-1, and gpd-2 promoters only during mitochondrial dysfunction which coincided with increased transcripts, (Figures 1J-L), suggesting ATFS-1 directly regulates expression of these genes.
Alternatively, 311 genes were induced during mitochondria stress but were not bound by ATFS-1 (Table S1-S2), suggesting indirect regulation. For example, over twenty genes involved in reactive oxygen species (ROS) detoxification were induced in an atfs-1-dependent manner (Nargund et al., 2012), but ATFS-1 did not interact with any of the corresponding promoters (Table S1). However, during mitochondrial stress ATFS-1 did bind and activate the promoter of the gene encoding SKN-1, the transcription factor previously shown to directly regulate induction of many ROS-detoxifying genes (Figure S1E and (Oliveira et al., 2009)).
ATFS-1 binds a UPRmtE to induce mitochondrial protective gene transcription
To identify promoter regulatory sequences, or UPRmt elements (UPRmtEs), we performed motif analysis of the genomic regions to which ATFS-1 bound in the promoters of mitochondrial protective genes transcriptionally induced during mitochondrial stress. Four potential elements were identified, but we focused on a 14 base pair sequence (Figures 2A and S2A-D) that was the most enriched, suggesting a role for this sequence in ATFS-1-dependent gene regulation.
To examine the function of the putative UPRmtE, transgenic worms expressing GFP via the hsp-60 promoter were employed (Nargund et al., 2012). As expected, ATFS-1 interacted with the multi-copy transgenic promoter during mitochondrial stress as determined by ChIP-seq and ChIP-qPCR (Figures 2B-C). And, as previously documented, the wild-type promoter was activated, as indicated by hsp-60 mRNA induction and increased GFP fluorescence, when the worms were raised on spg-7(RNAi) (Figures 2D-E). However, deletion of one of the three UPRmtEs in the hsp-60 promoter impaired hsp-60pr∷gfp induction during stress (Figure 2E), similar to atfs-1-deletion. Of note, deletion of one UPRmtE impaired, but did not completely block, hsp-60pr∷gfp induction as longer exposures to mitochondrial stress resulted in modest hsp-60pr∷gfp induction (Figures 2E, S2E), consistent with all three UPRmtEs contributing to hsp-60 regulation. Combined, these data suggest that ATFS-1 binds to this 14 base pair sequence to regulate mitochondrial protective gene induction during mitochondrial stress. UPRmtEs were not present in the promoter of hsp-4, consistent with the ER chaperone not being induced during mitochondrial stress (Figure 1M).
ATFS-1 interacts with the promoters of genes in multiple metabolic pathways
In addition to those genes identified previously, ChIP-seq indicated that ATFS-1 bound to the promoters of 851 additional genes (Figure 1A), suggesting that ATFS-1 may regulate a much broader set of transcripts than previously appreciated (Nargund et al., 2012). The five most enriched pathways identified by Gene Enrichment and Functional Annotation tool provided by the Database of Annotation, Visualization, and Integrated discovery (DAVID) (Dennis et al., 2003), were glycolysis/gluconeogenesis, ribosome, oxidative phosphorylation, the tricarboxylic acid (TCA) cycle and autophagy (Figure 3A, Table S3) suggesting ATFS-1 modulates multiple metabolic pathways.
Fig. 3. ATFS-1 interacts with multiple metabolic gene promoters.
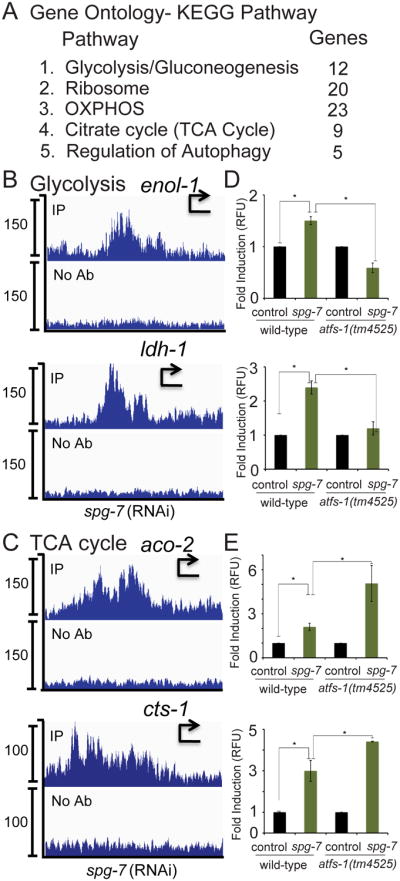
A. Gene ontology-KEGG Pathway analysis of the genes obtained by ATFS-1 ChIP-seq indicating the five most enriched pathways.
B-C. ChIP-seq profiles of representative genes in the glycolysis (B) and TCA cycle (C) pathways of wild-type worms raised on spg-7(RNAi), using ATFS-1 antibody or no antibody. The y-axis is the number of sequence reads and the x-axis is approximately 2.5 kilobases with the start codon marked with an arrowhead.
D-E. Expression levels of the glycolysis transcripts (D) enol-1 and ldh-1, and the TCA cycle transcripts (E) aco-2 and cts-1 in wild-type or atfs-1(tm4525) worms raised on control or spg-7(RNAi) determined by qRT-PCR (N = 3, ± SD, p* (student t-test) < 0.05).
See also Figure S3 and Table S3.
We next sought to determine if atfs-1 regulates transcription or transcript accumulation of the genes enriched in our ChIP-seq data. atfs-1-deletion did not affect the accumulation of the ribosomal or autophagy transcripts (Figures S3C-D). And as expected, atfs-1 was required for the induced expression of multiple glycolysis genes as well as lactate dehydrogenase during mitochondrial stress (Figures 3B and 3D). TCA cycle mRNAs were also induced during mitochondrial stress. But interestingly, they were further induced in atfs-1-deletion worms suggesting ATFS-1 negatively regulates TCA cycle transcript accumulation (Figure 3E).
