

Abstract
N -myristoylation is an essential fatty acid modification that governs the localization and activity of cell signaling enzymes, architectural proteins, and immune regulatory factors. Despite its importance in health and disease, there are currently no methods for reversing protein myristoylation in vivo. Recently, the Shigella flexneri protease IpaJ was found to cleave myristoylated glycine of eukaryotic proteins, yet the discriminatory mechanisms of substrate selection required for targeted demyristoylation have not yet been evaluated. Here, we performed global myristoylome profiling of cells treated with IpaJ under distinct physiological conditions. The protease is highly promiscuous among diverse N-myristoylated proteins in vitro, but is remarkably specific to Golgi associated ARF/ARL family GTPases during Shigella infection. Reconstitution studies revealed a mechanistic framework for substrate discrimination based on IpaJ’s function as a GTPase “effector” of bacterial origin. We now propose a concerted model for IpaJ function that highlights its potential for programmable demyristoylation in vivo.
Introduction
Survival of microbial pathogens during infection relies on their ability to evade immune recognition and to establish a replicative niche inside the host organism. To this end, pathogens evolve numerous strategies to adapt to the host environment. Several Gram-negative bacteria utilize a dedicated type III secretion system (T3SS) to deliver “effector” proteins directly into the infected host cell (Cornelis and Van Gijsegem, 2000). These effector proteins promote bacterial infection by subverting crucial cellular systems including immune responses, the cytoskeleton network and membrane trafficking (Alto and Orth, 2012). At the molecular level, many secreted effectors post-translationally modify (PTM) host proteins (Ribet and Cossart, 2010). The unconventional activities of these enzymes provide unique insights into the function of various PTMs and their roles in host cell signaling (Cui and Shao, 2011). Therefore, better characterization of pathogenic strategies relying on PTMs is critical to understand the mechanism of bacterial infections and the regulation of human health and disease.
Recently we found that the invasion plasmid antigen J (IpaJ), a T3SS effector protein secreted by Shigella flexneri, is a cysteine protease that catalyzes the hydrolysis of N-myristoylated glycine at the amino terminus of host proteins (Burnaevskiy et al., 2013). Bioinformatics studies suggest that nearly 1% of the human proteome harbors an N-myristoyl modification that mediates important protein and lipid interactions involved in cellular architecture and signal transduction (Farazi et al., 2001a; Maurer-Stroh et al., 2004). In addition, N-myristoylation is a potential target for therapeutic intervention because of its role in oncogenic signaling (Magnuson et al., 1995) as well as microbial pathogenesis. For example, rheovirus, picornavirus, and lentivirus (e.g. HIV) require C14:0 fatty acylation for cellular entry and egress (Harper and Gilbert, 1995; Maurer-Stroh and Eisenhaber, 2004; Raulin, 2000). N-myristoylation systems are also required for the physiology of human parasites including Plasmodium falciparum, Trypansoma brucei, and Leishmania species the causative agents of Malaria, African sleeping sickness, and Leishmaniasis respectively (Frearson et al., 2010; Tate et al., 2013). Thus, inhibiting mechanisms of protein N-myristoylation has been considered for anti-cancer, anti-viral and anti-parasitic therapies. Until recently however, the covalent attachment of myristic acid to an NH2-terminal glycine residue was thought to be an irreversible reaction (Farazi et al., 2001a).
We previously reported that IpaJ inhibits protein trafficking by cleaving the N-myristoylated glycine of ADP-ribosylation factor 1 (ARF1), a critical regulator of the Golgi apparatus structure and function (Burnaevskiy et al., 2013). ARF1 is a small molecular weight GTPase that cycles between GDP- and GTP-bound states (Kahn, 2009). The cycles of guanine nucleotide exchange and hydrolysis induce profound conformational rearrangements of the ARF protein that has two major consequences. First, in GDP-inactive conformation, the N-myristoyl group is masked to solvent, which renders the protein soluble (Kahn, 2009). GTP-exchange induces the so-called “myristoyl switch”, exposing the acylated N-terminal helix to promote association of ARF1 with the ER and Golgi membrane (Goldberg, 1998; Kahn, 2009). Second, the transition from the GDP- to GTP-bound (active) state allows ARF1 to recruit target proteins to membranes, thereby triggering numerous biological processes including transport vesicle formation (Kahn, 2009). Hence, the myristoyl switch provides temporal coordination of ARF1 localization with the recruitment of downstream substrates. By removing the myristoyl group, IpaJ releases ARF1 from the Golgi membranes leading to the inhibition of the General Secretory Pathway (Burnaevskiy et al., 2013). Although previous findings revealed the function of the protease, several important questions that have not yet been addressed. For example, it is unclear how IpaJ specifically recognizes N-myristoylated substrates. In addition, the timing and location of ARF1 cleavage by IpaJ have not yet been resolved.
In addition to ARF1, IpaJ was also found to cleave numerous N-myristoylated proteins in vitro (Burnaevskiy et al., 2013). The large number of potential substrates indicates that IpaJ might also cleave multiple proteins during infection, thereby affecting diverse host cell processes essential for Shigella pathogenesis. Furthermore, the ability of IpaJ to recognize distinct N-myristoylated proteins also suggests a unique mechanism of substrate specificity. Interestingly, bioinformatics analysis of secondary structures had assigned IpaJ to a poorly characterized C39 peptidase-like family of domains of unknown function (DUF3335) (Burnaevskiy et al., 2013). Putative enzymes of this family are found in more than 200 bacterial species including pathogenic Pseudomonas aeruginosa and Vibrio cholera (Pfam database, family DUF3335), but IpaJ remains the only member of this group with confirmed enzymatic activity. Thus, determining the mechanism of IpaJ specificity will shed light on the functions of the entire protease family.
In this study, we performed an unbiased profiling of the cellular ‘myristoylome’ and specifically evaluated potential targets of the IpaJ protease under differential expression conditions. IpaJ cleaved the majority of N-myristoylated proteins when incubated with whole cell lysates, yet demonstrated remarkable specificity for ARF and ARL GTPase families in the context of Shigella infection. To understand the mechanism of substrate selectivity, we further reconstituted the proteolytic reaction using a minimal substrate and purified components. Findings from these studies indicate that IpaJ recognizes two diverse structural elements, the first of which is common among all myristoylated proteins whereas the second is only found in GTPase family members. Further cellular studies provide a concerted model of IpaJ-mediated Golgi destabilization and suggest the potential for using IpaJ as a template to design programmable myristoyl protein inhibitors in vivo.
