

Abstract
DJ1 is a multifunctional protein whose mutations cause autosomal recessive early-onset Parkinson disease (PD). DJ1 loss of function disrupts mitochondrial function, but the signaling pathway whereby it interferes with energy metabolism is unknown. Here, we found that mouse embryonic fibroblasts obtained from DJ1-null (dj1-/-) mice showed higher glycolytic rate than those from wild type DJ1 (dj1+/+). This effect could be counteracted by the expression of the full-length cDNA encoding the wild type DJ1, but not its DJ1-L166P mutant form associated with PD. Loss of DJ1 increased hypoxiainducible factor-1α (Hif1α) protein abundance and cell proliferation. To understand the molecular mechanism responsible for these effects, we focused on PTEN-induced protein kinase-1 (Pink1), a PD-associated protein whose loss was recently reported to up-regulate glucose metabolism and to sustain cell proliferation (Requejo-Aguilar, Lopez-Fabuel, Fernandez, Martins, Almeida and Bolaños, 2014, Nature Communications 5, 4514). Noticeably, we found that the alterations in glycolysis, HIF1α and proliferation of DJ1-deficient cells were abrogated by the expression of Pink1. Moreover, we found that loss of DJ1 decreased pink1 mRNA and Pink1 protein levels, and that DJ1, by binding with Foxo3a transcription factor, directly interacted with pink1 promoter stimulating its transcriptional activity. These results indicate that DJ1 regulates cell metabolism and proliferation through Pink1.
Keywords: DJ1, Pink1, Foxo3a, glycolysis, Hif1, Parkinson's disease
Introduction
DJ1 was initially identified as an oncogene [1] and it has been attributed antioxidant [2, 3], chaperone [4, 5], transcriptional [6, 7] and RNA regulatory [8] properties. Mutations in DJ1, such as L166P, cause loss of mitochondrial integrity [9, 10] and contribute to the development of autosomal recessive early-onset Parkinson's disease (PD) [10]. Remarkably, the function of DJ1 has been linked with those of Pink1 and Parkin, two proteins also found mutated in autosomal recessive early-onset forms of PD [8]. Whilst PD relevant mutations in DJ1, Pink1 and Parkin lead to mitochondrial dysfunction [11], their metabolic consequences are not fully understood, and the mechanistic link between these proteins is still a matter of debate. Thus, it has been found that the physical interaction of these proteins protects cells against oxidative stress and promotes the degradation of unfolded proteins [12, 13]. However, other laboratories could not find such a physical interaction, and propose that DJ1 would either act on the same upstream pathway, or in parallel to Pink1/Parkin [14, 15] at protecting mitochondrial integrity [16, 17].
In view that DJ1 loss-of-function causes mitochondrial energy failure [18], here we hypothesized whether DJ1 deficiency altered glucose metabolism by interacting with PTEN-induced protein kinase-1 (Pink1), a protein whose loss was recently reported to up-regulate glucose metabolism to sustain cell proliferation [19]. We found that loss of DJ1 increased the glycolytic rate and cell proliferation, and that these effects were counteracted by the expression of Pink1. Furthermore, we found that DJ1 physically interacted with Foxo3a to activate pink1 promoter. These results indicate that DJ1 represses glucose metabolism and cell proliferation by transcriptionally activating Pink1.
Experimental
Statement regarding the ethical use of animals
All animals used in this work were bred at the Animal Experimentation Unit of the University of Salamanca, in accordance with Spanish legislation (RD 1201/2005) under license from the Spanish Ministry of Science and Innovation. Protocols were approved by the Bioethics Committee of the University of Salamanca.
Cell cultures
Mouse embryonic fibroblasts (MEF) were prepared from fetal (E13.5) dj1-/- and dj1+/+ (wild type; WT) offspring, derived from crossing dj1-/- mice at 11th generation under a C57Bl6/J background, generously donated by Wolfgang Wurst (Institute of Developmental Genetics, Helmholtz Zentrum München, Neuherberg, Germany) [20], with wild type C57Bl6/J mice. Cells were seeded (105 cells/cm2) in high glucose (25 mM) DMEM (Sigma, Madrid, Spain) with 10% fetal calf serum (FCS; Roche Diagnostics, Heidelberg, Germany), L-glutamine (4 mM) and 1% penicillin-streptomycin-amphoteryicin (Sigma), and incubated at 37°C in a humidified 5% CO2-containing atmosphere. MEF were used at passages 10-20.
Cells transfection
For plasmid transfections, 1.6 μg (per 103 cells) of the plasmid was mixed with 1 μM polyethylenimine (PEI, Sigma) and pre-incubated for 10 min at room temperature in Optimem medium (Invitrogen). Cells were then incubated with this PEI/DNA-containing mixture at 37 °C for 1 h, followed by washing with PBS and further incubated in the corresponding culture medium for 24 h (glycolytic flux and western blotting analyses) or 48 h (luciferase assay).
Assessment of cell proliferation
This was achieved by bromo-deoxy-uridine (BrdU) incorporation into DNA followed by flow cytometric analysis after a 3 h-pulse with BrdU (10 mg/mL) using the APC BrdU Flow Kit (Becton Dickinson Biosciences, Franklin Lakes, NJ, USA) following a previously described protocol [21]. The proportions of cell cycle phases were also determined by flow cytometric analyses of 7-AAD-stained cells. Cell proliferation was also confirmed by direct counting under light microscopy.
