

Abstract
Triple-negative breast cancer (TNBC), the most aggressive breast cancer subtype, occurs in younger women and is associated with poor prognosis. Gain-of-function mutations in TP53 are a frequent occurrence in TNBC and have been demonstrated to repress apoptosis and up-regulate cell cycle progression. Even though TNBC responds to initial chemotherapy, resistance to chemotherapy develops and is a major clinical problem. Tumor recurrence eventually occurs and most patients die from their disease. An urgent need exists to identify molecular-targeted therapies that can enhance chemotherapy response. In the present study, we report that targeting PELP1, an oncogenic co-regulator molecule, could enhance the chemotherapeutic response of TNBC through the inhibition of cell cycle progression and activation of apoptosis. We demonstrate that PELP1 interacts with MTp53, regulates its recruitment, and alters epigenetic marks at the target gene promoters. PELP1 knockdown reduced MTp53 target gene expression, resulting in decreased cell survival and increased apoptosis upon genotoxic stress. Mechanistic studies revealed that PELP1 depletion contributes to increased stability of E2F1, a transcription factor that regulates both cell cycle and apoptosis in a context-dependent manner. Further, PELP1 regulates E2F1 stability in a KDM1A-dependent manner, and PELP1 phosphorylation at the S1033 residue plays an important role in mediating its oncogenic functions in TNBC cells. Accordingly, depletion of PELP1 increased the expression of E2F1 target genes and reduced TNBC cell survival in response to genotoxic agents. PELP1 phosphorylation was significantly greater in the TNBC tumors than in the other subtypes of breast cancer and in the normal tissues. These findings suggest that PELP1 is an important molecular target in TNBC, and that PELP1-targeted therapies may enhance response to chemotherapies.
Keywords: PELP1, p53, DNA damage response, oncogene, coregulator, E2F1, TNBC
Introduction
Approximately 30,000 women in the United States are diagnosed with triple-negative breast cancer (TNBC) annually. TNBC is characterized by the absence of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) (1). Therefore, TNBC patients do not benefit from commonly used anti-estrogen- and herceptin-based therapies, and TNBC is an independent risk factor for poor survival, recurrence, and distant metastases (2–4). Recent studies revealed that TNBC comprises a heterogeneous group of tumors encompassing several molecular subtypes such as basal-like, normal breast-like, and the claudin-low subtype (5). Approximately, 62% of the basal-like TNBC and 43% of the non-basal-like TNBC have mutations in the tumor suppressor p53 (MTp53) (6). The majority of the p53 mutations observed in tumor tissues are point mutations in the DNA-binding domain, which causes the mutants to lose their ability to bind p53 response elements and regulate the transcription of p53 target genes (7). However, mutations confer the p53 protein with oncogenic gain-of-functions via interactions with other transcription factors (8,9) and are shown to promote deregulation of cell cycle progression through its interaction with the NF-Y complex (10). Even though TNBC tumors initially respond to chemotherapy, resistance develops and recurrence eventually occurs, resulting in chemotherapy failure.
The transcriptional factor E2F1 can promote cell cycle progression or apoptosis in a context-dependent manner (11). E2F1 has been implicated in inducing apoptotic target gene expression in response to DNA damage (12,13). Various post-translational modifications, such as acetylation, phosphorylation, and methylation, regulate the stability of E2F1 protein and switch its activity in response to DNA damage (14–16). E2F1 promotes apoptosis through the activation of key target gene expression, such as p73, Bim, PUMA, and NOXA (15,17). In MTp53 TNBC cells, E2F1 plays a key role in regulating apoptosis in response to DNA damage (18). However, the mechanism(s) promoting E2F1-mediated apoptosis in TNBC remains elusive and is an active area of investigation.
Proline-, Glutamic acid-, and Leucine-rich Protein-1 (PELP1) is an oncogenic co-regulator molecule for many nuclear receptors (19,20), is overexpressed in many cancers including TNBC, and is an independent prognostic marker for poor survival (21). In ER+ breast cancer, PELP1 regulates the transcription of ER target genes, and promotes hormone-independent activation of ER target genes and therapy resistance (20). In TNBC, PELP1 plays important roles in regulating proliferation, epithelial-to-mesenchymal transition, and metastasis (22). PELP1 also functions as a co-regulator molecule for a number of other transcription factors such as E2F1 (23) and p53 (24). PELP1 is a substrate of the cyclin-dependent kinases, regulates E2F1 target gene expression, and promotes the G1-S transition during the cell cycle progression (23). Recent evidence also suggests that PELP1 is substrate of the DNA damage response kinases (DDR), and PELP1 regulates WTp53-mediated apoptosis in response to DNA damage (24). PELP1 phosphorylation at the S1033 residue by the DDR kinase is vital for its WTp53 co-regulator functions (24). However, it was unknown whether PELP1 regulates the MTp53 oncogenic functions.
In the present study, we discovered that PELP1 functions as a co-regulator for the MTp53 oncogenic functions. We found that PELP1 depletion in TNBC cells causes enhanced sensitivity to genotoxic stress. Mechanistic studies revealed that PELP1 interacts with MTp53 and E2F1, and differentially regulates their target gene expression. PELP1 phosphorylation was increased in TNBCs. These findings suggest that PELP1 is an important molecular target in TNBC, and that PELP1-targeted therapies may enhance a patient’s response to chemotherapy.
Materials and Methods
Cell lines and Reagents
TNBC model cells (MDA-MB-231 and MDA-MB-468) were purchased from the American-Type Culture Collection (ATCC) and maintained in RPMI 1640 and DMEM medium supplemented with 10% fetal bovine serum, respectively (Sigma-Aldrich, St. Louis, MO). HEK-293T cells were obtained from ATCC and maintained in DMEM supplemented with 10% fetal bovine serum. MDA-MB-231 cells expressing control- or PELP1-shRNA were generated as described earlier (22). Non-targeted and SMARTpool PELP1siRNA were obtained from Dharmacon Inc. (Lafayette, CO). The p53 (mouse monoclonal) and E2F1 mouse monoclonal antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The PELP1 antibody was purchased from Bethyl laboratories (Montgomery, TX). Cleaved Caspase-3, cleaved PARP-1, and p53 antibody was purchased from Cell Signaling Technologies (Beverly, MA). The phospho-PELP1 antibody was generated as described previously (24). GFP-TRAP beads were obtained from ChromoTek (Planegg-Martinsried, Germany). Cisplatin (cis-Diamineplatinum (II) dichloride), carboplatin, and camptothecin were purchased from Sigma-Aldrich (St. Louis, MO, USA). For the radiation treatment, the exponentially growing cells were exposed to various doses of 137Cs -rays at a dose rate of 0.93 Gy/min at room temperature using a Gamma Cell-40 irradiator (Atomic Energy of Canada Ltd, Montreal, Canada).
qRT-PCR
MDA-MB-231 cells expressing control- or PELP1-shRNA were treated with camptothecin or gamma irradiation. Total RNA from these cells was isolated using the Trizol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. cDNA synthesis was performed using Superscript III RT-PCR kit (Invitrogen, Carlsbad, CA). Real-time PCR was done using a Cepheid Smartcycler II (Sunnyvale, CA) or Illumina Eco Real-time PCR machine (San Diego, CA) with the specific real-time PCR primers for CyclinA2, Cdc25c, Bim, Noxa, p73, and Actin. The sequences of the primers are as follows: Actin: F-GTGGGCATGGGTCAGAAG, Actin: R-TCCATCACGATGCCAGTG, CDC25C: F-GTATCTGGGAGGACACATCCAGGG, Cdc25c: R-CAAGTTGGTAGCCTGTTGGTTTG, Cyclin A2: F-AGCAGCCTGCAAACTGCAAAGTTG, Cyclin A2: R-TGGTGGGTTGAGGAGAGAAACAC, Bim: F-TGGCAAAGCAACCTTCTGATG, Bim: R-GCAGGCTGCAATTGTCTACCT, Noxa: F-GCTGGAAGTCGAGTGTGCTA, Noxa:R-CCTGAGCAGAAGAGTTTGGA, p73: F-GGATTCCAGCATGGACGTCTT, p73: R- GCGCGGCTGCTCATCT. Results were normalized to actin transcript levels, and the difference in fold expression was calculated using delta-delta-CT method. Student’s t test was used to analyze all the qRT-PCR data using Graphpad PRISM software.