Results
Mass spectrometry identification of IpaJ substrates in vitro
Advances in bioorthogonal ligation methods have provided new opportunities for proteomic analysis of protein acylation using azide or alkyne functionalized fatty acid chemical reporters (Grammel and Hang, 2013; Hang and Linder, 2011; Tate et al., 2014). In particular, the ability to track changes over the entire cellular myristoylome under differential conditions could provide an unbiased approach to identify IpaJ substrates in human cells. To develop this system, HeLa cells were metabolically labeled with alk-12, an alkyne-functionalized synthetic analog of myristic acid that is metabolized as the natural fatty acid for myristoylation of newly translated proteins (Figure 1A) (Charron et al., 2009; Wilson et al., 2011). To facilitate the visualization of proteins that incorporate alkyne-myristic acid alk-12, a rhodamine-azide fluorescent probe was attached to the alkyne group through Cu(II)-catalyzed cycloaddition reaction (click chemistry) in whole cell lysates and then directly visualized by an in-gel fluorescence assay (Figure 1A). As shown in Figure 1B, highly abundant N-myristoylated proteins could be detected by this method (left lane). Incubation of cell lysates with recombinant IpaJ resulted in the proteolytic elimination of the majority of N-myristoyl modifications under these conditions (Figure 1B, right lane).
Figure 1. IpaJ cleaves majority of myristoylated proteins in vitro.

(A) Design of the experiment to analyze protein cleavage by recombinant IpaJ in vitro. Alk-12 labeled myristoylated proteins were conjugated azido-rhodamine for in-gel visualization or with azido-biotin for purification and mass-spectrometric analysis.
(B) In-gel fluorescent visualization (left) of myristoylome profile of HeLa cell extracts from untreated or IpaJ treated samples. Total protein is shown (right). * Indicates proteins cleaved by IpaJ.
C. Scatter Plot of results from differential mass-spectrometric analysis. Proteins that are known to be N-myristoylated are colored red. Y-axis shows the fold-increase or decrease of protein abundance in IpaJ-treated sample compared to untreated-treated samples. Green shading marks the area of 2-fold deviation of protein abundance in IpaJ-treated lysates. For visualization purpose more than 10 fold over- or under-representation is shown as approximately 10 or −10, respectively.
D. Identified N-myristoylated proteins showing the fold increase or decrease of peptide abundance between IpaJ treated and untreated samples.
Over 250 human proteins are predicted to be N-myristoylated (Maurer-Stroh et al., 2002), an estimated half of which are expressed in cell-type dependent manner (Thinon et al., 2014). To specifically identify IpaJ substrates among the HeLa cell population, alkyne-labeled myristoylated proteins were treated with recombinant IpaJ and then conjugated with an azide-functionalized biotin tag. Biotinylated proteins were purified on streptavidin beads, digested with trypsin and resulting peptides were analyzed by mass-spectrometry to identify alkyne-labeled myristoylated proteins (Silva et al., 2006). Since the biotin tag is attached to the myristoyl group, the relative abundance of these peptides should correlate with the myristoylation status of a given protein. Indeed, we identified 102 proteins harboring the NH2-terminal glycine that represents a potential site for the myristoyl modification in untreated controls samples (Table S1). Importantly, the majority of these proteins were underrepresented in the IpaJ-treated sample (Figure 1C). In agreement with our previous finding that MARCKS and Src are cleaved by IpaJ in vitro (Burnaevskiy et al., 2013), both proteins were underrepresented in the IpaJ treated sample (Table S1). Furthermore, 34 out of 45 known N-myristoylated proteins were reduced at least two-fold after protease treatment (Figure 1C and Table S1). The list of in vitro IpaJ substrates comprises proteins of diverse molecular functions including kinases, E3 ubiquitin ligases, and α-subunits of guanine nucleotide-binding proteins (Figure 1D and Table S1) and of crucial signaling pathways such as apoptosis (apoptosis-inducing factor 2), autophagy (tyrosine-protein kinase ABL1), and immune response (tyrosine-protein kinase Lyn). In summary, the majority of the identified N-myristoylated proteins are sensitive to IpaJ in vitro, suggesting a unique, yet currently uncharacterized mechanism of substrate recognition and cleavage.
Reconstitution of the proteolytic demyristoylation reaction using minimal components
To elucidate the general mechanism of substrate recognition by IpaJ observed above, we set out to reconstitute myristoyl-glycine proteolysis using purified peptides in a homogenous non-ionic micelle environment (β-maltoside detergent). Initially, we synthesized a peptide corresponding to the NH2-terminus of ARF1 residues 2–17 (Gly2-Glu17) and this peptide was chemically myristoylated after synthesis (myr-Gly2-Glu17). In addition, a control peptide representing the expected IpaJ cleavage product of ARF1 residues 3–17 (Asn3-Glu17) was synthesized (Figure 2A). Each peptide was PEGylated to increase solubility and labeled with fluorescein isothiocyanate (FITC) for detection by analytical fluorescence Size-Exclusion Chromatography (fSEC). As shown in Figure 2A, The myr-Gly2-Glu17 peptide displayed a retention time of 10.8 minutes by fSEC and could be readily distinguished from the Asn3-Glu17 peptide (retention time of 12.3 minutes). Therefore, cleavage of myr-Gly2-Glu17 peptide by IpaJ is predicted to result in a decrease of peak intensity at 10.8 minutes and appearance of a peak at 12.3 minutes. Indeed, incubation of recombinant IpaJΔ50 with the myristoylated peptide resulted in an elution profile indistinguishable from Asn3-Glu17. Consistent with an enzymatic mechanism of reaction, the non-catalytic cysteine mutant of IpaJ (IpaJ C64A) had no effect on peptide cleavage (Figure 2A). In addition, IpaJ cleaved an excess of myr-Gly2-Glu17 in a time-dependent manner (Figure 2B). Thus, IpaJ proteolysis can be reconstituted in solution using peptide substrate and a minimal lipid environment.
Figure 2. Myristoylated N-terminus is necessary and sufficient for cleavage by IpaJ.

(A) Diagrams of the peptides corresponding to myr-Gly2-Glu17 and Asn3-Glu17 control are shown. fSEC analysis of myristoylated peptides after 30 minute of the cleavage reaction. Trace of the Asn3-Glu17 (blue) control compared myr-Gly2-Glu17 peptide in untreated (green), IpaJ treated (orange), and IpaJ C64A treated (red) samples are shown.
(B) Time course of the myr-Gly2-Glu17 cleavage by IpaJ. The peptide (1 μM) was incubated with MBP-IpaJΔ50 (50 nM) for indicated period of time (left panel). Areas under the curves were quantified to estimate percent of cleaved substrate (right panel).
(C) fSEC analysis of decanoyl and palmitoyl peptide cleavage. Trace of untreated Gly2-Glu17 harboring 10-carbon decanoyl (left) and 16-carbon palmitoyl (right) is shown in green and cleavage by IpaJ is shown in purple.
(D) Full-length human ARF1-6xHis was expressed alone (left) or co-expressed with IpaJ (right) in E. coli BL-21. Expected (theoretical) mass for full-length ARF1 protein is 22757 Da (minor species), and 22625 Da for methionine processed form (major species).