Each of the five OXPHOS complexes consists of multiple subunits and all but the succinate dehydrogenase complex are composed of both nuclear and mtDNA-encoded components. ChIP-seq indicated that ATFS-1 bound to several promoters of genes that encode components of each complex including nuo-4 (NADH ubiquinone oxidoreductase, I), sdha-1 (succinate dehydrogenase, II), ucr-2.1 and cyc-2.1 (cytochrome c reductase, III), cco-1 (cytochrome c oxidase, IV) and atp-3 (ATP synthase, V) (Figures 4A-F, Table S1). Similar to the TCA cycle mRNAs, these OXPHOS transcripts were also increased in atfs-1-deletion worms during mitochondrial stress (Figures 4G-L), indicating atfs-1 also negatively regulates OXPHOS transcript accumulation.
Fig. 4. ATFS-1 limits the accumulation of nuclear-encoded OXPHOS transcripts.

A-F. ChIP-seq profiles of representative OXPHOS genes nuo-4, sdha-1, ucr-2.1, cyc-2.1, cco-1, and atp-3 of wild-type worms raised on spg-7(RNAi), using ATFS-1 antibody or no antibody. The y-axis is the number of sequence reads and the x-axis is approximately 2.5 kilobases with the start codon marked with an arrowhead.
G-L. Expression levels of nuo-4, sdha-1, ucr-2.1, cyc-2.1, cco-1, and atp-3 mRNA in wild-type or atfs-1(tm4525) worms raised on control or spg-7(RNAi) determined by qRT-PCR (N = 3, ± SD, p* (student t-test) < 0.05) (right panels).
See also Figure S4 and Table S4.
Interestingly, ATFS-1 did not interact with many OXPHOS gene promoters during mitochondrial stress including nuo-2, nuo-6, sdhb-1, isp-1, cyc-2.2, cco-2, atp-4, and atp-5 (Table S1). However, these transcripts were also further increased in atfs-1-deletion worms (Figures S4A-H) indicating that atfs-1 is also a negative regulator of these transcripts although the mechanism is likely indirect (see Discussion). We examined the promoters of TCA cycle and OXPHOS genes bound by ATFS-1 for potential regulatory sequences but were unable to identify any consensus elements consistent with binding by a bZip transcription factor (Figure S4I) (Buske et al., 2010).
spg-7(RNAi) was utilized throughout these studies to induce mitochondrial stress, however similar results were obtained using the clk-1(qm30) allele; a deletion that impairs ubiquinone biogenesis and previously demonstrated to activate the UPRmt, albeit to a lesser degree than spg-7(RNAi) (Baker et al., 2012; Nargund et al., 2012). In clk-1(qm30) worms, atfs-1(RNAi) decreased hsp-60 mRNA (Figure S4J) while causing an increase in nuclear-encoded OXPHOS transcripts (Figure S4K-L) indicating that ATFS-1 negatively regulates OXPHOS transcript levels during multiple forms of mitochondrial stress.
ATFS-1 associates with mtDNA during mitochondrial stress
ChIP-seq also revealed that during mitochondrial stress, ATFS-1 bound to the non-coding region of mtDNA (Figure 5A), suggesting a role for ATFS-1 in mtDNA regulation. Consistent with multiple copies of mtDNA per cell, the number of ChIP-seq reads in mtDNA was greater than all nuclear interactions (Figures 5A and 1B-D). Because mtDNA sequences also exist in the C. elegans nuclear genome, we eliminated those sequences acquired by ATFS-1 ChIP-seq that mapped to multiple regions of the genome. Even with the increased stringency, ATFS-1 binding was still significantly enriched in the mtDNA non-coding region (Figure S5A).
Fig. 5. ATFS-1 limits mtDNA-encoded transcript accumulation and directly binds to the non-coding region of mtDNA.

A. ChIP-seq profiles of the entire mitochondrial genome (mtDNA) of wild-type worms raised on spg-7(RNAi), using ATFS-1 antibody or no antibody. The y-axis is the number of sequence reads and the x-axis is the 13,794 nucleotide mitochondrial genome. The protein coding genes are marked with blue arrows, the rRNA genes with purple arrows, the non-coding region in green, and the putative UPRmtE is marked with a red box.
B. ChIP of the mtDNA non-coding region in the presence or absence of ATFS-1 antibody from wild-type worms raised on control or spg-7(RNAi) as measured by qPCR (N = 3, ± SD, p* (student t-test) < 0.05).
C. mtDNA non-coding region sequence with the putative UPRmtE in red.
D. EMSA using recombinant ATFS-1 and wild-type D-loop oligonucleotides or D-loop oligonucleotides with the 9 base pair UPRmtE scrambled. Unbound oligos (black square), ATFS-1-DNA complex (black arrow) and the loading well (gray arrow) are indicated.
E. Immunoblots of lysates from worms raised on control or spg-7(RNAi) following fractionation into total lysate (T), postmitochondrial supernatant (S) and mitochondrial pellet (M) (Nargund et al., 2012). Endogenous NDUFS3 serves as a mitochondrial marker and hsp-60pr∷gfp as a stress-induced cytosolic marker. Full-length (black arrow) and the mitochondrial forms (gray arrows) of ATFS-1 are indicated.
F. Relative mtDNA copy number in wild-type and atfs-1(tm4525) worms raised on control or spg-7(RNAi) as determined by qPCR (N = 3, ± SD, p* (student t-test) < 0.05).