Immunoprecipitation (IP) and Chromatin Immunoprecipitation (ChIP)
For immunoprecipitation, total cellular lysates were prepared using NP40 lysis buffer (50 mM Tris-Hcl-pH7.5, 0.5 mM EDTA, 0.5% NP-40, 150 mM NaCl, 25 mM NaF, 0.1 mM sodium orthovanadate, and phosphatase and protease inhibitor cocktails). The lysates were diluted twofold using dilution buffer (50 mM Tris-Hcl-pH7.5, 150 mM NaCl, and 0.5 mM EDTA). Proteins were immunoprecipitated overnight using PELP1, p53, or the isotype-matched IgG antibody. Antibody complexes were pulled down using Protein-A Sepharose beads, and the immunoprecipitates were separated on an 8% SDS-PAGE gels. A GFP-TRAP pull-down assay was performed as described in the manufacturer’s protocol (ChromoTek, Munich Germany). ChIP and re-ChIP were done as described previously (25,26). Briefly, formaldehyde was added to the cells to crosslink the protein–DNA complexes, then the cell and nuclear membranes were disrupted and the DNA was fragmented by sonication. The lysates were subjected to ChIP using PELP1, p53, E2F1, H3K9Ac, or IgG antibodies. The protein–DNA complexes were reverse crosslinked followed by Proteinase K treatment. The immunoprecipitated DNA was quantified using real-time PCR, and the recruitment was calculated as percentage input. ChIP primer sequences: Cyclin A2: F-GCGCTTTCATTGGTCCATTT, Cyclin A2: R-GCGGCTGTTCTTGCAGTTC, Cdc25c: F-GAATGGACATCACTAGTAAGGCGCG, Cdc25c: R-GCAGGCGTTGACCATTCAAACCTTC, Bim: F-CTAAGGCTTGTGTCCGGAAG, Bim: R-CTTCGTCGGAGGATTTGAG. Statistical significance of ChIP data was done using student’s t test.
Western Blot Analysis
Cells expressing control- or PELP1-shRNA or PELP1-siRNA were treated with various DNA damaging agents. The cells were lysed using modified Radio-Immuno Precipitation Assay (RIPA) buffer (150 mM NaCl, 50 mM Tris-HCl, 50 mM NaF, 5 mM EDTA, 0.5% [wt/vol] sodium deoxycholate, and 1% Triton X-100) containing phosphatase and protease inhibitor cocktails. Equal amounts of protein were separated using 10% SDS-PAGE gels, and then transferred overnight to nitrocellulose membrane. Following the transfer, the membrane was blocked in 5% non-fat dry milk solution in TBST for 4 h. The membrane was then incubated with primary antibody, followed by secondary antibody, and developed using ECL kit (Thermo Fisher Scientific, Rockford, IL).
Cell Viability and Apoptosis Assays
Cells expressing control- or PELP1-shRNA or siRNA were plated at a density of 2×103 cells per well in a 96-well tissue culture plates and allowed to attach overnight. The cells were then treated with different doses of indicated drugs for 72 h, and cell viability was measured using CellTiter-Glo® luminescent cell viability assay (Promega, Madison, WI) as per manufacturer’s protocol. Statistical significance was analyzed using one-way ANOVA, followed by multiple comparisons using Graphpad PRISM software.
Tumor Tissue Microarray
Breast invasive ductal carcinoma tissue arrays that contain 75 cases/150 cores of breast cancer (BR-1504a TMAs) were purchased from US Biomax Inc (Rockville, MD). Twenty-eight TNBC tumor tissues with paired normal tissues were obtained in collaboration with UT Southwestern Medical Center (Dallas, TX). Immunohistochemistry was performed on the tumor array as described previously (25). Briefly, tumor sections were incubated overnight with the primary antibodies for phospho- Ser1033-PELP1 (1:250) or PELP1 (Bethyl laboratories, Montgomery, TX) (1:250) and then incubated with secondary antibody for 1 hour. The staining was visualized using the 3,3'-diaminobenzidine (DAB) substrate and counterstained with hematoxylin QS (Vector Lab, Burlingame, CA, USA). Allred scoring system was used to score the tumor staining. Statistical differences in the IHC among breast cancer subtypes were determined using Kruskal–Wallis test, followed by multiple comparisons using Dunn’s post test.
Results
PELP1 interacts with mutant p53 and regulates its target gene transcription
Recent studies demonstrated that PELP1 is a novel substrate of DDR kinases and functions as a coactivator for WTp53 and that PELP1-depleted cells exhibit resistance to apoptosis (24). PELP1 signaling plays a critical role in the proliferation of TNBC cells (22), and p53 is commonly mutated in TNBC tumors (6). We tested whether PELP1 signaling crosstalk occurs with MTp53 using two MTp53 TNBC cell lines, MDA-MB-231 and MDA-MB-468, which possess R280K and R273H mutations, respectively (27,28). To determine whether PELP1 interacts with MTp53, we treated MDA-MB-231 cells with camptothecin for 6 h and then immunoprecipitated the cells with control IgG, PELP1, or p53 antibodies. Western blot analyses of the immunoprecipitates revealed complex formation of endogenous PELP1 and MTp53 (Fig 1A). We validated the findings using a different MTp53 background cell line, MDA-MB-468 (Fig 1B). We further validated the PELP1–MTp53 interaction using exogenously expressed epitope-tagged GFP-p53MT and GST-PELP1 plasmids in 293T cells. After 48 h, the cells were treated with vehicle or camptothecin for 2 h, and the interactions were confirmed using GFP-TRAP immunoprecipitation. Western blot analyses of the immunoprecipitates revealed that GFP-MTp53, but not GFP, forms a complex with GST-PELP1 (Fig 1C).
Figure 1.