(E) In-gel fluorescence assay (top panel) showing Alexa 647-labeled myristoylated ARF1 expressed in E. coli BL-21 after untreated (control) or IpaJ treated samples. Coomassie staining was used to confirm equal loading of ARF1 protein (middle panel).
Protein myristoylation is necessary for peptide bond hydrolysis by IpaJ
Experimental alterations of myristoylated proteins have been challenging because N-myristoyl transferase (NMT) does not accommodate alternative fatty acids (e.g., palmitoyl) or tolerate amino acid substitution of Glycine or specific residues at the amino terminus of target proteins (Bhatnagar et al., 1998; Bhatnagar et al., 1997; Towler et al., 1987). The ability to organically synthesize acylated peptide substrates, allowed us to ask what common features of the ARF peptide (Gly2-Glu17) might be essential for IpaJ hydrolysis. To determine if the hydrocarbon chain length is important for IpaJ cleavage of acylated substrates, the 14-carbon myristoyl (C14:0) group of the ARF1 peptide was substituted for 16-carbon palmitoyl (C16:0) or 10-carbon decanoyl (C10:0). The resulting peptides (palm-Gly2-Glu17 and dec-Gly2-Glu17) have retention time close to 10.8 minutes, and therefore could be resolved from the non-acylated form and tested in the cleavage assay. While IpaJ cleaved the myr-Gly2-Glu17 completely (see Figure 2A), only small fractions of palm-Gly2-Glu17 and dec-Gly2-Glu17 were deacylated over a 30-minute time course (Figure 2C). These data suggest that the 14-carbon acyl group is a necessary component of IpaJ substrate recognition and membrane-based protein cleavage reaction (see below). To confirm that N-myristoylation is required for amide bond cleavage by IpaJ, we expressed and purified 6X-Histidine tagged ARF1 protein from E. coli, which does not possess an endogenous protein N-myristoylation system. The majority of ARF-His protein was processed by Methionine aminopeptidase, resulting in the full-length non-acylated form of ARF1 GTPase (MW = 22,625 Da). Importantly, co-expression with IpaJΔ50 had no effect on ARF1, suggesting that N-myristoylation is necessary for Gly2 cleavage (Figure 2D). To confirm that N-myristoylated glycine could be cleaved in the E. coli expression system, full-length ARF1 was co-expressed with yeast N-myristoyl transferase (yNMT) in the presence of azide-myristic acid. Purified azide-myristoylated ARF1 was incubated with recombinant IpaJΔ50 (or control), and the reaction products were labeled with alkyne-Alexa647 (Burnaevskiy et al., 2013). In-gel fluorescence confirmed that recombinant IpaJΔ50 cleaved in vitro myristoylated human ARF1 in context of the E. coli expression system (Figure 2E). Together, these data indicate that N-myristoylation of glycine-2 is essential for IpaJ proteolysis.
Defining the minimal substrate sequence for IpaJ proteolysis
Examination of substrates identified in myristoylome profile (Figure 1) revealed a great diversity in the N-terminal amino acid sequences (Figure 3A). The ability of IpaJ to cleave these different substrates in vitro suggests that IpaJ may recognize the myristoyl-glycine, but not the rest of the N-termini. To test this idea, we synthesized minimal IpaJ peptide substrate that only included myristoyl group, Gly2, and a lysine in position 3 (Lys3) followed by PEG3 and FITC (myr-Gly2-Lys3). As a control for cleavage, we conjugated PEG3/FITC directly to Lysine (Lys3), which would mimic the potential hydrolytic product. The myr-Gly2-Lys3 peptide had retention time of 12 minutes and could be distinguished from Lys3 that eluted as two peaks at 15 and 15.8 minutes (Figure 3B). Importantly, incubation of myr-Gly2-Lys3 with IpaJ resulted in a significant decrease of the 12 minutes peak and appearance of two peaks at 15 and 15.8 minutes (Figure 3C). This cleavage was not observed in the presence of IpaJC64A (Figure 3C).
Figure 3. Defining the N-myristoyl-glycine as the minimal IpaJ substrate.
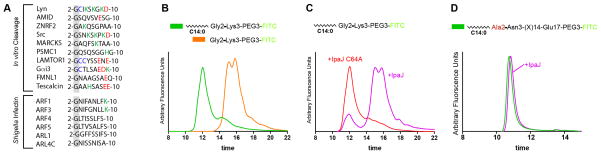
(A) Multiple sequence alignment of N-myristoylated proteins from selected in vitro substrates (top) and ARF/ARL proteins identified in vivo (bottom). Positively charged residues (green), negatively charged residues (red), and sites for protein palmitoylation (blue) are shown.
(B and C) fSEC analysis of the minimal myr-Gly-Lys peptide. (B) Trace of myr-Gly-Lys (green) and Lys peptide (orange) are shown. (C) myr-Gly-Lys treated with recombinant wild type IpaJ (purple) or IpaJ C64A mutant (red) are shown.
(D) fSEC analysis of the myr-Ala2-Glu17. Trace of untreated (green) and IpaJ (purple) treated myr-Ala2-Glu17 peptide is shown. The trace of the IpaJ sample was manually offset by 10 seconds for visualization purposes.
The structure of N-myristoyl transferase (NMT), the enzyme that catalyzes protein N-myristoylation, in complex with peptide substrate suggests that rotational flexibility of Gly2 is essential for transferring the myristoyl group, thereby making glycine the only residue that can be N-myristoylated (Bhatnagar et al., 1998; Bhatnagar et al., 1997; Farazi et al., 2001a). To test if Gly2 is essential in the recognition by IpaJ, we substituted Glycine for Alanine, which harbors a methyl side chain (Figure 3D). IpaJ did not cleave myr-Ala2-Glu17 peptide in solution, indicating that the small side chain of glycine or its rotational flexibility allows IpaJ to access the substrate peptide bond. Although we cannot exclude that certain NH2-terminal sequences contribute to the efficacy of IpaJ substrate recognition, these results indicate that myristoylated glycine is the minimal element required for IpaJ proteolysis.
IpaJ is highly specific in the context of Shigella infection
The in vitro myristoylome profiling data and reconstitution studies indicate that recombinant IpaJ has the potential to recognize and cleave the glycine of all N-myristoylated proteins. However, our previous experiments suggest that IpaJ may encode mechanisms of selectivity in vivo (Burnaevskiy et al., 2013). For example, ARF1/2p GTPases dominated the pool of potential IpaJ substrates identified by yeast genetics. Furthermore, we found that mammalian ARF1 is released from Golgi membranes prior to other N-myristoylated proteins in cells ectopically expressing IpaJ. To advance these insights through unbiased analyses, we performed whole cell myristoylome profiling to identify physiological substrates of T3SS translocated IpaJ during Shigella infection.