G-I. Expression levels of the mtDNA-encoded mRNAs nd6, cox2 and atp6 in wild-type or atfs-1(tm4525) worms raised on control or spg-7(RNAi) determined by qRT-PCR (N = 3, ± SD, p* (student t-test) < 0.05).
J. Expression level of the mtDNA-encoded ctb-1 mRNA in wild-type or atfs-1(tm4525) worms expressing no transgenes (-) or transgenic wild-type ATFS-1, ATFS-1Δ1-32.myc or ATFS-1ΔNLS raised on control or spg-7(RNAi) determined by qRT-PCR (N = 3, ± SD, p* (student t-test) < 0.05).
K. Schematics of wild-type ATFS-1, ATFS-1 with an impaired mitochondrial targeting sequence (MTS) (ATFS-1Δ1-32.myc), and ATFS-1 with a mutated nuclear localization sequence (NLS) (ATFS-1ΔNLS).
L. Fluorescent photomicrographs of atfs-1(tm4525);hsp-60pr∷gfp worms expressing transgenic ATFS-1, ATFS-1Δ1-32.myc, or ATFS-1ΔNLS raised at 20°C. Scale bar, 0.5 mm.
See also Figure S5.
ATFS-1 binding to mtDNA was specifically enriched during mitochondrial stress (Figure 5B), which required ATFS-1-specific antibodies (Figure 5B) and was reduced in atfs-1(tm4525) worms (Figure S5B). While the regulatory segments of C. elegans mtDNA have not been extensively characterized, ATFS-1 binding was most enriched in an A-T-rich segment lacking protein or RNA coding genes (Figure 5A) suggested to be the regulatory region or D-loop (Bratic et al., 2010). Interestingly, this segment contained a UPRmtE in the region where ATFS-1 binding was most enriched (Figures 5A and 5C). Similar to the ChIP experiments, an electrophoretic mobility shift assay (EMSA) indicated that recombinant ATFS-1 bound to double stranded oligonucleotides identical to the C. elegans mtDNA non-coding region that contained the UPRmtE (Figure 5D and Table S7). Importantly, ATFS-1 binding was reduced when the UPRmtE was scrambled (Figure 5D), suggesting that ATFS-1 binds the mtDNA UPRmtE directly.
During mitochondrial stress, ATFS-1 import is impaired allowing a percentage of ATFS-1 to traffic to the nucleus (Nargund et al., 2012). However, ChIP-seq (Figure 5A) and ChIP-qPCR (Figures 5B and S5B) indicated that ATFS-1 also accumulates in mitochondria during stress. Consistent with this data, subcellular fractionation indicated that multiple isoforms or fragments of ATFS-1 accumulated in mitochondria during stress (Figure 5E), similar to our previous report (Nargund et al., 2012). The mechanism(s) that contribute to the increased accumulation of ATFS-1 within mitochondria during stress are not entirely clear. However, ATFS-1 interacted with the atfs-1 promoter and atfs-1 transcription was significantly induced (Figures S5C-E) consistent with an increase in ATFS-1 expression during mitochondrial stress (Figure 5E). Two splice variants of atfs-1 have been identified that encode proteins of 472 and 488 amino acids but with the same MTS (Figure S5F). Interestingly, only the shorter isoform was transcriptionally induced during mitochondrial stress (Figures S5G-H) suggesting this variant lacking a 48 base pair intron that encodes a 16 amino acid sequence may preferentially interact with mtDNA. Unfortunately, our ATFS-1 antibodies do not distinguish between the splice variants. Thus, we cannot exclude potential effects of altered ATFS-1 protein processing within mitochondria contributing to its stabilization.
atfs-1 represses the accumulation of mtDNA-encoded transcripts during the UPRmt
We next sought to determine if mitochondrial-localized ATFS-1 regulates mtDNA during mitochondrial dysfunction. mtDNA levels were not significantly altered during mitochondrial stress in wild-type or in atfs-1(tm4525) worms (Figure 5F), suggesting that ATFS-1 does not regulate mtDNA replication or turnover.
To determine if atfs-1 affects mtDNA-encoded transcripts, the steady state levels of multiple mtDNA-encoded mRNAs were examined in wild-type and atfs-1(tm4525) worms. Consistent with a compensatory response to regenerate OXPHOS complexes during mitochondrial dysfunction, mtDNA-encoded transcripts were modestly induced during stress (Figures 5G-J and S5I-K). However, in atfs-1-deletion worms, the mtDNA-encoded transcripts were further increased (Figures 5G-J and S5I-K) suggesting that atfs-1 negatively regulates the accumulation of mtDNA-encoded OXPHOS transcripts similar to the nuclear-encoded OXPHOS transcripts. All mtDNA-encoded transcripts examined were further increased in atfs-1-deletion worms consistent with increased transcription of a polycistronic transcript, however we cannot exclude the effects of atfs-1 on mRNA turnover. mtDNA-encoded transcripts were also increased when clk-1(qm30) worms were raised on atfs-1(RNAi) (Figures S5L-M). In sum, these results indicate that ATFS-1 accumulates within mitochondria and that atfs-1 is a negative regulator of mtDNA-encoded OXPHOS transcript accumulation.
Mitochondrial localization of ATFS-1 is required to limit the accumulation of mtDNA-encoded mRNAs during stress
Despite the accumulation of ATFS-1 within mitochondria and the interaction of ATFS-1 with mtDNA, it remained possible that an ATFS-1-induced nuclear-encoded protein mediated mtDNA-encoded transcript accumulation. We first considered the three nuclear-encoded genes required for mtDNA transcription (Bestwick and Shadel, 2013; Falkenberg et al., 2007). The mitochondrial RNA polymerase rpom-1 was induced in an atfs-1-dependent manner during stress while the TFAM (hmg-5) and TFB2M (tfbm-1) encoding genes were either not affected or not increased (Figures S5N-P), indicating that transcriptional induction of these genes in the absence of atfs-1 does not contribute to the increased mtDNA-encoded transcripts.