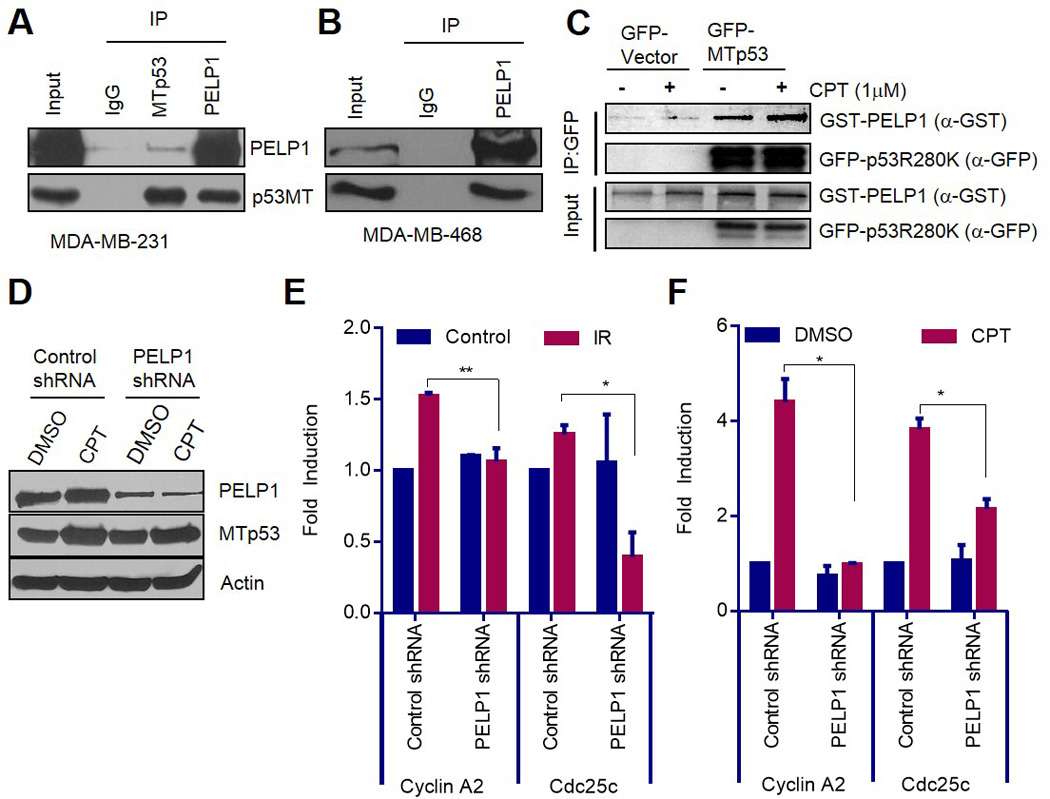
PELP1 interacts with MTp53 and regulates its target gene expression. (A, B) MDA-MB-231 cells (A) and MDA-MB-468 cells (B) were treated with 1µM of camptothecin for 6 h, and immunoprecipitation was done using IgG, PELP1, or p53 antibodies. Immunoprecipitates were subjected to Western blot analysis using PELP1 or p53 antibodies. (C) HEK-293T cells were transfected with GFP or GFP-MTp53 (R280K) along with GST-PELP1-expressing plasmids. After 48 h, cells were treated with 0.1% DMSO or 1µM of camptothecin for 2 h. GFP-trap IP and Western analysis was subsequently performed using GFP or GST antibodies. (D) MDA-MB-231 cells stably expressing control-shRNA or PELP1-shRNA were treated with 1 µM of camptothecin for 24 h and the expression of PELP1 and MTp53 was analyzed using Western analysis. (E, F) MDA-MB-231 cells were either exposed to 5gy gamma irradiation, and allowed to recover for 9 h (E) or treated with DMSO or 1µM of camptothecin for 24 h (F). cDNA synthesis and qRT-PCR analysis were subsequently performed to determine MTp53 target gene (Cdc25c and Cyclin A2) expression. *, P<0.05; **, P<0.01.
Hot-spot mutations in the DNA-binding domain of p53 render the apoptotic functions of the tumor suppressor dysfunctional. MTp53 acquires oncogenic gain-of-function that activates the transcription of transcription factor NF-Y-mediated G2/M genes (10). Since PELP1 interacted with MTp53, we determined whether PELP1 regulates transcription of MTp53 target genes. For this analysis, we have used MDA-MB-231 cells that stably express control- or PELP1-shRNA. PELP1-shRNA-expressing cells showed ~80% reduction in endogenous PELP1. Further, knockdown of PELP1 did not affect MTp53 protein levels compared to control-shRNA-expressing cells (Fig 1D). To determine whether PELP1 knockdown affects MTp53 target gene expression, control and PELP1-depleted MDA-MB-231 cells were exposed to gamma-radiation or camptothecin, and the transcription of the NF-Y target genes was analyzed using qRT-PCR. PELP1-depleted MDA-MB-231 cells had significant less expression of NF-Y target genes, Cyclin A2, and Cdc25c in response to genotoxic stress than the control-shRNA cells (Fig 1E, F). These results suggest that PELP1 has the potential to interact with MTp53, and that PELP1 participates in MTp53-mediated NF-Y target gene expression upon genotoxic stress.
PELP1 modulates MTp53 recruitment to target promoters and epigenetic changes
Next, we examined whether PELP1 regulates MTp53 target gene expression through its recruitment to the promoter region. ChIP analysis using IgG or PELP1 antibody in MDA-MB-231 cells after exposure to camptothecin revealed that PELP1 was recruited to the promoter region of Cdc25c and Cyclin A2 (Fig 2A). To examine whether PELP1 and MTp53 are co-recruited to the target gene promoters, we have performed sequential chip analysis. The sequential ChIP showed that PELP1 and MTp53 are co-recruited to the promoter region of Cdc25c and Cyclin A2 (Fig 2B). Further, MTp53 recruitment to the promoter region of the NF-Y target genes was significantly reduced in PELP1-shRNA-expressing cells in response to genotoxic stress compared to the control-shRNA-expressing cells (Fig 2C). Earlier studies showed that PELP1 regulates target gene transcription through the recruitment of histone-modifying enzymes, such as Histone Acetyl Transferases (HATs), that cause the acetylation of histone tails, leading to gene transcription (29,30). Since PELP1 depletion reduced MTp53 recruitment to the target gene promoter, we examined whether PELP1 depletion causes a concurrent decrease in active histone marks in the promoter regions. Upon genotoxic stress, a significant reduction in the H3K9Ac mark occurred in the promoter regions of Cyclin A2 and Cdc25c in the PELP1-depleted cells than in the control cells (Fig 2D). Collectively, these results suggest that PELP1 plays an important role in the modulation of MTp53 functions by regulating its recruitment to the promoter of target genes and by promoting active histone modifications at MTp53 target genes.
Figure 2.
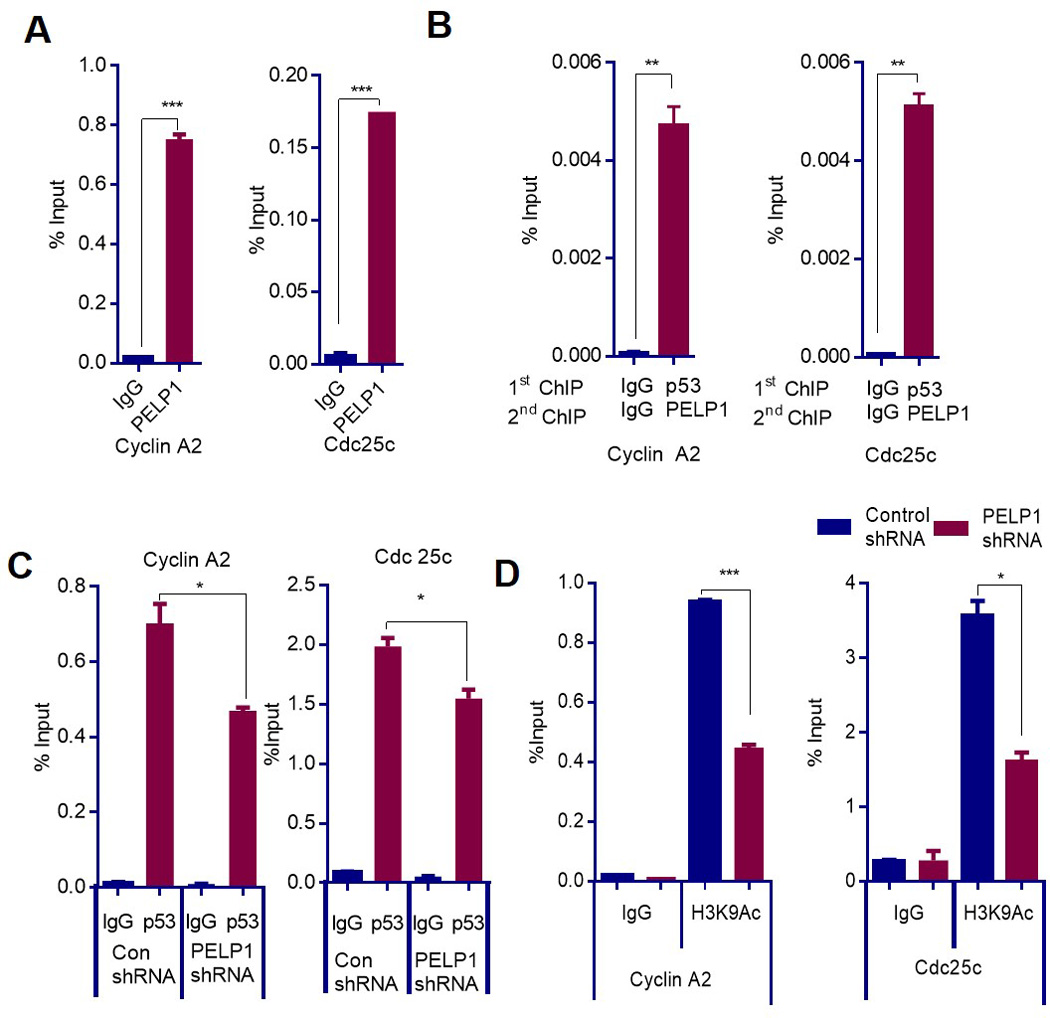
PELP1 recruits and regulates histone modifications on MTp53 target gene promoters. (A) MDA-MB-231 cells were treated with camptothecin for 6 h and were subjected to ChIP assay using the PELP1 antibody. PELP1 recruitment was analyzed by qRT-PCR with primers specific for NF-Y binding elements on the Cdc25c and Cyclin A2 promoters. *** P<0.001. (B) Sequential ChIP was performed on MDA-MB-231 cells that were treated with camptothecin for 6 h. Initial ChIP was performed using IgG or p53 antibody followed by re-ChIP using IgG or PELP1. qRT-PCR was performed in duplicates with primers specific for Cyclin A2 and Cdc25c. **P < 0.01. (C) MDA-MB-231 cells expressing control- or PELP1-shRNA were treated with camptothecin for 6 h and were subjected to ChIP assay using p53 antibody. Recruitment of MTp53 was analyzed using qRT-PCR with primers specific for NF-Y binding elements on the Cdc25c and Cyclin A2 promoters. *, P<0.05; (D) MDA-MB-231 cells expressing control- or PELP1-shRNA were treated with camptothecin for 6 h, chromatin was isolated and subjected to ChIP assay using the H3K9Ac antibody. The status of the H3K9Ac mark was analyzed using qRT-PCR with primers specific for NF-Y binding elements on the Cdc25c and Cyclin A2 promoters.*, P<0.05; *** P<0.001.