Initially, HeLa cells were labeled with alkyne-myristic acid alk-12 for 24 hours and then infected with either wild-type or ΔipaJ Shigella strain (Figure 4A). In-gel fluorescence revealed remarkable selectivity of IpaJ in the context of bacterial infection. Only one visible band (~20 kDa) had decreased intensity upon infection with wild-type Shigella, whereas the ΔipaJ strain had no noticeable effect (Figure 4B). This profile was notably different from the profile observed on cell lystates treated with recombinant IpaJ (Figure 1B). Mass-spectrometry was then used to compare the myristoylation profile of HeLa cells after infection with either wild type or the ΔipaJ Shigella strain. Subsequent analysis was focused on proteins that i) harbored a site for potential myristoylation and that, ii) were decreased reproducibly in two experimental repetitions (Table S2, S3). Consistent with the in-gel fluorescence profile (Figure 4B), only a limited number of proteins were affected by IpaJ during infection (Figures 4C and 4D). Specifically, ARF1 GTPase decreased upon infection with the wild type Shigella but not ΔipaJ mutant strain. However, ARF1 was not the only substrate, since other ARF isoforms, namely ARF3, ARF4 and ARF5, all decreased reproducibly to a similar extent (Table S2, S3). In addition, IpaJ also targeted the members of the related ARL (ARF-like) family. Specifically, ARL1 was identified as a reproducible hit, and ARL4C, while not recovered in the first run, was found to be affected to a similar extent as ARL1 in the repeated experiment (Table S2, S3). Intriguingly, we also noticed that E3 ubiquitin ligase ZNRF2 was reproducibly and strongly affected by the wild type Shigella to the extent of falling below detection limit, while not significantly changed by the ΔipaJ strain (Table S2, S3). To summarize, we found that in contrast to in vitro setting, IpaJ is highly specific in the context of Shigella infection. Furthermore, unbiased interrogation of the myristoylome profile revealed several ARF and ARL isoforms as well as E3 ubiquitin ligase ZNRF2 as potential new substrates of IpaJ.
Figure 4. IpaJ has limited number of substrates during infection.

(A) Design of the experiment to analyze protein cleavage by IpaJ during Shigella infection. Alk-12 labeled myristoylated proteins were conjugated azido-rhodamine for in-gel visualization or with azido-biotin for purification and mass-spectrometric analysis.
(B) In-gel fluorescent visualization of myristoylome profile of uninfected, Shigella WT, or Shigella ΔipaJ infected cells (left). Total protein is shown (right). Arrow indicates proteins cleaved by IpaJ.
(C) Representative scatter Plot of results from differential mass-spectrometric analysis. Proteins with N-terminal Glycine are shown in red. Y-axis shows the fold-increase or decrease of protein abundance in WT Shigella infected samples compared to Shigella ΔipaJ treated cells. Green shading marks the area of 2-fold deviation of protein abundance. Myristoylated proteins falling below the green shading are cleaved by IpaJ. For visualization purpose more than 10 fold over- or under-representation is shown as approximately 10 or −10, respectively.
(D) Identified N-myristoylated proteins in one representative sample showing the fold increase or decrease of peptide abundance between WT Shigella and Shigella ΔipaJ infected cells.
IpaJ functions as an ARF/ARL effector of bacterial origin
At first glance, results from the myristoylome profiling experiments appear to be inconsistent. That is, the diversity of in vitro substrates (Figure 1) contrasts dramatically with the small number of proteins affected by IpaJ during Shigella infection (Figure 4). Seeking to understand the mechanism of specificity for ARF/ARL proteins, we asked if additional interactions beyond the myristoylated glycine recruit IpaJ to its major physiological substrates. For example, the canonical GTPase domain of ARF1 binds upstream and downstream regulators (e.g. GEFs and GAPs) and signaling substrates (“effectors”) in a GTP-dependent manner. To test if IpaJ interacts directly with the GTPase domain, we incubated recombinant Maltose Binding Protein (MBP) tagged IpaJΔ50 with Glutathione S-transferase (GST) tagged ARF1ΔN17. As shown in Figure 5A, IpaJ was precipitated with glutathione immobilized GST-ARF1ΔN17, but not GST control protein.
Figure 5. IpaJ functions as an ARF substrate.

(A) Glutathione-pulldown of GST (control) or GST-ARF1Δ17 in the presence of MBP-IpaJΔ50. Proteins by separated with SDS PAGE and visualized by Coomassie staining.
(B) Glutathione-pulldown of GST-ARF1Δ17 in the nucleotide free (free), GTP, or GDP bound state in the presence of MBP-IpaJΔ50 (left) or the GAT domain of GGA1 (right). Proteins were separated by SDS PAGE and visualized by Coomassie staining.
(C) Glutathione-pulldown of GST-ARF5Δ17 in the nucleotide free (free), GTP, or GDP bound state in presence of MBP-IpaJΔ50 (left) and visualized as in (A).
(D and E) Crystal structure of GTP-bound ARF1 (green) in complex with the coatamer subunit gamma-1 (red) (PDB ID: 3TJZ). Binding interface comprises switch I and interswitch regions of ARF1. (E) Specifically, Ile49 and Trp66 (see Figure 5G) participate in this interaction.
(F and G) Glutathione-pulldown of GST-ARF1Δ17 harboring the indicated mutations in the presence of MBP-IpaJΔ50. Proteins were visualized as in (A).
(H) Homologous competition experiments between GTP-loaded ARF1 GTPase domain (ARF1Δ17) and the indicated effector substrates. Dissociation (Kd) constants are shown.
ARF proteins rapidly cycle between GDP-bound inactive and GTP-bound active states. We found that IpaJ does not interact with the GDP-bound or nucleotide free ARF1 or ARF5 GTPase domain (Figure 5B and 5C). These data are consistent with experiments showing that the myristoylated NH2-terminus of ARF1 (GDP) is protected from hydrolysis by IpaJ (Figure S1). In addition, the interaction between IpaJ and ARF GTPases is unlikely to resemble Sec7-domain GEF interactions that are stabilized in the nucleotide free state. Rather, the pattern of association between IpaJ and ARFs was similar to the natural GTPase effector Gamma ear protein Gamma Adaptin (GGA) that binds ARF1 in GTP-dependent manner (Figure 5B). ARF1 GTPase domain exhibited submicromolar affinity for IpaJ (Kd=0.28 μM) and low micromolar affinity for GGA (Kd=1.81 μM) indicating that the host-pathogen protein interaction falls within a physiological binding range (Figure 5H).