We next sought to determine if mitochondrial localization of ATFS-1 is required to regulate mtDNA-encoded transcript accumulation during stress. We utilized previously described atfs-1 deletion strains that express transgenic wild-type ATFS-1 or ATFS-1 lacking the MTS (ATFS-1Δ1-32.myc) (Figure 5K) (Nargund et al., 2012). Of note, while ATFS-1Δ1-32.myc cannot be imported into mitochondria it does accumulate within nuclei (Nargund et al., 2012) and induced nuclear transcription similar to that observed in wild-type worms during stress (Figure 5L).
As expected, mitochondrial stress caused a modest induction of the mtDNA-encoded complex III gene ctb-1, which was further increased in atfs-1(tm4525) worms (Figure 5J, columns 1-3). And, transgenic expression of wild-type ATFS-1 limited the accumulation of the mtDNA-encoded transcript (Figure 5J, column 4), similar to that observed in wild-type worms. Interestingly, expression of ATFS-1Δ1-32.myc, which cannot be imported into mitochondria, failed to limit the accumulation mtDNA-encoded mRNAs (Figure 5J, column 5) indicating that mitochondrial localization of ATFS-1 is required. Because ATFS-1Δ1-32.myc activates nuclear transcription (Figure 5L), a downstream nuclear-encoded target of ATFS-1 is not likely required for the negative regulation of mtDNA-encoded transcripts. Additionally, transgenic expression of ATFS-1ΔNLS, which has attenuated nuclear activity (Figure 5J, column 6) but still traffics to mitochondria (Nargund et al., 2012), also limited the accumulation of the mtDNA-encoded transcripts in atfs-1-deletion worms further demonstrating that the mitochondrial activity of ATFS-1 can be separated from its nuclear function. Thus, in addition to inducing transcription of genes that promote mitochondrial proteostasis, ATFS-1 also limits the accumulation of nuclear and mtDNA-encoded OXPHOS transcripts during stress.
ATFS-1 promotes OXPHOS complex assembly during mitochondrial dysfunction
In addition to mitochondrial molecular chaperones and proteases, which promote OXPHOS complex assembly (Mimaki et al., 2012 Rugarli and Langer, 2012), ATFS-1 also bound the promoters of OXPHOS complex assembly factors and iron-sulfur biogenesis components which are required for OXPHOS complex biogenesis (Ghezzi and Zeviani, 2012) (Table S1). Specifically, ATFS-1 bound the promoters of nuaf-1 and Y17G9B.5 (ECSIT), two NADH ubiquinone oxidoreductase assembly factors, and lpd-8 (NFU1), a Fe-S cluster biogenesis component (Figures 6A-C). These genes were induced during mitochondrial stress in an atfs-1-dependent manner (Figures 6D-F) suggesting a role for ATFS-1 in OXPHOS complex assembly.
Fig. 6. ATFS-1 promotes OXPHOS complex assembly during mitochondrial dysfunction.

A-C. ChIP-seq profiles of nuaf-1, lpd-8, and Y17G9B.5 of wild-type worms raised on spg-7(RNAi), using ATFS-1 antibody or no antibody. The y-axis is the number of sequence reads and the x-axis is approximately 2.5 kilobases with the start codon marked with an arrowhead.
D-F. Expression levels of nuaf-1, lpd-8, and Y17G9B.5 mRNA in wild-type or atfs-1(tm4525) worms raised on control or spg-7(RNAi) determined by qRT-PCR (N = 3, ± SD, p* (student t-test) < 0.05) (right panels).
G. SDS-PAGE immunoblots of extracts from wild-type and atfs-1(tm4525) worms raised on control or spg-7(RNAi). COX1 is a component of the cytochrome c oxidase complex (IV) and tubulin was used as a loading control.
H. SDS-PAGE immunoblots of extracts from wild-type and atfs-1(tm4525) worms raised on control or spg-7(RNAi) (upper three panels). The lower two panels are blue-native PAGE immunoblots of the same extracts. NDUFS3 is a component of the NADH ubiquinone oxidoreductase complex, ATP5A is an ATP synthase component and tubulin was used as a loading control.
I. Oxygen consumption in wild-type or atfs-1(tm4525) worms raised on control or spg-7(RNAi) (N = 3, ± SD, p* (student t-test) < 0.05).
We next examined the abundance of individual OXPHOS proteins as well as OXPHOS complexes in wild-type and atfs-1-deletion worms. We were unable to detect altered steady-state levels of a mtDNA-encoded protein during mitochondrial stress (Figure 6G) consistent with post-transcriptional regulation of OXPHOS components during mitochondrial dysfunction (Mick et al., 2011). However, the accumulation of two nuclear encoded OXPHOS proteins was increased during mitochondrial stress. Consistent with atfs-1 limiting OXPHOS mRNA accumulation (Figures 4 and 5), atfs-1(tm4525) worms expressed higher levels of NDUFS3 (complex I) and ATP5A (complex V) when raised on spg-7(RNAi) (Figure 6H, top three panels). Despite the increased nuclear and mtDNA-encoded OXPHOS mRNAs and nuclear-encoded protein components in atfs-1-deletion worms, assembly of complexes I and V was reduced, as determined by blue-native PAGE gels (Figure 6H, lower two panels). Consistent with reduced respiratory chain and ATP synthase assembly, atfs-1-deletion worms consumed less oxygen during mitochondrial stress (Figure 6I), suggesting ATFS-1 promotes OXPHOS.