PELP1-depleted TNBC cells exhibit increased sensitivity to genotoxic stress
p53 gain-of-function mutations deregulate the cell’s response to genotoxic stress by enhancing the expression of cell cycle genes despite the presence of DNA damage. Knockdown of MTp53 in cancer cells reduces cell proliferation and tumorigenicity in vitro and in vivo (31,32). Since PELP1 functions as a coactivator of MTp53 oncogenic functions, we examined whether PELP1 depletion contributes to the increased sensitivity to DNA damaging agents. To test this hypothesis, we used MDA-MB-231 cells that stably express control- or PELP1-shRNA. The cells were exposed to various doses of camptothecin (Fig 3A) or carboplatin (Fig 3B) or cisplatin (Fig 3C) for 72 h, and the cell survival percentage was determined. PELP1 depletion significantly enhanced the sensitivity to chemotherapeutic drugs. Similar results were obtained with MDA-MB-468 cells that were transiently transfected with control- or PELP1-siRNA followed by treatment with different doses of camptothecin (Fig 3D). Overall, these results suggest that PELP1 depletion could cause an increased sensitivity of TNBC cells to chemotherapeutics.
Figure 3.
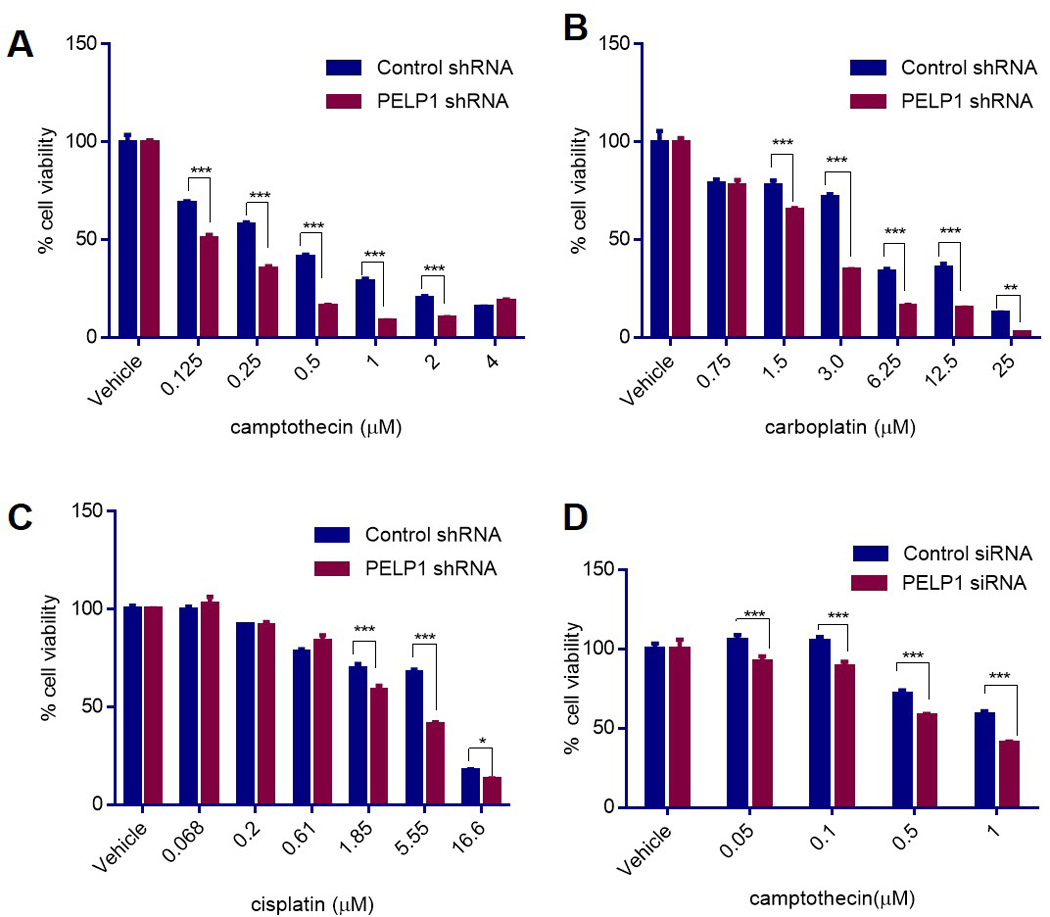
PELP1-depleted TNBC cells exhibit increased sensitivity to genotoxic stress. (A, B, C) MDA-MB-231 cells expressing control- or PELP1-shRNA were treated with DMSO or with different doses of (A) camptothecin, (B) carboplatin, or (C) cisplatin for 72 h and then analyzed for survival. *, P<0.05; **, P<0.01; *** P<0.001. (D) MDA-MB-468 cells transfected with control- or PELP1-siRNA were treated with DMSO or different doses of camptothecin for 72 h and then analyzed for survival as described in Methods. *** P<0.001
PELP1 interacts with E2F1 and regulates E2F1-mediated apoptosis
Next, we determined whether the reduced survival in PELP1-depleted TNBC cells was due to an increase in apoptosis. Western analyses indicated that PELP1 knockdown cells had more cleaved caspase-3 and cleaved PARP-1 than the control cells upon treatment with camptothecin (Fig 4A). Since one of the key mediators of apoptosis, p53, is mutated in the TNBC cell lines that we used in our assays, we hypothesized that PELP1 depletion could cause an increase in apoptosis through the E2F1 pathway. Earlier studies showed that E2F1 can promote cell cycle progression or apoptosis in a context-dependent manner (11). Similarly, PELP1 has been shown to regulate E2F1-mediated cell cycle progression (23); however, whether PELP1 regulates E2F1 activities in response to genotoxic stress remains unknown. First, we determined whether PELP1 knockdown alters E2F1 target genes that are considered critical for apoptosis such as p73 and Bim (15,17). MDA-MB-231 cells expressing control- or PELP1-shRNA were treated with camptothecin for 24 h, and then analyzed for p73, Bim, and Noxa using qRT-PCR. Compared to the control cells, PELP1-depleted cells expressed significantly higher levels of the apoptotic target genes of E2F1 when treated with camptothecin (Fig 4B). PELP1 knockdown significantly increased the recruitment of E2F1 to the target promoter upon genotoxic stress (Fig 4C). Overall, these results suggest that PELP1 depletion can increase apoptosis through the activation of the E2F1 pathway in MTp53-expressing TNBC cells.
