

Abstract
Trialkylstannanes are versatile precursors for chemical transformations, including radiolabeling with a variety of halogens, particularly iodine. In the present work a convenient, Pd-mediated stannylation method is presented that can be performed in an open flask. The method is selective for aryl iodides allowing selective stannylations in the presence of other halogen atoms. The reaction conditions are mild, making the method compatible with chemically sensitive bioactive compounds.
Keywords: Stannylation, Radioiodination, Opioid, Sigma receptor, Radioligand binding
Trialkyltin groups participate in vital chemical transformations, such as radiohalogenation, carbon-carbon and carbon-heteroatom bond forming reactions (Scheme 1). These versatile precursors undergo a wide range of electrophilic substitution reactions, which tolerate a broad spectrum of functional groups.1–11
Scheme 1.

Diverse transformations of alkylstannyl precursors
Radiohalogen-labeled bioactive compounds are important parts of modern diagnostic imaging (F-18, I-131, I-123, I-124, and Br-76), pharmacological and biochemical research, and cancer therapy (I-125, I-131, and At-211). Radiotracers range from simple small molecules to complex bioactive ligands such as alkaloids, carbohydrates and peptides. These radiohalogenated small molecules are prepared through indirect and direct radiohalogenation routes.12 Indirect methods include halodeboronation and halodestannylation.13–16 In general, for radiolabeling of complex, bioactive molecules halodestannylation is preferred, partly owing to the facile separation of the radiohalogenated product and the metalated precursor.17–19
Traditional methods to access trialkylstannanes typically require harsh reaction conditions and use flammable reagents as in halogen-metal exchange.20 Therefore, these methods are infeasible to carry out in laboratories not equipped for organic synthesis. Furthermore, excessive heating involved in most metal-catalyzed stannylation methods limits their applicability, especially in cases where chemically sensitive drug molecules need to be stannylated.21 Although milder stannylation conditions have been reported, those have not been demonstrated on molecules with unprotected functional groups.22,23 Therefore, the aim was to elaborate a stannylation method that works efficiently under ambient conditions on a multitude of complex radioiodination targets.
Herein, we report a simple, Pd-catalyzed stannylation method selective for iodoarenes. The reaction is compatible with a wide array of iodine-containing aromatic compounds including simple iodobenzene derivatives, heterocycles, and complex, multifunctional semi-synthetic and synthetic bioactive molecules. The presented stannylation method is carried out at rt and does not require the use of dry solvents or inert gases, making it accessible to researchers not specializing in organic synthesis. Using our method, CNS receptor ligands of high importance were successfully stannylated. To the best of our knowledge, single-step stannylations of 4-iodoclonidine (alpha-2 receptor agonist) and IPAG (sigma-1 receptor antagonist) have not been described in the literature before.24
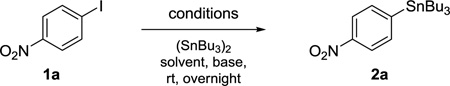
Reaction conditions were optimized using 1-iodo-4-nitrobenzene (1a). The primary goal was to find a Pd-catalyst and reaction conditions, which allow for efficient stannylation with (Bu3Sn)2 at room temperature using common laboratory grade solvents in an open flask. Initially, Pd-catalysts were screened in i-PrOH using various equivalents of (Bu3Sn)2 (Table 1). Hünig’s base (DIEA) was added to neutralize any acidic species possibly forming under non-anhydrous conditions, as even small amounts of acid could lead to substantial protodestannylation of the product. The best yield was achieved with Pd2(dba)3 (entry 5), while the versatile Pd(0) catalyst tetrakis(triphenylphosphine) Pd(0) led to greatly reduced yields (entry 1), owing to its instability under non-anhydrous, non-air free reaction conditions. Catalysts containing Pd(II) gave inferior results (entries 2 and 3), presumably because reduction of the Pd(II) to Pd(0) at ambient temperature is not facile. After careful optimization (not shown) it was found that the catalyst loading could be reduced to 2.5 mol%, and 1.1 equiv (Bu3Sn)2 was sufficient.
Table 1.