Mitochondrial and nuclear accumulation of ATFS-1 promotes respiration and development during mitochondrial dysfunction
A challenge to determining the biological significance of atfs-1-dependent OXPHOS gene regulation is the difficulty in separating this activity of ATFS-1 from its role in promoting proteostasis by upregulating transcription of mitochondrial chaperone and assembly factors (Figures 1 and 6D-F). We utilized worms expressing ATFS-1Δ1-32myc, which regulates nuclear transcription similar to wild-type ATFS-1 during mitochondrial stress (Figure 7A), but does not traffic to mitochondria and cannot regulate mtDNA-encoded transcripts (Figure 5J). Interestingly, worms expressing ATFS-1Δ1-32myc had less assembled complexes than worms expressing wild-type ATFS-1 (Figure 7B) and consumed less oxygen (Figure 7C) during mitochondrial stress, demonstrating the importance of mitochondrial localized ATFS-1. As expected, atfs-1-deletion worms expressing ATFS-1ΔNLS displayed defects in OXPHOS complex assembly (Figure 7D) and had reduced oxygen consumption (Figure 7E), consistent with reduced nuclear activity (Figure 5L). Combined, these results indicate that like nuclear-localized ATFS-1, mitochondrial-localized ATFS-1 promotes OXPHOS complex assembly and function during mitochondrial dysfunction.
Fig. 7. Mitochondrial and nuclear accumulation of ATFS-1 promotes respiration and development during mitochondrial dysfunction.

A. Photomicrographs of wild-type hsp-60pr∷gfp and afts-1(tm4525);hsp-60pr∷gfp worms expressing transgenic ATFS-1, or ATFS-1Δ1-32myc raised on spg-7(RNAi). Scale bar, 0.5 mm.
B. SDS-PAGE immunoblots of extracts from atfs-1(tm4525) worms expressing transgenic ATFS-1, or ATFS-1Δ1-32myc raised on spg-7(RNAi) (upper three panels). The lower two panels are blue-native PAGE immunoblots of the same extracts.
C. Oxygen consumption in atfs-1(tm4525) worms expressing transgenic ATFS-1 or ATFS-1Δ1-32myc raised on spg-7(RNAi) (N = 3, ± SD, p* (student t-test) < 0.05).
D. SDS-PAGE immunoblots of extracts from atfs-1 (tm4525) worms expressing transgenic ATFS-1 or ATFS-1ΔNLS raised on spg-7(RNAi) (upper three panels). The lower two panels are blue-native PAGE immunoblots of the same extracts.
E. Oxygen consumption in atfs-1(tm4525) worms expressing transgenic ATFS-1 or ATFS-1ΔNLS raised on spg-7(RNAi) (N = 3, ± SD, p* (student t-test) < 0.05).
F. Representative photomicrographs of synchronized afts-1(tm4525);hsp-60pr∷gfp worms expressing transgenic ATFS-1, ATFS-1Δ1-32myc or ATFS-1ΔNLS allowed to develop for 72 hours on spg-7(RNAi). Note the increased progeny in the ATFS-1WT panel relative to the ATFS-1Δ1-32myc and ATFS-1ΔNLS panels. Scale bar, 0.5 mm.
G. Representative photomicrographs of synchronized wild-type or afts-1(tm4525) worms expressing transgenic ATFS-1 Δ1-32myc. Scale bar, 0.5 mm.
H. Signaling model depicting ATFS-1's function during mitochondrial stress.
See also Figure S6.
Furthermore, transgenic worms expressing ATFS-1Δ1-32myc developed slower than transgenic worms expressing wild-type ATFS-1 (Figure 7F). This developmental defect was partially rescued when ATFS-1Δ1-32myc was expressed in atfs-1 wild-type worms in which endogenous ATFS-1 accumulates in mitochondria (Figure 7G). Because ATFS-1Δ1-32myc expressing worms activate the nuclear response, but fail to limit mtDNA encoded transcripts (Figure 5J), these results indicate that ATFS-1's role in limiting mtDNA-encoded OXPHOS transcripts is a physiologically important activity of the UPRmt.
Discussion
The pathologies associated with mitochondrial dysfunction are numerous and include neurodegenerative (Rugarli and Langer, 2012) and mitochondrial diseases (Vafai and Mootha, 2012), bacterial infection (Pellegrino et al., 2014), as well as general aging in otherwise healthy individuals (Park and Larsson, 2011). However, the cellular mechanisms and pathways in place to survive mitochondrial stress and ultimately promote recovery are only beginning to be understood. Our data suggest that regenerating or rebuilding defective mitochondria is different and potentially more tightly coordinated than mitochondrial biogenesis in developing healthy cells. The current study provides a unique and unexpected role for ATFS-1 during mitochondrial stress and UPRmt activation. In addition to mediating the transcriptional induction of a mitochondrial protective response that increases the organelle's proteostasis capacity, ATFS-1 restricted expression of mitochondrial and nuclear-encoded OXPHOS transcripts, ultimately promoting respiratory recovery from mitochondrial stress, suggesting that ATFS-1 coordinates the rate of OXPHOS complex biogenesis to match the complex assembly capacity of the defected organelles.
Our studies evolved from a ChIP-seq experiment to determine where in the genome ATFS-1 bound during stress to complement our previous studies that identified a number of atfs-1-dependent genes induced during mitochondrial stress including mitochondrial proteostasis (Nargund et al., 2012). ChIP-seq indicated that ATFS-1 bound directly to the promoters of many of these genes during mitochondrial stress suggesting that induction is mediated by direct ATFS-1 binding. Importantly, the ChIP-seq analysis identified many additional genes potentially regulated by ATFS-1 including every glycolysis gene, as well as multiple TCA cycle and OXPHOS genes suggesting that during the UPRmt, ATFS-1 plays a role in metabolic regulation or adaptation to promote recovery from mitochondrial dysfunction. As described, we found that ATFS-1 was a positive regulator of glycolysis gene transcription during mitochondrial stress suggesting the UPRmt promotes an alternative form of ATP production (Nargund et al., 2012). Unlike glycolysis genes, TCA cycle and OXPHOS genes were further increased in worms lacking atfs-1 indicating atfs-1 is a negative regulator of TCA cycle and OXPHOS mRNA accumulation. atfs-1-deletion also resulted in a further increase of mtDNA-encoded transcripts indicating that atfs-1 also negatively regulates mtDNA transcript accumulation.