Figure 4.
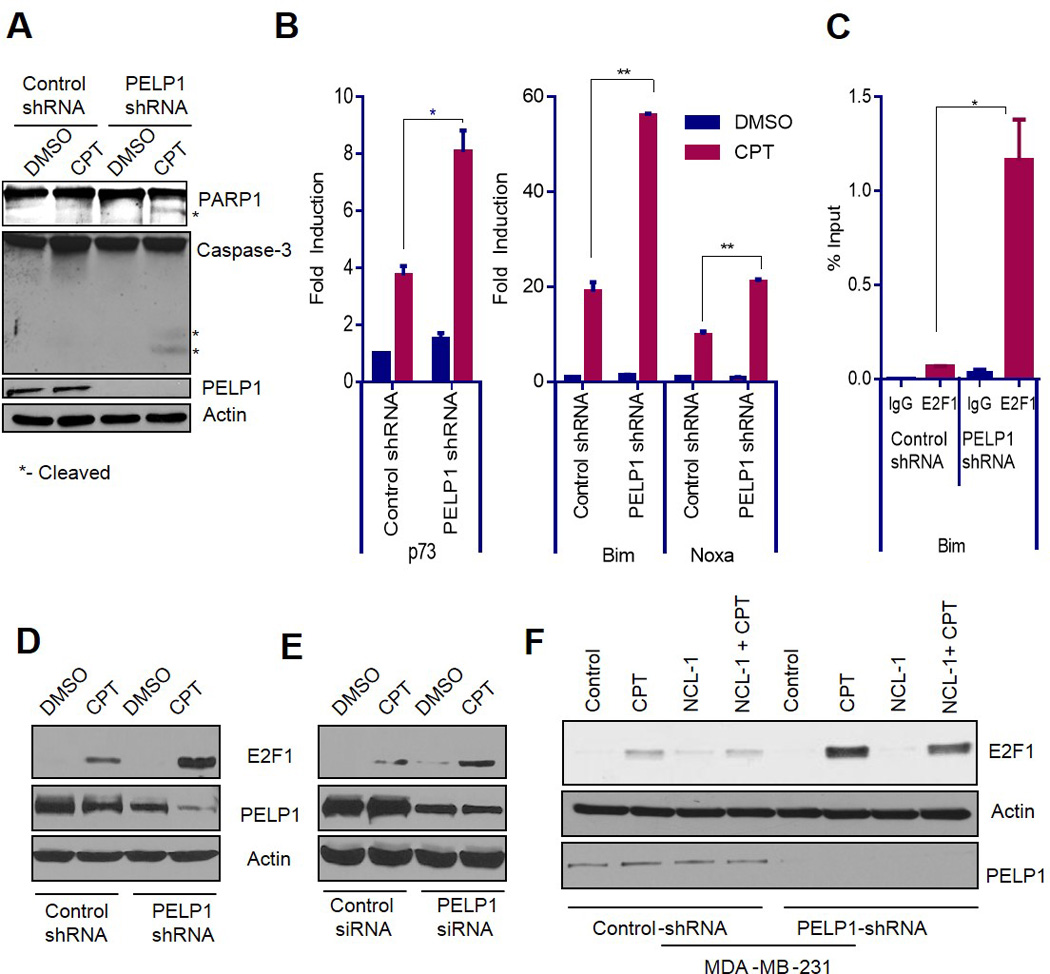
PELP1 regulates apoptosis by modulating E2F1 target gene expression and regulates E2F1 stability in a KDM1A-dependent manner. (A) MDA-MB-231 cells expressing control- or PELP1-shRNA were treated with DMSO or 1µM of camptothecin for 24 h and then analyzed by Western blotting with the caspase-3, PARP-1, PELP1, or actin antibodies. (B) MDA-MB-231 cells expressing control- or PELP1-shRNA were treated with DMSO or 1µM of camptothecin for 24 h. Subsequently, cDNA synthesis and qRT-PCR analyses of E2F1 target genes (p73, Bim, and Noxa) expression were performed. *, P<0.05. **, P<0.01. (C) MDA-MB-231 cells expressing control-shRNA or PELP1-shRNA were treated with 1µM camptothecin for 24 h followed by ChIP analysis. E2F1 recruitment to Bim promoter was analyzed using qRT-PCR. *, P<0.05. (D, E) MDA-MB-231 cells stably expressing control- or PELP1-shRNA (D), or transiently transfected with control-siRNA or PELP1-siRNA (E), were treated with DMSO or 1µM of camptothecin for 24 h followed by Western blotting with the E2F1, PELP1, or actin antibodies. (F) MDA-MB-231 cells expressing control- or PELP1-shRNA were treated with DMSO or the KDM1 inhibitor (NCL-1, 10µM) for 12h, and then treated with DMSO or 1µM of camptothecin for 24 h. Cells lysates were prepared and analyzed by Western blotting with the PELP1 or actin or E2F1 antibodies.
PELP1 regulates E2F1 stability in a KDM1A-dependent manner
To determine whether the increased recruitment of E2F1 to the promoter region is because of increased protein levels, we carried out Western analysis of MDA-MB-231 cells stably expressing control-shRNA or PELP1-shRNA after treatment with camptothecin. E2F1 protein levels were dramatically higher in PELP1 knockdown than in the control cells upon camptothecin treatment (Fig 4D). Similar results were obtained when PELP1 was knocked down transiently in these cells (Fig 4E). E2F1 protein levels under conditions of stress are primarily regulated by post-translational modification such as phosphorylation, acetylation, and methylation. E2F1 methylation by Set9 causes protein degradation. Upon genotoxic stress, E2F1 is demethylated by KDM1A (LSD1), which stabilizes the protein (16,33). Previous studies have indicated that PELP1 interacts with KDM1A and regulates substrate specificity of KDM1A from H3K4 to H3K9 (34). We hypothesized that PELP1 regulates E2F1 stability in a KDM1A-dependent manner. Due to the lack of commercial antibodies for E2F1-lys185 methylation, we used the KDM1A inhibitor, NCL-1 (35), to determine the importance of KDM1A in regulating E2F1 stability in PELP1 knockdown cells. MDA-MB-231 cells expressing control- or PELP1-shRNA were treated with DMSO or NCL-1 for 12 h, and then treated with DMSO or camptothecin. Western analysis of the cell lysates indicated that E2F1 levels were stabilized in PELP1-shRNA cells, and the increase in stability was dramatically reduced upon NCL-1 treatment (Fig 4F).