Optimization of the stannylation on 1a.
 | ||||
|---|---|---|---|---|
| entry | solvent | catalyst (equiv) | base (equiv) | yield (%)c |
| 1a | i-PrOH | Pd(PPh3)4 (0.1) | DIEA (2.5) | <10 |
| 2a | i-PrOH | PdCl2(PPh3)2 (0.1) | DIEA (2.5) | 0 |
| 3a | i-PrOH | PdCl2(PTol3)2 (0.1) | DIEA (2.5) | 19 |
| 4a | i-PrOH | Pd2(dba)3 (0.1) | DIEA (2.5) | 92 |
| 5b | i-PrOH | Pd2(dba)3 (0.025) | DIEA (2.5) | 97 |
| 6b | toluene | Pd2(dba)3 (0.025) | DIEA (2.5) | 37 |
| 7b | THF | Pd2(dba)3 (0.025) | DIEA (2.5) | 44 |
| 8b | hexanes | Pd2(dba)3 (0.025) | DIEA (2.5) | 14 |
| 9b | MeOH | Pd2(dba)3 (0.025) | DIEA (2.5) | 62 |
| 10b | i-PrOH | Pd2(dba)3 (0.025) | DIEA (1.0) | 94 |
| 11b | i-PrOH | Pd2(dba)3 (0.025) | K2CO3 (2.5) | 73 |
| 12b | i-PrOH | Pd2(dba)3 (0.025) | pyridine (2.5) | 0 |
| 13b | i-PrOH | Pd2(dba)3 (0.025) | no base | 68 |
2 Equiv (Bu3Sn)2.
1.1 Equiv (Bu3Sn)2.
Isolated yields.
Next, the role of solvent and base was investigated. Changing the solvent from i-PrOH conferred no advantage (entries 6–9). The reaction yielded the desired stannylation product with a variety of bases, albeit in lower yields than with DIEA (entries 11, 12). When no base was added to the reaction, product yield was significantly diminished (entry 13).
With the optimal reaction conditions in hand, the reaction scope was investigated, initially on simple aryl halides (Figure 1A). Under these mild conditions, the reaction is mostly restricted to electron-deficient aromatic systems, which is in agreement with previous literature reports.25,26 The best yields were achieved with nitro-derivatives 2a and 2b, while decreasing yields were observed in the presence of other electron-withdrawing groups (2c, 2d, 2g). An electron rich morpholine derivative gave the desired product (2f) in a significantly lowered yield. Under these conditions, chlorine and bromine-containing substrates such as 1-chloro and 1-bromo-4-nitrobenzene failed to undergo stannylation. This highlights the excellent selectivity of this reaction for iodine, which is preferred from a biology or medicinal chemistry standpoint, since several of the relevant stannylation targets (ie. 4-iodoclonidine 1o, AM251 1q) contain halogen atoms other than iodine as well.
Figure 1.

Substrate scope. A) Simple aryl iodides. B) Heterocycles. C) bioactive molecules.
Next, heterocyclic compounds were investigated (Figure 1B). Substituted pyridine derivatives reacted readily and afforded the corresponding products (2h and 2i) in good yields. The vinyl iodine afforded the desired tributylstannyl compound 2j in good yield. Although the conversion was low, the reaction worked on the naphthyridine derivative (2k) highlighting the functional group tolerance of the method.
Finally, our reaction conditions were applied to a wide range of iodine-containing bioactive molecules (Figure 1C). These compounds are clinically used drugs (1l) or important receptor ligands (1m–r) extensively used by laboratories for radioactive imaging and receptor binding studies. Their wide application in radiolabeling necessitates a method that enables the convenient and efficient generation of the stannylated radiolabeling precursors. Our protocol is compatible with a variety of unprotected functional groups: alcoholic hydroxyl (2l, 2p), imide (2l), guanidine (2o, 2r), phenolic hydroxyl (2p), hydrazide (2q), sulfonamide (2n), and amide (2m, 2p) are tolerated under reaction conditions. Stannylation of IBNtxA (1p)—a powerful opioid analgesic labeling the recently discovered 6TM/E11 opioid site with a safer side-effect profile than morphine—was achieved in a single step, simplifying the previously reported three-step procedure.6,27 Furthermore, stannylation of IBNtxA using the traditional conditions showed no product formation.