The identification of a nuclear transcription factor that also interacts with mtDNA is not unprecedented (Szczepanek et al., 2012). A recent analysis of multiple mammalian ChIP-seq datasets suggests that a number of nuclear transcription factors also bind mtDNA although the potential regulatory roles of each were not examined (Marinov et al., 2014). Additionally, the estrogen receptors alpha and beta have been found to accumulate in the nucleus and mitochondria and promote mitochondrial biogenesis (Chen et al., 2004). However, the role of the mitochondrial-localized estrogen receptor has proved difficult to elucidate (Klinge, 2008). Because ATFS-1 has clearly defined mitochondrial and nuclear localization sequences, we were able to separate the mitochondrial and nuclear activities of ATFS-1 to demonstrate the physiologic relevance of the mitochondrial-localized fraction. Worms expressing ATFS-1 lacking the MTS induce transcription of nuclear-encoded genes similar to wild-type worms during mitochondrial stress, but fail to limit the accumulation of mtDNA-encoded mRNAs. Interestingly, these worms have OXPHOS complex assembly defects, reduced oxygen consumption and are developmentally impaired indicating a protective role for the mitochondrial fraction of ATFS-1.
During stress, mitochondrial import defects cause ATFS-1 to traffic to the nucleus. A percentage of ATFS-1 also accumulates within mitochondria, binds mtDNA, and limits mtDNA-encoded mRNA accumulation although the mechanism is unclear. Intriguingly, ATFS-1 binds its own promoter, which coincides with increased transcription of an atfs-1 splice variant lacking a 16 amino acid sequence near the MTS suggesting the splice isoform may preferentially interact with mtDNA or regulate mtDNA-encoded transcripts. Alternatively, mitochondrial stress-dependent processing of ATFS-1 may contribute to its stability within mitochondria. However, inhibition of the Lon protease, which degrades ATFS-1 within mitochondria in the absence of stress, did not impair mtDNA-encoded transcript accumulation (Figures S6A-D). While our data are consistent with ATFS-1 impairing transcription, we cannot rule out effects of ATFS-1 on mitochondrial mRNA turnover.
The discovery that atfs-1 affects transcript levels of OXPHOS genes encoded by separate genomes provides an interesting perspective on mitochondrial adaptation during mitochondrial dysfunction. Maintaining and ultimately recovering mitochondrial function during mitochondrial stress is likely a complicated process requiring the coordination of multiple signaling pathways and cellular functions. While ATFS-1 antagonizes OXPHOS transcript accumulation during mitochondrial dysfunction, our data indicate that a separate stress induced compensatory pathway is activated to induce OXPHOS gene transcription (for example, Figures 3E, 4F, and 5G; lanes 1 and 2). The identification of this unknown transcription factor and the potential mechanism by which it is antagonized by ATFS-1 is currently under investigation.
What differentiates the positive regulation of mitochondrial chaperone, OXPHOS complex assembly factors, and glycolysis genes and the negative regulation of OXPHOS genes by ATFS-1 is unclear. Negative regulation of some nuclear-encoded OXPHOS genes by ATFS-1 involves direct promoter binding by ATFS-1 (Figure 4), but the promoters of these genes do not contain obvious UPRmtEs suggesting that ATFS-1 when bound as a repressor may have a distinct binding site. As ATFS-1 was the only bZip protein found to be required for the induction of mitochondrial chaperone genes, we hypothesize it functions as a homo-dimer (Haynes et al., 2010). However, bZip proteins can also form hetero-dimers to regulate transcription (Reinke et al., 2013). Because atfs-1 also negatively regulates OXPHOS transcripts that are not bound by ATFS-1 (Fig S4A-H), we favor a model where ATFS-1 forms a hetero-dimer with the stimulatory transcription factor impairing its activity. Of course, we cannot exclude the potential effects of chromatin structure or markings on OXPHOS transcript regulation by ATFS-1.
In sum, our data suggests that ATFS-1 optimizes OXPHOS complex assembly by at least two mechanisms (Figure 7H). Once activated, ATFS-1 traffics to the nucleus and activates transcription of a number of genes that promote mitochondrial proteostasis and OXPHOS complex assembly. Simultaneously, ATFS-1 coordinately reduces OXPHOS transcript accumulation from both genomes to match OXPHOS complex biogenesis rates to the protein folding and complex assembly capability of the defective organelles, ultimately promoting efficient respiratory recovery during mitochondrial dysfunction.
Experimental Procedures
Worm strains and plasmids
The reporter strain hsp-60pr∷gfp(zcIs9)V, the atfs-1 (tm4525) allele and he hsp-60pr∷gfp plasmid have been described previously (Baker et al., 2012; Nargund et al., 2012; Yoneda et al., 2004). To generate the hsp-60pr*∷gfp plasmid that lacks a UPRmtE, the ATGATGCAA sequence located 533 base pairs upstream of the start codon was mutated to GATATCCAA using standard molecular biology techniques. The hsp-60pr*∷gfp plasmid (5 ng/μl) was co-injected with a lin-15 rescuing plasmid and pBluescript (65 ng/μl) into lin-15(n765ts) worms generating multiple stable extra-chromosomal arrays. The plasmids expressing transgenic ATFS-1 have been described previously (Nargund et al., 2012).