Genotoxic stress induces PELP1 phosphorylation at Ser1033 in TNBC model cells
Earlier studies showed that PELP1 is phosphorylated at Serine 1033 (S1033) by the DDR kinases (24). To examine whether such regulation also occurs in TNBC model cells, MDA-MB-231 and MDA-MB-468 cells were treated with camptothecin and PELP1 phosphorylation was analyzed by Western blotting using phospho-specific antibody. The results showed that PELP1 is phosphorylated in both TNBC cell lines upon genotoxic stress (Fig 5A, B). We then determined whether PELP1 phosphorylation by DDR kinases is important for the anti-apoptotic function of PELP1. We used a specific siRNA to deplete endogenous PELP1 in MDA-MB-231 cells, then transfected the cells with siRNA-resistant WT-GFP-PELP1 or MT (S1033A) GFP-PELP1-expressing plasmids. The cells were treated with DMSO or camptothecin, and the effect of PELP1 mutation on E2F1 stability was analyzed using Western blotting, and E2F1 apoptotic target gene expression was analyzed by qRT-PCR. Western analysis showed clear down regulation of endogenous PELP1 and equal expression of GFP-tagged WT as well as MT-PELP1 proteins (Fig 5C). Further analysis revealed that PELP1 S1033A mutant (MT)-expressing cells expressed significantly higher levels of E2F1 (Fig 5D) and enhanced apoptotic target gene transcription compared to the cells expressing WT-PELP1 (Fig 5E). Expression of the pro-survival MTp53 target, cdc25c, was also significantly down-regulated in the MT-PELP1-expressing cells (Fig 5F). These results suggest that PELP1 phosphorylation plays important roles in repressing E2F1-mediated apoptosis and in assisting MTp53 oncogenic functions.
Figure 5.
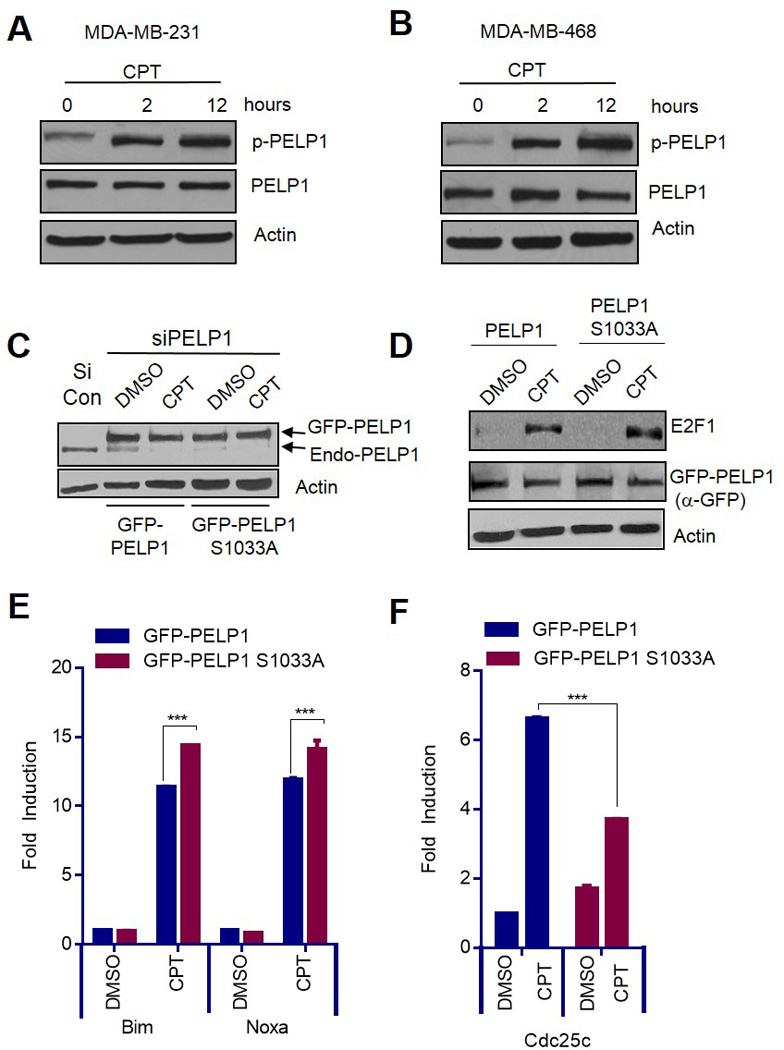
PELP1 is phosphorylated upon DNA damage at Ser1033 in TNBC cells. (A, B) MDA-MB-231 cells (A) and MDA-MB-468 cells (B) were treated with 1µM of camptothecin for 2 and 12 h, and total lysates were analyzed for PELP1 phosphorylation using phospho-PELP1 ser1033 specific antibody (p-PELP1). (C - F) MDA-MB-231 cells were transfected with stealth siRNA specific to PELP1 to reduce endogenous PELP1. After 48 h, the cells were transfected with siRNA-resistant GFP-PELP1 or GFP-PELP1S1033A-expressing plasmids. After 24 h, the cells were treated with DMSO or 1µM of camptothecin for 24 h. Knockdown of endogenous PELP1 was determined by Western blotting (C). GFP-PELP1, E2F1, and Actin levels were determined by Western blotting (D). qRT-PCR was used to determine Bim, Noxa (E), and Cdc25c (F) expression. ***, p<0.001.
PELP1 phosphorylation is increased in TNBC subtype of breast cancer
PELP1 is phosphorylated at Serine 1033 (p-PELP1) by the DDR kinases (23), and recent studies showed the significance of DDR kinase ATM deregulation in metastatic tumors (36,37). To examine the physiological importance of PELP1 phosphorylation in TNBC, we carried out immunohistochemistry analysis of PELP1 phosphorylation using human breast tumor tissue array (TMA). We found significantly increased levels of PELP1 S1033 phosphorylation in TNBC tumors than in luminal ER+ and Her2+ breast cancer subtypes (Fig 6A, C, and Table 1). Further IHC analysis using antibody that recognizes WT PELP1 did not reveal any significant differences among ER+ve, Her2+ve, and TNBC subtypes (Fig 6A, B, and Table 1). However, we did observe increased expression of WT PELP1 expression in ER+Her2+ breast subtype (Fig 6A, B, and Table 1). Taken together, WT and pPELP1 IHC results suggest that PELP1 phosphorylation is uniquely elevated in TNBC subtype and increase is not due to enhanced expression of PELP1 (Table 1). We further validated the TMA results using an additional TMA array that contained 28 TNBC tumors along with paired normal control breast tissues. Similar to the TMA results, PELP1 S1033 phosphorylation was significantly higher in the TNBC tumor tissues than in the normal tissues (Table 2). Collectively, these results suggest that hyper-activated DDR-PELP1-MTp53 pathway may play a role in TNBC progression.
Figure 6.
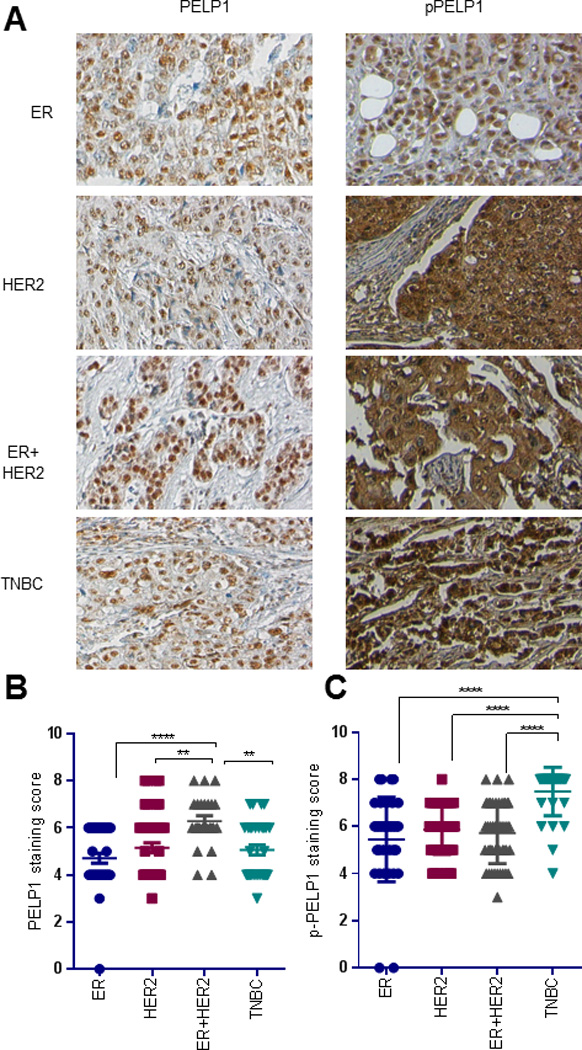
(A) Immunohistochemical staining of total PELP1 and pPELP1 (ser1033) on breast cancer tissues. Representative image of TMA stained with total PELP1 and p-PELP1 for each tumor type is shown. (B) Total PELP1 staining on tumor array was scored using Allred score system, and the average score for each tumor type was plotted on a graph. (C) p-PELP1 staining on tumor array was scored using Allred score system, and the average score of each tumor type was plotted on a graph. Statistical significance of differences in IHC scores was determined using Kruskal–Wallis test, followed by multiple comparisons using Dunn’s post test.