Recognition of the GTP-bound ARFs indicated that IpaJ might interact with structural elements and residues utilized by ARF effector proteins. Specifically, switch I, interswitch, switch II, and helix 3 of ARF1 undergo major conformational rearrangements upon guanine-nucleotide exchange, thereby allowing GTP-dependent interaction with downstream targets (Donaldson and Jackson, 2011; Kahn, 2009). To test if IpaJ recognized these conserved elements, we introduced point mutations to ARF1 GTPase domain and tested its interaction with recombinant IpaJΔ50. For this analysis we focused on residues that were previously shown by mutational screens and/or X-ray crystal structures to interact with regulators including GTPase activating proteins (GAPs) or downstream effectors such as POR1, MKLP1, GGA1, LTA1, AP-1, and COP subunits (Table S4). ARF1 double mutants I49T/F51Y, W66H/D72A, and W78C/R79G were significantly impaired in their ability to bind IpaJ (Figure 5F). To interrogate the interaction site more precisely, we introduced single mutations into ARF1 and tested them in the same assay. Three mutants (I49T, W66H, and W78C) completely abolished the interaction between ARF1 and IpaJ, whereas D72A had mild effect (Figure 5G). I49 and W66 belong to switch I and interswitch region of ARF1 and are found at the binding interface with the effector γCOP (Figures 5D and 5E) (Yu et al., 2012). W78 is a component of switch II that stabilizes the GTP-bound conformation. Consistent with these structural elements mediating binding to IpaJ, ARF1 W66H and ARF1 W78C had 10–20 fold lower affinities for IpaJ then the WT GTPase (Figure 5H). Taken together, we conclude that IpaJ acts as a GTPase “effector” of bacterial origin.
IpaJ displays selectivity toward Golgi associated GTPases
Our unbiased analysis of the myristoylome has implicated ARF6 as the only member of ARF family that is not cleaved efficiently during Shigella infection. To gain a better insight into ARF selectivity, we first tested the interaction between ARF6 and IpaJ in vitro. Surprisingly, and similarly to ARF1 and ARF5, the GTP-bound form of ARF6ΔN13 interacted directly with IpaJΔ50 in GST pulldown experiments (Figure 6A). Therefore, the inability of IpaJ to cleave ARF6 in cells cannot be attributed to differential GTPase domain recognition. We then tested if the protease recognized ARF isoforms at different locations in mammalian cells. When expressed alone, ARF1, ARF5, and ARL1 were localized to the Golgi, and ARF6 near the plasma membrane, consistent with previous reports (Figure 6C) (D’Souza-Schorey et al., 1995; Peters et al., 1995). IpaJ induced a complete redistribution of ARF1, ARF5 and ARL1 from Golgi membranes into the cytosol, whereas a significant fraction of ARF6 remained enriched on the plasma membrane (Figure 6C). In-gel fluorescence assay further supported the idea that ARF6 is a poor substrate for IpaJ in cells (Figure 6B, right panel). These data suggest that IpaJ selectively cleaves Golgi-associated GTPases. In support of this hypothesis, we found that the catalytically inactive mutant IpaJ C64A is trapped on Golgi membranes (Figure 6D, inset 1). This stable complex is likely mediated by ARF GTPase domain interaction since WT IpaJ is cytosolic (Figure S2A) and since Brefeldin A (a potent ARF inhibitor) prevented IpaJ C64A binding to disrupted Golgi membranes (Figure 6D inset 2). While interpretation of these two experiments are limited by gross changes in Golgi structure, we also found that IpaJ C64A co-localized with ARF1 (Figure 6D) as well as Golgi-associated ARF5, and ARL1 (Figure S2B) but not plasma membrane associated ARF6 (Figure 6E). From the totality of these findings, we now propose that selective cleavage of Golgi associated ARF-family members is largely mediated through IpaJ interaction with the GTP-bound ARF GTPase domain. In addition, plasma membrane localized ARF6 and potentially other N-myristoylated substrates are likely to be limited by other, yet to be identified, factors that may include the subcellular localization of the protease or its substrates.
Figure 6. IpaJ cleaves only Golgi-localized GTPases.

(A) Glutathione-pulldown of GST-ARF6Δ13 in the nucleotide free (free), GTP, or GDP bound state in presence of MBP-IpaJΔ50 (left) and visualized as in Figure 5A.
(B) In-gel fluorescence showing myristoylated ARF1 or ARF6 after co-expression with IpaJ or IpaJC64A in HeLa cells (top panel). Equal loading of ARF1/ARF6 (middle panel) and expression of IpaJ (lower panel) were confirmed by western blot analysis. Graph shows the quantification of fluorescent signal (in arbitrary units (au) +/− SD) from three experimental repetitions using ImageJ.
(C) Fluorescent Microscopy of EGFP-tagged ARF1-, ARF5-, ARL1-, and ARF6 when co expressed with mCherry vector (control) or IpaJ-mCherry (IpaJ). Inset box shows ARF protein localization (middle panel) and cis-Golgi (right panel) visualized by gm130 immunofluorescence. The bottom two rows focus on the plasma membrane localization of ARF6.
(D) Fluorescent Microscopy mCherry tagged IpaJC64A and EGFP-tagged ARF1 in HeLa cells left untreated (top row) or treated with Brefeldin A (bottom row). Cis-Golgi was visualized with gm130 antibodies.
(E) Fluorescent Microscopy mCherry tagged IpaJC64A and EGFP-tagged ARF6 in HeLa cells. Inset (1) is focused on the cis-Golgi and inset (2) is focused on plasma membrane.
Discussion
While the importance of N-myristoylation in cellular signaling is widely recognized, the specific functional role of this modification often remains unclear largely owing to technical challenges of studying lipidated proteins in vivo and the irreversibility of this modification. We had previously identified the Shigella T3SS effector protein IpaJ as the first N-myristoylation reversing enzyme. The present study extends our earlier work in several ways. First, by performing unbiased proteomic analysis of N-myristoylated proteins, we find that IpaJ is a highly selective protease that primarily recognizes Golgi-associated ARF/ARL family GTPases. Further reconstitution of IpaJ proteolysis using artificial peptide substrates has revealed a combination of specificity determinants that explains how IpaJ can display broad substrate recognition in vitro, but is highly selective upon Type III secretion. Based on the combination of in vivo myristoylome profiling, biochemical interactions studies, and cellular analyses we now propose a concerted model of IpaJ proteolysis in context of ARF GTPase cycle on Golgi membranes during Shigella infection (Figure 7).
Figure 7. Concerted Model of IpaJ Function at the Golgi apparatus.

(A) Model of ARF GTPase function in Golgi regulation. Top panel: GEF-catalyzed nucleotide exchange activates ARF and recruits it onto membrane via the myristoyl switch. GTP-bound ARF recruits downstream effectors to promote formation of transport vesicles. Bottom panel: Type III secreted IpaJ recognizes GTP-ARF and inactivates it by irreversibly releasing it from the membranes via Myristoyl-Glycine Cleavage.
(B) Concerted model showing the proposed sequence of events for IpaJ cleavage of myristoylated ARF proteins at the Golgi apparatus.