Microscopy
C. elegans were imaged using a Zeiss AxioCam MRm mounted on a Zeiss Imager.Z2 microscope.
ChIP-Sequencing and Bioinformatics
ChIP assays were performed as described (Rechtsteiner et al., 2010). In brief, synchronized worms were raised in liquid under the described conditions and harvested at the L4 stage by sucrose flotation. Cross-linking of DNA and protein was performed by treating the worms with 11% formaldehyde at 4°C for one hour. The worms were then lysed via Teflon homogenizer and sonicated to obtain 500-1000 base pair DNA fragments. ATFS-1 was then immunoprecipitated from 500 μg of lysate using ATFS-1 antibodies (Nargund et al., 2012) and protein A sepharose beads (Invitrogen). The cross-links were reversed by incubation at 65°C for 5 hours. The DNA fragments were then ethanol-precipitated and purified using QiaQuick Spin Columns (Qiagen). The DNA fragments were sequenced using the Hi-Seq Illumina platform. The raw sequence data obtained by sequencing was first clipped for adapter sequences and then mapped to the C. elegans genome (ce6 from UC Santa Cruz) using BWA MEM (version 0.7.5). The output SAM files were processed and sorted with the Picard tools and then peaks were determined using MACS version 1.4 with the no-model parameter. Alternatively, the DNA fragments were quantified via qPCR using the promoter specific primers (Table S5).
To support the binding of ATFS-1 to mtDNA, additional analysis was performed to remove sequences that mapped to mtDNA as well as to the nuclear genome. The output mapping files (BAM files) were filtered with SAMtools to remove any read that had a mapping quality less than 10: SAMtools view −b −q 10 input.bam > output.bam.
To identify candidate UPRmtE motifs, several promoter regions that were enriched in the ChIP-seq using anti-ATFS-1 such as atfs-1, hsp-60, mthsp-70, dnj-10, tim-17, tim-23, ymel-1, drp-1, gpd-2, mff-2, ldh-1, dct-1, F56C11.3, T09F3.2, and F25B4.4 were examined using the MEME motif analysis program (http://meme.sdsc.edu) (Bailey et al., 2009). To identify motifs potentially involved in repressing nuclear-encoded TCA cycle and OXPHOS genes the promoters of the nuo-3, nuo-4, nduf-2.2, nduf-6, nduf-7, Y51H1A.3, sdha-1, cyc-2.1, cco-1, cco-2, atp-2, atp-3, H28O16.1, F58F12.1, aco-2, cts-1 and mdh-1 genes enriched in the ATFS-1 ChIP-seq were similarly examined using the MEME motif analysis program.
RNA isolation and quantitative RT-PCR
Total RNA was isolated using the RNA STAT reagent (Tel-Test) and treated with DNAse (Ambion). cDNA was then synthesized from total RNA using the iScript™ cDNA Synthesis Kit (Bio-Rad Laboratories). qPCR was used to determine the expression levels of the indicated genes using iQ™ sybr green supermix and MyiQ™2 Two-Color Real-Time PCR Detection System (Bio-Rad). Gene specific primers are listed in Table S6. Actin was used as a control. Fold changes in gene expression were calculated using the comparative CtΔΔCt method.
Mitochondrial genome quantitation
mtDNA quantitation was measured by qPCR as described (Bratic et al., 2009; Bratic et al., 2010).
Electrophoretic mobility shift assay (EMSA)
GST-ATFS-1 was expressed in E. coli (BL21) as described (Nargund et al, 2012). GST-ATFS-1 was affinity purified using GST-spin trap columns (GE Healthcare). The EMSA was performed as described (Sano et al, 2001). 3 μg of purified ATFS-1 was incubated with pre-annealed IRDye700 labeled oligonucleotides (Integrated DNA technologies) composed of the mtDNA non-coding region including the UPRmtE (Table S7). Identical duplexed DNA with a scrambled UPRmtE was used to verify the specificity of the DNA-protein complex. The mixture was then separated on a 4% native polyacrylamide gel and imaged using an Odyssey Infrared Imager (Li-Cor Biosciences). EMSA was performed multiple times.
Oxygen consumption
Oxygen consumption assays were performed as described (Haynes et al., 2010) using a Clark type electrode (Braeckman et al., 2002).
Protein analysis and antibodies
Whole worm lysates as well as cellular fractionation were performed as previously described (Nargund et al., 2012). Antibodies against α-tubulin were purchased from Calbiochem, NDUFS3 (nuo-2 in C. elegans, complex I) antibodies from MitoSciences, ATP5A (H28O16.1 in C. elegans, complex V) and COX1 (Anti-MTCO1) from Abcam. Immunoblots were visualized using an Odyssey Infrared Imager (Li-Cor Biosciences). All western blot experiments were performed multiple times.
Blue Native PAGE
Blue native PAGE was performed essentially as described (Pujol et al., 2013; van den Ecker et al., 2010). Briefly, wild-type and atfs-1(tm4525) worms were synchronized by bleaching, raised on either control or spg-7(RNAi) and harvested at the L4 stage. Worm lysates were obtained using a Teflon homogenizer. 50 μg of lysate were solubilized using 1% digitonin and subsequently, 25 μg of total protein was separated on 3-12% native gel (Native-PAGE Novex 3-12% gel from Invitrogen) using Xcell SureLock Mini Cell (Invitrogen). Western blots were performed rather than the standard coomassie staining as it was difficult to obtain high enough quantities of purified mitochondria from the spg-7(RNAi) treated atfs-1(tm4525) worms. For the western blots, the antibodies described above were used along with secondary antibodies conjugated to horseradish peroxidase.