There is increasing evidence that sigma receptor antagonists could be useful in decreasing cell proliferation and be used for cancer therapy.28,29 IPAG (1r), a sigma receptor antagonist is a valuable probe for investigating and exploiting sigma action. Single-step synthesis of stannylated IPAG (2r) was possible for the first time using our method, whereas in a previous report, a three-step route was utilized.24
Similarly, a stannylated precursor of radiolabeled AM251 (2q), and a stannylated 4-I-clonidine (2o), an adrenergic (α2) receptor antagonist were synthesized. AM251 is a cannabinoid (CB1) receptor inverse agonist with structural similarity to rimonabant, clonidine (non-iodinated form of 1o) is used clinically in the treatment of ADHD, high blood pressure, and pain.30,31 To our knowledge, this is the first report of stannylated clonidine. To validate the experimental usefulness of radioiodinated IPAG, clonidine, and AM251, the stannylated precursors were labeled with 125I using standard protocols.6 These radioiodinated analogs were then assayed in radioligand binding assays in mouse brain (Table 2). The binding affinities (KD) are consistent with literature values, demonstrating that analogs made by our method bind their targets without loss of affinity. The stannylated precursors can be used for radioiodination with other important iodine isotopes (124I, 131I) as well.
Table 2.
Validation of radioiodinated drugs by radioligand saturation studies using mouse brain homogenates.
| compound | yielda | KD (nM) | literature KD (nM) |
|---|---|---|---|
| [125I]IPAG | 29 | 10.3 ± 4.4 | 8.7 – 11.21h |
| [125I]4-I-Clonidine | 11 | 0.11 ± 0.03 | 0.46 ± 0.0612b |
| [125I]AM251 | 33 | 0.46 ± 0.01 | 0.23 – 0.6212a |
average radiochemical yield based on two repetitions.
In conclusion, a simple and efficient, Pd-catalyzed stannylation method selective for iodoaryl compounds was developed. Substrates ranging from simple iodoaryl and iodoheteroaryl compounds to complex multifunctional bioactive molecules underwent stannylation to yield valuable radiolabeling precursors in moderate to good yields. The reaction tolerates a wide range of unprotected functional groups. The major advantage of our method is the compatibility with ambient conditions and open flask setup; no temperature control, anhydrous solvents or inert gases are required to successfully carry out the reaction, making it accessible to researchers looking to synthesize radioiodination precursors. First synthesis of stannylated IPAG and p-tributyltin-clonidine is reported, along with a simplified synthesis of stannylated IBNtxA after traditional approaches to stannylate these molecules were unsuccessful. The protocol allows aryl iodine-containing drugs or probes to be stannylated, providing valuable precursors to imaging agents or radioligands for receptor binding studies. Our method thus provides a valuable additional tool to the toolbox of radio/medicinal chemists interested in elucidating the biochemical and pharmacological mechanisms of drug action.
Supplementary Material
Acknowledgments
The authors are grateful for financial support from NIDA (T32DA07274) to JEP, (DA034106-01) to SM, (DA06241, DA02165, & DA07242) to GWP, a Predoctoral Fellowship from the PhRMA Foundation to SGG, and a Core Grant from NCI to MSKCC (CA08748) is gratefully acknowledged. The authors would like to thank George Sukenick (MSKCC) for assistance with NMR experiments, Rong Wang (MSKCC) and the UC Riverside HRMS Facility (NSF grant CHE-0541848) for HRMS measurements.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Stille JK. Angew. Chem. Int. Ed. 1986;25:508–524. [Google Scholar]
- 2.Andersson Y, Langstrom B. Journal of the Chemical Society, Perkin Transactions. 1995;1:287–289. [Google Scholar]
- 3.Beletskaya IP. J. Organomet. Chem. 1983;250:551–564. [Google Scholar]