(C) Comparison of the target motifs for IpaJ and the C39 peptidase family. Arrows indicate the protease cleavage sites. Glycines are colored red and myristoyl group colored blue.
A concerted model of IpaJ function during Shigella infection
Step 1: Recognition of N-myristoylated ARF GTPase substrates
Our infection studies have revealed that Golgi associated ARFs are specific substrates of IpaJ cleavage. Two lines of evidence support the idea that the association of IpaJ with the GTPase domain is the initial event of ARF recognition during Shigella infection. First, we found that IpaJ directly interacts with the GTPase domain of ARF family members and that this interaction is GTP dependent. Structural elements involved in GTP-dependent host effector interactions (e.g GGA1 and γCOP) are also critical for IpaJ interaction indicating that the protease functions as a GTPase effector of bacterial origin. The effector-like mechanism of action implies that IpaJ may need to compete with host proteins for recognition and cleavage of ARF GTPases. Interestingly, IpaJ exhibits near 10 fold stronger affinity for ARF1 than GGA1, suggesting that it may compete effectively with the natural signaling system. However, we can not conclude rule out the possibility that IpaJ interacts non-competitively with pre-assembled ARF-effector complexes similar to other virulence mechanisms (Selyunin et al., 2014). Nevertheless, these studies provide the molecular basis for selective recognition of ARF GTPases at the Golgi. Second, and perhaps most importantly, subcellular localization studies indicate that the catalytically inactive IpaJ mutant (IpaJ C64A) associates with Golgi membranes at least partially in an ARF-dependent manner. These data strongly suggest that GTPase domain binding occurs independently and prior to peptide bond hydrolysis.
Step 2: Orientation of the catalytic core through myristoyl Glycine substrate recognition
Once IpaJ is targeted to the Golgi through ARF GTPase interactions (step 1), the catalytic residues (e.g. C64 nucleophile) need to be properly oriented toward the carbonyl carbon substrate of Gly2 prior to catalysis. We propose that IpaJ binding to the myristoylated hydrocarbon fulfills this requirement (step 2). Mass spectrometry data indicated that the myristoyl group is absolutely essential for Gly2 cleavage by IpaJ (Figure 2). We also found that C14:0 labeled ARF peptide was a much better substrate than C10- or C16-labeled peptides (Figures 2A and 2C) and that myristoylated Glycine plus one additional residue (myr-Gly2-X3) constitutes the minimal IpaJ substrate. Taken together, these data suggest that IpaJ possess a hydrophobic pocket or groove that accommodates 14 carbon saturated fatty acid chains. This microbial designed interaction would be remarkably similar to other myristoyl modifying enzymes, including N-myristoyl transferase (e.g., NMT) and Sirtuin 6 (Sirt6) that specifically accommodate 14-carbon fatty acid chains through large hydrophobic interactions (Bhatnagar et al., 1998; Jiang et al., 2013).
While these findings indicate that myristoyl recognition is necessary for IpaJ function, two additional pieces of information, one based on a series of experimental observations and the other from circumstantial evidence, support the idea that myristoyl group properly orients the substrate for peptide bond hydrolysis. First, we found that a myristoylated Alanine peptide (myr-Ala2-Glu17 peptide) could not be cleaved by IpaJ (Figure 3D). Addition of the methyl side chain may sterically inhibit the access of IpaJ to its substrate. Alternatively, and similarly to NMT (Farazi et al., 2001b), IpaJ may require the rotational flexibility of Glycine to orient Cysteine 64 for nucleophilic attack. Substitution of Alanine for Glycine would restrict this conformational flexibility, preventing efficient cleavage of the substrate. Beyond this evidence, we previously assigned IpaJ to the C39 peptidase-like family (DUF3335) based on conserved secondary structural elements and a high degree of amino acid identity around the C-H-D catalytic triad (Burnaevskiy et al., 2013). Interestingly, the closely related C39 peptidase family of bacteriocin processing enzymes recognizes di-Glycine motifs of substrate leader peptides (Havarstein et al., 1995). As shown in Figure 7C, myristoylated Glycine is chemically analogous to di-Glycine, suggesting that the substrate recognition motif of IpaJ is analogous to the C39-peptidase family. Thus, it is likely that IpaJ protease has evolved from these ancestral proteases to recognize the myristoylated-Glycine moiety independently of other specificity determinants.
Step 3: Peptide bond hydrolysis
Mutational data supports a classic Cysteine protease reaction mechanism including deprotonation of Cys64 thiol group by His218 followed by nucleophilic attack on substrate carbonyl carbon (Figure 7B). This rate-limiting event is followed by release of Asn3-ARF GTPase and hydrolysis of the thiol ester intermediate to generate myr-Gly2 carboxylic acid cleavage product (not shown).
Step 4: Release from ARF GTPase and recycling of IpaJ
Because IpaJ functions as an ARF “effector” (step 1), the enzyme must then be released from the GTPase following peptide bond hydrolysis. We speculate that substrate release by IpaJ coincides with the GTP hydrolysis, since IpaJ does not interact with the nucleotide free or the GDP-bound ARF form. However, it is not yet clear whether ARF retains GTP after cleavage. It is possible that cleavage of the myristoylated Glycine promotes a conformational change in ARF that cannot be accommodated by IpaJ (e.g., the nucleotide free state). Alternatively, IpaJ could remain associated with the GTPase domain of ARF after hydrolysis. If IpaJ binds and cleaves ARF without competition with host effector proteins (see step 1), the stabilization of an IpaJ/ARF/effector complex on Golgi membranes may persist until GTP hydrolysis is stimulated by host GTPase activating proteins (GAPs). The stable association of IpaJ C64A on Golgi membrane is consistent with the idea that protease may interact with ARF non-competitively throughout the GTPase cycle. In either event, ARF is eventually released from the membrane and cannot be recruited again, leading to exhaustion of the functional ARF pool and inhibition of vesicular trafficking.
Manipulating and monitoring cellular lipidomes using bacterial agents
The ability of IpaJ to cleave the myristoylated glycine of numerous proteins in vitro, yet exhibit high selectivity for the ARF family GTPases during Shigella infection suggests that the protease could be reprogrammed to cleave novel substrates in vivo. By attaching a protein-binding domain to IpaJ that associates with a specific myristoylated target, our findings indicate that IpaJ could be redirected from ARF-family GTPase members to potentially novel substrates in cells. While further work is needed to address this possibility, the modular nature of the substrate binding and cleaving mechanism of IpaJ, as well as the ability to reprogram other enzymes-substrate interactions in a similar manner, holds great promise for the design of highly specific N-myristoylation inhibitors. It is also important to highlight that IpaJ belongs to growing family of functionally related bacterial proteases, including Yersinia YopT and Legionella RavZ, which proteolytically eliminate lipid groups from host proteins as a mechanism of pathogenesis (Choy et al., 2012; Shao et al., 2002). By analogy to IpaJ, it might therefore be possible to use bacterial proteases to study protein acylation in a variety of cellular contexts. Taken together, the combination of bacterial modifiers of protein acylation coupled to the growing availability of click-compatible probes for acyl-based proteomics should facilitate further investigations on protein lipodomics in human health and disease.