Table 1.
Comparison of Mean IHC Scores of P-PELP1 and total PELP1 among various subtypes of breast cancer
| Subtype | ER | HER2 | ER+HER2 | TNBC | p value |
|---|---|---|---|---|---|
| IHC*/n | 19 | 23 | 12 | 15 | |
| p-PELP1 | 5.44±0.28 | 5.86±0.15 | 5.92±0.28 | 7.48±0.18 | <0.0001 |
| PELP1 | 4.71±0.20 | 5.15±0.20 | 6.29±0.22 | 5.03±0.21 | 0.0001 |
Mean ± standard error of mean.
Statistical differences in the staining between breast cancer subtypes were determined using Kruskal–Wallis test.
Table 2.
Comparison of Mean Scores of P-PELP1 Expression between Triple-negative Tumors and Adjacent Normal Breast Tissues (N=28)
Mean ± standard error of mean.
p<0.05, significant difference, two-tailed paired t test
Discussion
p53 is an important tumor suppressor gene that safeguards cells from malignant transformation. TNBC tumors commonly exhibit gain-of-function mutations in p53 (MTp53), and patients with MTp53 have a poor response to chemo/radiation therapy, poor prognosis, and reduced survival (38,39). PELP1 is a recently discovered substrate of the ATM kinase, and plays a critical role in WTp53-mediated apoptosis in ER+ breast cancer (24) but its role in MTp53 expressing TNBC subtype of breast cancer remains unknown. In the present study, we found in MTp53-expressing TNBC cells that (i) PELP1 interacted with MTp53, regulated transcription of MTp53 target genes, facilitated MTp53 recruitment, and altered epigenetic changes at the target gene promoter, (ii) PELP1 depletion enhanced sensitivity of MTp53-expressing TNBC cells to chemotherapeutics and induced apoptosis, (iii) PELP1 interacted with E2F1, regulated E2F1 apoptotic target gene expression, and PELP1 depletion increased E2F1 stability in a KDM1A-dependent manner, and (iv) PELP1 phosphorylation by DDR kinases contributed to the oncogenic functions of PELP1, and PELP1 phosphorylation was significantly greater in the TNBC subtype than in the other receptor subtypes and the normal tissue. Collectively, these results suggest that PELP1 may be targeted in MTp53-expressing TNBC to obtain greater response to chemotherapy.
MTp53 can repress apoptosis and promote cell proliferation, metastasis, and genetic instability. Unlike, WTp53 whose protein levels are tightly modulated by MDM2 and post-translational modifications, MTp53 protein levels are highly stable (8). The majority of p53 mutations are missense and are present in the DNA-binding domain. These mutations inactivate p53, and confer the protein with oncogenic gain-of-functions, and MTp53 interacts and deregulates the functions of other transcription factors (8) . For example, MTp53 interacts with p63 and p73 and sequesters them in the cytoplasm (9). MTp53 causes deregulation of cell cycle progression through its interaction with the NF-Y complex and causes increased transcription of target genes in response to DNA damage (10). In our study, we found that PELP1 interacts with MTp53 and PELP1 knockdown significantly reduced the activation of MTp53-NF-Y target genes. Mechanistic studies indicated the PELP1 is needed for optimal recruitment of MTp53 and for promoting conducive epigenetic marks at the MTp53-NF-Y target genes. Collectively, these emerging studies suggest that MTp53 interactions with proto-oncogene PELP1 could influence its oncogenic functions and may have potential implications in chemotherapy response, and overall prognosis of patients.
E2F1-mediated apoptosis is triggered in response to genotoxic stress (14). Recent studies have delineated the key post-translational modifications that switch E2F1 functions from cell cycle regulation to apoptosis especially in MTp53-expressing cells. KDM1A-mediated demethylation is the upstream activation mark for E2F1 stabilization and for apoptotic target gene activation (16,33). Earlier studies showed that PELP1 functions as a cofactor of E2F1 (23), and PELP1 promotes epigenetic changes by modulating the activity of histone demethylase KDM1A (34). Our results suggest that PELP1 plays an important role in modulating E2F1 stability via KDM1A pathway in MTp53-expressing cells under conditions of stress. Since PELP1 interacts with KDM1A and regulates its substrate specificity, it is not surprising that PELP1 regulates KDM1A-mediated demethylation of E2F1. Upon treatment with the KDM1A inhibitor, E2F1 stability in response to genotoxic stress is substantially reduced. We also observed that inhibition of PELP1 phosphorylation causes substantial increase in E2F1 stability activity with a concomitant reduction in cell survival. PELP1 phosphorylation by the ATM kinase appears to be important for the repression of E2F1-mediated apoptotic functions and for MTp53 oncogenic activities.
We recently showed that PELP1 functions as a coactivator for WTp53 (24). In WTp53-expressing breast cancer cells (such as ZR-75 and MCF-7), PELP1 binds to p53, regulates p53 target gene transcription. PELP1 knockdown reduces apoptotic gene expression, and PELP1 knockdown cells exhibit resistance to apoptosis (24). In this study, we made an unexpected discovery that PELP1 knockdown enhanced apoptosis of MTp53-expressing cells. Our results from targeting PELP1 in WTp53 and MTp53 cells are suggestive of a beneficial effect when PELP1-targeted therapy is combined with chemotherapeutics agents for treating TNBC. We postulate that this combination therapy might protect the normal WTp53 cells from undergoing apoptosis, but at the same time could sensitize the MTp53 TNBC cells to chemotherapeutic agents. This combination treatment could have implications in reducing the toxic side-effects from chemotherapy drugs.
PELP1 expression is commonly deregulated in breast cancer, and overexpression of PELP1 is considered as a marker of poor prognosis (21). A recent report suggests that hyperactive ATM (ATM S1981-phosphorylation) is associated with lymph node metastasis in TNBC (37). Since PELP1 is a substrate of DDR kinases including ATM, we tested the physiological importance of PELP1 phosphorylation (Ser 1033) by immunohistochemical staining of breast cancer tumor tissue microarrays with p-PELP1 antibody. We found that PELP1 phosphorylation was greater in TNBC subtype than in other breast cancer subtypes. These studies utilized a small number of tumor samples and therefore represent a proof of principle study and suggest a possibility that PELP1 phosphorylation may serve as a biomarker for potential therapy using PELP1 axis-targeted drugs. However, additional studies are clearly needed with a large number of breast tumor samples to determine the prognostic significance of PELP1 in TNBC.
Collectively, our results have deciphered a novel role for PELP1 in regulating a chemotherapy response in MTp53-expressing TNBC cells. Molecular studies revealed that the oncogenic activities of PELP1 are accentuated in the presence of MTp53 and that targeting PELP1 axis in this subtype of breast cancer can enhance the sensitivity of TNBC cells to chemotherapeutic drugs. Further, MTp53 status could serve as a pivotal factor that predicts outcome when PELP1 is targeted in combination with chemotherapy, and could be used as a biomarker to select the intended patient population.