- 4.Coenen HH, Moerlein SM. J. Fluorine Chem. 1987;36:63–75. [Google Scholar]
- 5.Huang C, Liang T, Harada S, Lee E, Ritter T. J. Am. Chem. Soc. 2011;133:13308–13310. doi: 10.1021/ja204861a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majumdar S, Burgman M, Haselton N, Grinnell S, Ocampo J, Pasternak AR, Pasternak GW. Bioorg. Med. Chem. Lett. 2011;21:4001–4004. doi: 10.1016/j.bmcl.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mandolesi SD, Vaillard SE, Podestá JC, Rossi RA. Organometallics. 2002;21:4886–4888. [Google Scholar]
- 8.Meyer N, Sivanathan S, Mohr F. J. Organomet. Chem. 2011;696:1244–1247. [Google Scholar]
- 9.Reich HJ, Goldenberg WS, Sanders AW, Jantzi KL, Tzschucke CC. J. Am. Chem. Soc. 2003;125:3509–3521. doi: 10.1021/ja028301r. [DOI] [PubMed] [Google Scholar]
- 10.Vaidyanathan G, Affleck DJ, Alston KL, Zalutsky MR. J. Label. Compd. Radiopharm. 2007;50:177–182. [Google Scholar]
- 11.van Klink GPM, de Boer HJR, Schat G, Akkerman OS, Bickelhaupt F, Spek AL. Organometallics. 2002;21:2119–2135. [Google Scholar]
- 12.Seevers RH, Counsell RE. Chem. Rev. 1982;82:575–590. [Google Scholar]
- 13.Kabalka GW, Akula MR, Zhang J. Nucl. Med. Biol. 2002;29:841–843. doi: 10.1016/s0969-8051(02)00344-x. [DOI] [PubMed] [Google Scholar]
- 14.Kabalka GW, Akula MR, Zhang J. Nucl. Med. Biol. 2003;30:369–372. doi: 10.1016/s0969-8051(02)00443-2. [DOI] [PubMed] [Google Scholar]
- 15.Kabalka GW, Mereddy AR. Nucl. Med. Biol. 2004;31:935–938. doi: 10.1016/j.nucmedbio.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Yong L, Yao ML, Green JF, Kelly H, Kabalka GW. Chem. Commun. 2010;46:2623–2625. doi: 10.1039/b925258d. [DOI] [PubMed] [Google Scholar]
- 17.Corcoran EB, Williams AB, Hanson RN. Org. Lett. 2012;14:4630–4633. doi: 10.1021/ol302076d. [DOI] [PubMed] [Google Scholar]
- 18.Chiosis G. 7,834,181. U.S. Patent. 2011
- 19.Burnett DA, Caplen MA, Domalski MS, Browne ME, Davis JHR, Clader JW. Bioorg. Med. Chem. Lett. 2002;12:311–314. doi: 10.1016/s0960-894x(01)00750-8. [DOI] [PubMed] [Google Scholar]
- 20.Cherry K, Lebegue N, Leclerc V, Carato P, Yous S, Berthelot P. Tetrahedron Lett. 2007;48:5751–5753. [Google Scholar]
- 21.Farina V. Pure Appl. Chem. 1996;68:73–78. [Google Scholar]
- 22.Mitchell TN. J. Organomet. Chem. 1986;304:1–16. [Google Scholar]
- 23.Qiu D, Meng H, Jin L, Wang S, Tang S, Wang X, Mo F, Zhang Y, Wang J. Angewandte Chemie International Edition. 2013;52:11581–11584. doi: 10.1002/anie.201304579. [DOI] [PubMed] [Google Scholar]
- 24.Kimes AS, Wilson AA, Scheffel U, Campbell BG, London ED. J. Med. Chem. 1992;35:4683–4689. doi: 10.1021/jm00103a005. [DOI] [PubMed] [Google Scholar]
- 25.McKean DR, Parrinello G, Renaldo AF, Stille JK. The Journal of Organic Chemistry. 1987;52:422–424. [Google Scholar]
- 26.Sandosham J, Undheim K. Acta Chem. Scand. 1989;43:684–689. [Google Scholar]
- 27.Majumdar S, Grinnell S, Le Rouzic V, Burgman M, Polikar L, Ansonoff M, Pintar J, Pan Y-X, Pasternak GW. Proc. Natl. Acad. Sci. U. S. A. 2011;108:19778–19783. doi: 10.1073/pnas.1115231108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim FJ, Schrock JM, Spino CM, Marino JC, Pasternak GW. Biochem. Biophys. Res. Commun. 2012;426:177–182. doi: 10.1016/j.bbrc.2012.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schrock JM, Spino CM, Longen CG, Stabler SM, Marino JC, Pasternak GW, Kim FJ. Mol. Pharmacol. 2013;84:751–762. doi: 10.1124/mol.113.087809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gatley SJ, Lan R, Pyatt B, Gifford AN, Volkow ND, Makriyannis A. Life Sci. 1997;61:PL191–PL197. doi: 10.1016/s0024-3205(97)00690-5. [DOI] [PubMed] [Google Scholar]
- 31.Höcker J, Weber B, Tonner PH, Scholz J, Brand P-A, Ohnesorge H, Bein B. Eur. J. Pharmacol. 2008;582:70–77. doi: 10.1016/j.ejphar.2007.12.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.