Supplementary Material
Acknowledgments
We thank Nicholas Socci, Aleksandra Trifunovic, Susan Strome and Yi-Fan Lin for advice, the National BioResource Project and the Caenorhabditis Genetics Center for worm strains and the Genomics and Bioinformatics Facilities at Memorial Sloan Kettering Cancer Center. This work was supported by the Alfred W. Bressler Scholar Fund, the Ellison Medical Foundation, and NIH grants R01AG040061 (CMH) and NRSA F32GM102974 (AMN).
Footnotes
Accession Numbers: The NCBI Gene Expression Omnibus accession number fro the ChIP-seq data reported is GSE63803.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic acids research. 2009;37:W202–208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM, Nargund AM, Sun T, Haynes CM. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS genetics. 2012;8:e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestwick ML, Shadel GS. Accessorizing the human mitochondrial transcription machinery. Trends in biochemical sciences. 2013 doi: 10.1016/j.tibs.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braeckman BP, Houthoofd K, Vanfleteren JR. Assessing metabolic activity in aging Caenorhabditis elegans: concepts and controversies. Aging cell. 2002;1:82–88. doi: 10.1046/j.1474-9728.2002.00021.x. discussion 102-103. [DOI] [PubMed] [Google Scholar]
- Bratic I, Hench J, Henriksson J, Antebi A, Burglin TR, Trifunovic A. Mitochondrial DNA level, but not active replicase, is essential for Caenorhabditis elegans development. Nucleic acids research. 2009;37:1817–1828. doi: 10.1093/nar/gkp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratic I, Hench J, Trifunovic A. Caenorhabditis elegans as a model system for mtDNA replication defects. Methods. 2010;51:437–443. doi: 10.1016/j.ymeth.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Buske FA, Boden M, Bauer DC, Bailey TL. Assigning roles to DNA regulatory motifs using comparative genomics. Bioinformatics. 2010;26:860–866. doi: 10.1093/bioinformatics/btq049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JQ, Eshete M, Alworth WL, Yager JD. Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. Journal of cellular biochemistry. 2004;93:358–373. doi: 10.1002/jcb.20178. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome biology. 2003;4:3. [PubMed] [Google Scholar]
- Falkenberg M, Larsson NG, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annual review of biochemistry. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, Zeviani M. Assembly factors of human mitochondrial respiratory chain complexes: physiology and pathophysiology. Advances in experimental medicine and biology. 2012;748:65–106. doi: 10.1007/978-1-4614-3573-0_4. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Molecular cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge CM. Estrogenic control of mitochondrial function and biogenesis. Journal of cellular biochemistry. 2008;105:1342–1351. doi: 10.1002/jcb.21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhard K, Guiard B, Pellecchia G, Tzagoloff A, Neupert W, Langer T. Membrane protein degradation by AAA proteases in mitochondria: extraction of substrates from either membrane surface. Molecular cell. 2000;5:629–638. doi: 10.1016/s1097-2765(00)80242-7. [DOI] [PubMed] [Google Scholar]
- Mick DU, Fox TD, Rehling P. Inventory control: cytochrome c oxidase assembly regulates mitochondrial translation. Nature reviews Molecular cell biology. 2011;12:14–20. doi: 10.1038/nrm3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. Understanding mitochondrial complex I assembly in health and disease. Biochimica et biophysica acta. 2012;1817:851–862. doi: 10.1016/j.bbabio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123:277–289. doi: 10.1016/j.cell.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Oliveira RP, Porter Abate J, Dilks K, Landis J, Ashraf J, Murphy CT, Blackwell TK. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging cell. 2009;8:524–541. doi: 10.1111/j.1474-9726.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CB, Larsson NG. Mitochondrial DNA mutations in disease and aging. The Journal of cell biology. 2011;193:809–818. doi: 10.1083/jcb.201010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, Haynes CM. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature. 2014 doi: 10.1038/nature13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol C, Bratic-Hench I, Sumakovic M, Hench J, Mourier A, Baumann L, Pavlenko V, Trifunovic A. Succinate dehydrogenase upregulation destabilize complex I and limits the lifespan of gas-1 mutant. PloS one. 2013;8:e59493. doi: 10.1371/journal.pone.0059493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechtsteiner A, Ercan S, Takasaki T, Phippen TM, Egelhofer TA, Wang W, Kimura H, Lieb JD, Strome S. The histone H3K36 methyltransferase MES-4 acts epigenetically to transmit the memory of germline gene expression to progeny. PLoS genetics. 2010;6:e1001091. doi: 10.1371/journal.pgen.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke AW, Baek J, Ashenberg O, Keating AE. Networks of bZIP protein-protein interactions diversified over a billion years of evolution. Science. 2013;340:730–734. doi: 10.1126/science.1233465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. The EMBO journal. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Ohyama A, Takase K, Yamamoto M, Machida M. Electrophoretic mobility shift scanning using an automated infrared DNA sequencer. BioTechniques. 2001;31:1056–1058. 1060, 1062. doi: 10.2144/01315st06. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends in endocrinology and metabolism: TEM. 2012;23:459–466. doi: 10.1016/j.tem.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6472–6477. doi: 10.1073/pnas.0308686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanek K, Lesnefsky EJ, Larner AC. Multi-tasking: nuclear transcription factors with novel roles in the mitochondria. Trends in cell biology. 2012;22:429–437. doi: 10.1016/j.tcb.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- van den Ecker D, van den Brand MA, Bossinger O, Mayatepek E, Nijtmans LG, Distelmaier F. Blue native electrophoresis to study mitochondrial complex I in C. elegans. Analytical biochemistry. 2010;407:287–289. doi: 10.1016/j.ab.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. Journal of cell science. 2004;117:4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.