Acknowledgments
This study was supported by the NIH/NCI grant CA095681 (RKV); CPRIT grant DP150096 (RKV; GR); CPRIT pre-doctoral fellow ship grant RP140105 (SK); CPRIT post-doctoral fellowship grant RP140105 (GSR); and the Cancer Therapy and Research Center at the University of Texas Health Science Center at San Antonio through the NCI Cancer Center Support Grant P30CA054174-17.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest
References
- 1.Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007;109(9):1721–8. doi: 10.1002/cncr.22618. [DOI] [PubMed] [Google Scholar]
- 2.Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–34. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 3.Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–81. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- 4.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–9. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright JD, Lim C. Mechanism of DNA-binding loss upon single-point mutation in p53. J Biosci. 2007;32(5):827–39. doi: 10.1007/s12038-007-0083-3. [DOI] [PubMed] [Google Scholar]
- 8.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2(2):a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, Reumers J, Couceiro JR, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol. 2011;7(5):285–95. doi: 10.1038/nchembio.546. [DOI] [PubMed] [Google Scholar]
- 10.Di AS, Strano S, Emiliozzi V, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10(3):191–202. doi: 10.1016/j.ccr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 11.DeGregori J, Johnson DG. Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Curr Mol Med. 2006;6(7):739–48. doi: 10.2174/1566524010606070739. [DOI] [PubMed] [Google Scholar]
- 12.Engelmann D, Putzer BM. The dark side of E2F1: in transit beyond apoptosis. Cancer Res. 2012;72(3):571–5. doi: 10.1158/0008-5472.CAN-11-2575. [DOI] [PubMed] [Google Scholar]
- 13.Phillips AC, Vousden KH. E2F-1 induced apoptosis. Apoptosis. 2001;6(3):173–82. doi: 10.1023/a:1011332625740. [DOI] [PubMed] [Google Scholar]
- 14.Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001;15(14):1833–44. [PMC free article] [PubMed] [Google Scholar]
- 15.Pediconi N, Ianari A, Costanzo A, et al. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat Cell Biol. 2003;5(6):552–8. doi: 10.1038/ncb998. [DOI] [PubMed] [Google Scholar]
- 16.Xie Q, Bai Y, Wu J, et al. Methylation-mediated regulation of E2F1 in DNA damage-induced cell death. J Recept Signal Transduct Res. 2011;31(2):139–46. doi: 10.3109/10799893.2011.552914. [DOI] [PubMed] [Google Scholar]
- 17.Hershko T, Ginsberg D. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J Biol Chem. 2004;279(10):8627–34. doi: 10.1074/jbc.M312866200. [DOI] [PubMed] [Google Scholar]
- 18.Tonsing-Carter E, Shannon HE, Bailey BJ, Mayo LD, Pollok KE. Blockade of MDM2-mediated signaling in context of DNA damage increases E2F1 expression and enhances cell death in triple-negative breast cancer cells. Cancer Research. 2013 Apr 15;279(10):8627–8634. [Google Scholar]
- 19.Gonugunta VK, Miao L, Sareddy GR, et al. The social network of PELP1 and its implications in breast and prostate cancers. Endocr Relat Cancer. 2014;21(4):T79–T86. doi: 10.1530/ERC-13-0502. [DOI] [PubMed] [Google Scholar]
- 20.Girard BJ, Daniel AR, Lange CA, Ostrander JH. PELP1: a review of PELP1 interactions, signaling, and biology. Mol Cell Endocrinol. 2014;382(1):642–51. doi: 10.1016/j.mce.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Habashy HO, Powe DG, Rakha EA, et al. The prognostic significance of PELP1 expression in invasive breast cancer with emphasis on the ER-positive luminal-like subtype. Breast Cancer Res Treat. 2010;120(3):603–12. doi: 10.1007/s10549-009-0419-9. [DOI] [PubMed] [Google Scholar]
- 22.Roy S, Chakravarty D, Cortez V, et al. Significance of PELP1 in ER-negative breast cancer metastasis. Mol Cancer Res. 2012;10(1):25–33. doi: 10.1158/1541-7786.MCR-11-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nair BC, Nair SS, Chakravarty D, et al. Cyclin-dependent kinase-mediated phosphorylation plays a critical role in the oncogenic functions of PELP1. Cancer Res. 2010;70(18):7166–75. doi: 10.1158/0008-5472.CAN-10-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nair BC, Krishnan SR, Sareddy GR, et al. Proline, glutamic acid and leucine-rich protein-1 is essential for optimal p53-mediated DNA damage response. Cell Death Differ. 2014;21(9):1409–18. doi: 10.1038/cdd.2014.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mann M, Cortez V, Vadlamudi R. PELP1 oncogenic functions involve CARM1 regulation. Carcinogenesis. 2013;34(7):1468–75. doi: 10.1093/carcin/bgt091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bararia D, Trivedi AK, Zada AA, et al. Proteomic identification of the MYST domain histone acetyltransferase TIP60 (HTATIP) as a co-activator of the myeloid transcription factor C/EBPalpha. Leukemia. 2008;22(4):800–7. doi: 10.1038/sj.leu.2405101. [DOI] [PubMed] [Google Scholar]
- 27.Bartek J, Iggo R, Gannon J, Lane DP. Genetic and immunochemical analysis of mutant p53 in human breast cancer cell lines. Oncogene. 1990;5(6):893–9. [PubMed] [Google Scholar]
- 28.Nigro JM, Baker SJ, Preisinger AC, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342(6250):705–8. doi: 10.1038/342705a0. [DOI] [PubMed] [Google Scholar]
- 29.Mann M, Cortez V, Vadlamudi RK. Epigenetics of estrogen receptor signaling: role in hormonal cancer progression and therapy. Cancers (Basel) 2011;3(3):1691–707. doi: 10.3390/cancers3021691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64(2):435–59. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bossi G, Lapi E, Strano S, et al. Mutant p53 gain of function: reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene. 2006;25(2):304–9. doi: 10.1038/sj.onc.1209026. [DOI] [PubMed] [Google Scholar]
- 32.Lim LY, Vidnovic N, Ellisen LW, Leong CO. Mutant p53 mediates survival of breast cancer cells. Br J Cancer. 2009;101(9):1606–12. doi: 10.1038/sj.bjc.6605335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kontaki H, Talianidis I. Lysine methylation regulates E2F1-induced cell death. Mol Cell. 2010;39(1):152–60. doi: 10.1016/j.molcel.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Nair SS, Nair BC, Cortez V, et al. PELP1 is a reader of histone H3 methylation that facilitates oestrogen receptor-alpha target gene activation by regulating lysine demethylase 1 specificity. EMBO Rep. 2010;11(6):438–44. doi: 10.1038/embor.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueda R, Suzuki T, Mino K, et al. Identification of cell-active lysine specific demethylase 1-selective inhibitors. J Am Chem Soc. 2009;131(48):17536–7. doi: 10.1021/ja907055q. [DOI] [PubMed] [Google Scholar]
- 36.Boohaker RJ, Cui X, Stackhouse M, Xu B. ATM-mediated Snail Serine 100 phosphorylation regulates cellular radiosensitivity. Radiother Oncol. 2013;108(3):403–8. doi: 10.1016/j.radonc.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 37.Sun M, Guo X, Qian X, et al. Activation of the ATM-Snail pathway promotes breast cancer metastasis. J Mol Cell Biol. 2012;4(5):304–15. doi: 10.1093/jmcb/mjs048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bertheau P, Lehmann-Che J, Varna M, et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast. 2013;22(Suppl 2):S27–S29. doi: 10.1016/j.breast.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 39.Dobes P, Podhorec J, Coufal O, et al. Influence of mutation type on prognostic and predictive values of TP53 status in primary breast cancer patients. Oncol Rep. 2014 doi: 10.3892/or.2014.3346. [DOI] [PubMed] [Google Scholar]