Materials and Methods: Methods
This manuscript contains supplementary information with extended materials and methods description.
Plasmids and cloning
All cloning was performed with either conventional PCR techniques or Gateway® compatible vectors according to manufacturer recommendations (Invitrogen). Site-directed mutations were generated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene).
In vitro cleavage of proteins and in-gel fluorescence
To assay in vitro substrates of IpaJ, HeLa cells were cultured in the presence of 20 uM Alk-12 for. For in vitro cleavage of ARF1 and ARF6, cells were also transfected with respective STREPtag-containing constructs. Cell lysate was incubated with MBP-IpaJΔ50 for 30 minutes. 100 ug of protein solutions were click-labeled with rhodamine-azide (Charron et al., 2009) and visualized in gel on Typhoon Trio imaging system. The remaining portions were used for azido-biotin coupling and mass-spectrometric analysis.
Bacterial infection of cultured cells
For Shigella infections, HeLa cells were cultured in the presence of 20 μM of alk-12 (Charron et al., 2009). Cell were infected with Shigella flexneri M90T for 6 hours and lysed for protein click-labeling and subsequent in-gel visualization and mass-spectrometry.
Proteomic Identification of alk-12 Labeled Proteins
Proteomic analysis of the acylated proteins was performed essentially as described previously (Wilson et al., 2011) with modifications (see supplementary methods). Briefly, 2 mg of protein was conjugated with azido-biotin. Protein samples were incubated with streptavidin beads (Thermo). The beads were then washed, resuspended in 25 mM ammonium bicarbonate (ABC) and incubated with trypsin at 37 °C overnight. The supernatant was collected, and prepared for mass-spectrometry. LC-MS analysis was performed with a Dionex 3000 nano-HPLC coupled to an LTQ-Orbitrap ion trap mass spectrometer (ThermoFisher). Acquired tandem MS spectra were extracted using ProteomeDiscoverer v. 1.4.0.288 (Thermo, Bremen, Germany) and queried against Uniprots complete human database. For a matched protein, its abundance was calculated based on the average area of the three most abundant peptides (Silva et al., 2006).
Synthesis and cleavage of the acylated peptides
Peptides were synthesized manually on a scale of 50 micromole using Rink-amide copoly-(styrene-divinylbenzene) resin (Novabiochem, CA) with a substitution value of 0.41 mmol/g, Fmoc protected amino acids and HATU activation in NMP. Purification was achieved on either a Vydac C4 or C18 (250×10mm) column using 220 nm wavelength detection. For enzymatic cleavage peptides were incubated with IpaJΔN50 at 37°C for indicated amount of time. Reaction was stopped by addition of methanol with 0.5M HCl. The supernatant was analyzed by HPLC gel filtration with TOSOH TSK-GEL® SuperSW3000 column.
Recombinant protein purification and in vitro binding
Recombinant proteins were expressed in BL21-DE3 E. coli strains and purified using gluthatione- or nickel-coupled resin. For in vitro binding, ARF proteins were nucleotide-exchanged using EDTA and were incubated with MBP-IpaJΔN50 or MBP-GGA176-215 and GST-agarose beads. Beads were washed and ARF -GST was eluted with 10 mM glutathione and ran on SDS-PAGE. Homologous competition assays were conducted as describe in the extended Material Methods accompanying this manuscript.
In vitro cleavage of recombinant ARF1
The cleavage of bacterially expressed ARF1 was performed as described previously (Burnaevskiy et al., 2013). Briefly, ARF1-His was expressed in E. coli BL-21 alone or co-expressed with yeast NMTp in the presence of azide myristic acid. MBP-IpaJΔN50 was expressed separately in E. coli BL-21. The lysates of the ARF1 and IpaJΔN50 expressing cells were mixed together and incubated at 37 °C. ARF1 was then purified using nickel agarose (Qiagen). N-myristoylated proteins were labeled with Alex Fluor® 647 Alkyne (Invitrogen) and visualized in gel. Cleavage of non-myristoylated ARF1 was analyzed by intact mass-spectrometry.
Supplementary Material
Acknowledgments
We would like to thank the Alto lab for helpful discussion preparation of this manuscript. We acknowledge the services of the UTSW proteomics core for intact mass-spectrometry analysis. We thank Haydn Ball of UT Southwestern Protein Chemistry Technology Core for the peptides synthesis. We thank Ryan Hibbs for his help with fSEC analysis and the Proteomics Resource Center at The Rockefeller University for mass spectrometry analysis. H.C.H. acknowledges support from NIH-NIGMS R01GM087544 grant and Starr Cancer Consortium I7-A717. L.E.R. acknowledges support from NIH-NIAID (NRSA F32 I-F32-AIO98384). This work was supported by grants from the National Institute of Health (NIAID; AI083359 and NIGMS; GM100486), the Welch Foundation (#I-1704), and the Burroughs Wellcome Fund to N.M.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alto NM, Orth K. Subversion of cell signaling by pathogens. Cold Spring Harb Perspect Biol. 2012;4:a006114. doi: 10.1101/cshperspect.a006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar RS, Futterer K, Farazi TA, Korolev S, Murray CL, Jackson-Machelski E, Gokel GW, Gordon JI, Waksman G. Structure of N-myristoyltransferase with bound myristoylCoA and peptide substrate analogs. Nat Struct Biol. 1998;5:1091–1097. doi: 10.1038/4202. [DOI] [PubMed] [Google Scholar]
- Bhatnagar RS, Schall OF, Jackson-Machelski E, Sikorski JA, Devadas B, Gokel GW, Gordon JI. Titration calorimetric analysis of AcylCoA recognition by myristoylCoA:protein N-myristoyltransferase. Biochemistry. 1997;36:6700–6708. doi: 10.1021/bi970311v. [DOI] [PubMed] [Google Scholar]
- Burnaevskiy N, Fox TG, Plymire DA, Ertelt JM, Weigele BA, Selyunin AS, Way SS, Patrie SM, Alto NM. Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature. 2013;496:106–109. doi: 10.1038/nature12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron G, Zhang MM, Yount JS, Wilson J, Raghavan AS, Shamir E, Hang HC. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc. 2009;131:4967–4975. doi: 10.1021/ja810122f. [DOI] [PubMed] [Google Scholar]
- Choy A, Dancourt J, Mugo B, O’Connor TJ, Isberg RR, Melia TJ, Roy CR. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. 2012;338:1072–1076. doi: 10.1126/science.1227026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis GR, Van Gijsegem F. Assembly and function of type III secretory systems. Annual review of microbiology. 2000;54:735–774. doi: 10.1146/annurev.micro.54.1.735. [DOI] [PubMed] [Google Scholar]
- Cui J, Shao F. Biochemistry and cell signaling taught by bacterial effectors. Trends in biochemical sciences. 2011;36:532–540. doi: 10.1016/j.tibs.2011.07.003. [DOI] [PubMed] [Google Scholar]
- D’Souza-Schorey C, Li G, Colombo MI, Stahl PD. A regulatory role for ARF6 in receptor-mediated endocytosis. Science. 1995;267:1175–1178. doi: 10.1126/science.7855600. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Jackson CL. ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat Rev Mol Cell Biol. 2011;12:362–375. doi: 10.1038/nrm3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farazi TA, Waksman G, Gordon JI. The biology and enzymology of protein N-myristoylation. The Journal of biological chemistry. 2001a;276:39501–39504. doi: 10.1074/jbc.R100042200. [DOI] [PubMed] [Google Scholar]
- Farazi TA, Waksman G, Gordon JI. Structures of Saccharomyces cerevisiae N-myristoyltransferase with bound myristoylCoA and peptide provide insights about substrate recognition and catalysis. Biochemistry. 2001b;40:6335–6343. doi: 10.1021/bi0101401. [DOI] [PubMed] [Google Scholar]
- Frearson JA, Brand S, McElroy SP, Cleghorn LA, Smid O, Stojanovski L, Price HP, Guther ML, Torrie LS, Robinson DA, et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature. 2010;464:728–732. doi: 10.1038/nature08893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg J. Structural basis for activation of ARF GTPase: mechanisms of guanine nucleotide exchange and GTP-myristoyl switching. Cell. 1998;95:237–248. doi: 10.1016/s0092-8674(00)81754-7. [DOI] [PubMed] [Google Scholar]
- Grammel M, Hang HC. Identification of lysine acetyltransferase substrates using bioorthogonal chemical proteomics. Methods in molecular biology. 2013;981:201–210. doi: 10.1007/978-1-62703-305-3_16. [DOI] [PubMed] [Google Scholar]
- Hang HC, Linder ME. Exploring protein lipidation with chemical biology. Chemical reviews. 2011;111:6341–6358. doi: 10.1021/cr2001977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper DR, Gilbert RL. Viral lipoproteins: the role of myristoylation. Biochemical Society transactions. 1995;23:553–557. doi: 10.1042/bst0230553. [DOI] [PubMed] [Google Scholar]
- Havarstein LS, Diep DB, Nes IF. A family of bacteriocin ABC transporters carry out proteolytic processing of their substrates concomitant with export. Mol Microbiol. 1995;16:229–240. doi: 10.1111/j.1365-2958.1995.tb02295.x. [DOI] [PubMed] [Google Scholar]
- Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013;496:110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn RA. Toward a model for Arf GTPases as regulators of traffic at the Golgi. FEBS Lett. 2009;583:3872–3879. doi: 10.1016/j.febslet.2009.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson BA, Raju RV, Moyana TN, Sharma RK. Increased N-myristoyltransferase activity observed in rat and human colonic tumors. Journal of the National Cancer Institute. 1995;87:1630–1635. doi: 10.1093/jnci/87.21.1630. [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh S, Eisenhaber B, Eisenhaber F. N-terminal N-myristoylation of proteins: prediction of substrate proteins from amino acid sequence. Journal of molecular biology. 2002;317:541–557. doi: 10.1006/jmbi.2002.5426. [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh S, Eisenhaber F. Myristoylation of viral and bacterial proteins. Trends in microbiology. 2004;12:178–185. doi: 10.1016/j.tim.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh S, Gouda M, Novatchkova M, Schleiffer A, Schneider G, Sirota FL, Wildpaner M, Hayashi N, Eisenhaber F. MYRbase: analysis of genome-wide glycine myristoylation enlarges the functional spectrum of eukaryotic myristoylated proteins. Genome Biol. 2004;5:R21. doi: 10.1186/gb-2004-5-3-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters PJ, Hsu VW, Ooi CE, Finazzi D, Teal SB, Oorschot V, Donaldson JG, Klausner RD. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. Journal of Cell Biology. 1995;128:1003–1017. doi: 10.1083/jcb.128.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raulin J. Lipids and retroviruses. Lipids. 2000;35:123–130. doi: 10.1007/BF02664760. [DOI] [PubMed] [Google Scholar]
- Ribet D, Cossart P. Pathogen-mediated posttranslational modifications: A re-emerging field. Cell. 2010;143:694–702. doi: 10.1016/j.cell.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyunin AS, Reddick LE, Weigele BA, Alto NM. Selective protection of an ARF1-GTP signaling axis by a bacterial scaffold induces bidirectional trafficking arrest. Cell reports. 2014;6:878–891. doi: 10.1016/j.celrep.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao F, Merritt PM, Bao Z, Innes RW, Dixon JE. A Yersinia effector and a Pseudomonas avirulence protein define a family of cysteine proteases functioning in bacterial pathogenesis. Cell. 2002;109:575–588. doi: 10.1016/s0092-8674(02)00766-3. [DOI] [PubMed] [Google Scholar]
- Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Molecular & cellular proteomics: MCP. 2006;5:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- Tate EW, Bell AS, Rackham MD, Wright MH. N-Myristoyltransferase as a potential drug target in malaria and leishmaniasis. Parasitology. 2013:1–13. doi: 10.1017/S0031182013000450. [DOI] [PubMed] [Google Scholar]
- Tate EW, Kalesh KA, Lanyon-Hogg T, Storck EM, Thinon E. Global profiling of protein lipidation using chemical proteomic technologies. Current opinion in chemical biology. 2014;24C:48–57. doi: 10.1016/j.cbpa.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinon E, Serwa RA, Broncel M, Brannigan JA, Brassat U, Wright MH, Heal WP, Wilkinson AJ, Mann DJ, Tate EW. Global profiling of co- and post-translationally N-myristoylated proteomes in human cells. Nature communications. 2014;5:4919. doi: 10.1038/ncomms5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towler DA, Adams SP, Eubanks SR, Towery DS, Jackson-Machelski E, Glaser L, Gordon JI. Purification and characterization of yeast myristoyl CoA:protein N-myristoyltransferase. Proc Natl Acad Sci U S A. 1987;84:2708–2712. doi: 10.1073/pnas.84.9.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JP, Raghavan AS, Yang YY, Charron G, Hang HC. Proteomic analysis of fatty-acylated proteins in mammalian cells with chemical reporters reveals S-acylation of histone H3 variants. Molecular & cellular proteomics: MCP. 2011;10:M110. doi: 10.1074/mcp.M110.001198. 001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Breitman M, Goldberg J. A structure-based mechanism for Arf1-dependent recruitment of coatomer to membranes. Cell. 2012;148:530–542. doi: 10.1016/j.cell.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.