Determination of the glycolytic rate
Suspensions of known amounts of cells (4-5×105 cells) obtained by smooth detaching from the cultures were incubated in sealed vials containing a central well containing 1 ml of water, which was used for 3H2O trapping. Cells were incubated in the presence of 5 μCi of D-[3-3H]glucose in a Krebs-Henseleit buffer (11 mM Na2HPO4, 122 mM NaCl, 3.1 mM KCl, 0.4 mM KH2PO4, 1.2 mM MgSO4, 1.3 mM CaCl2; pH 7.4) containing 5 mM D-glucose at 37 °C. In order to ensure an adequate O2 supply for oxidative metabolism by the cells throughout the 90 min incubation period, the gas phase of the vials containing the cells was supplied with extra O2 before the vials were sealed. The glycolytic flux was measured by assaying the rate of 3H2O production from [3-3H]glucose, as detailed previously [22]. Lactate released to the culture media was also determined to estimate the glycolytic rate. To do so, the increments in absorbance of the culture media samples were measured at 340 nm in a mixture containing 1 mM NAD+ and 22.5 U ml-1 lactate dehydrogenase in 0.25 M glycine/0.5 M hydrazine/1 mM EDTA at pH 9.5.
Plasmids and site-directed mutagenesis
pcDNA-DEST47-Pink1-C-GFP was purchased from Addgene (plasmid 13316) [23]. GFP-DJ1 (EX-10087-M03) was purchased from GeneCopoeia (Rockville, MD, USA). This GFP-DJ1 cDNA fusion construct was subjected to site-directed mutagenesis to change Leu166 to Pro166 using the QuikChange XL site directed mutagenesis kit (Stratagene, La Jolla, CA, USA) using the following forward and reverse primers, respectively: 5′-GGGACCAGCTTCGAGTTTGCGCCTGCAATTGTTGAAGCCCTGAATGGC-3′ and 5′-GCCATTCAGGGCTTCAACAATTGCAGGCGCAAACTCGAAGCTGGTCCC-3′ (mutated nucleotides underlined). For Pink1 promoter activity assays, we used pGL4-based luciferase reporter constructs harboring either the 0.4 kb regulatory region of Pink1 (pink1 wild type promoter) or its 3 single-nucleotide substitution mutations within the Foxo3a-binding site (pink1 FOXO3a-mutant promoter), which were generously donated by Y. Mei (Fujian, China) [24]. To investigate cholecystokinin (cck) promoter activity, we used the pGL3-CCK-1615 construct, kindly provided by H. Ariga (Sapporo, Japan) [25].
RT-qPCR analysis
This was performed in total RNA samples purified from MEF. Reverse transcription of pink1 was performed for 50 min at 48°C, and PCR conditions were 10 min at 95°C followed by 35 cycles of 30 s at 95°C plus 30 s at 54 °C and 30 s at 72 °C, using the Maxima SYBR Green qRT-PCR Master Mix (Fermantas) and the following primers (purchased from Thermo Scientific, Offenbach, Germany): forward, 5′-TCAGGAGATCCAGGCAATTTT-3′; reverse, 5′-GCATGGTGGCTTCATACACA-3′. Expression values were normalized using β-actin as housekeeping gene using the following primers: forward 5′-CGATGCCCTGAGGCTCTTTT-3′; reverse, 5′-CAACGTCACACTTCATGATG-3′. The mRNA abundance of pink1 transcript was normalized to the β-actin mRNA abundance obtained in the same sample. The resulting normalized values of the dj1-/-samples, with or without previous expression of dj1 or dj1 (L166P), were expressed as the fold change compared to the corresponding normalized values of the WT samples.
Luciferase assay
To analyze pink1 and cck promoter activity, WT and dj1-/- MEFs were transfected with either the wild type pink1, the Foxo3a-mutant pink1 or the cck promoters-driven luciferase reporter constructs [24, 25], together with empty vector (M03; GeneCopoeia), dj1-expressing vector (EX-10087-M03; GeneCopoeia) or dj1 (L166P) vector. After 24 h, cells were lysed and luciferase activity was determined using a Luciferase Assay System kit (Promega Biotech Iberica) following the manufacturers' instructions. Values were normalized to those found in either the WT or the dj1-/- samples, as indicated.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as described previously [26]. Formaldehyde cross-linked chromatin fragments were immunoprecipitated with either 10 μg of anti-DJ1 antibody (Abcam, Cambridge Science Park, Cambridge, UK) or 2 μg of anti-histone H3 antibody (Abcam) overnight at 4 °C. Control samples were treated in the same way except that no antibody was present. Immune-complexes were captured with protein A/G-agarose and washed with 150 mM NaCl, 13.5% sucrose, 1% Triton X-100, 0.2% SDS, 0.02 NaN3, 5 mM EDTA, 20 mM Tris–Cl pH 8.0, and then 500 mM NaCl, 1% Triton X-100, 0.1% deoxycholate, 0.2% NaN3, 1 mM EDTA and 50 mM HEPES pH 7.5, and finally Tris–EDTA (10 mM TRIS, 1 mM EDTA, pH 8.0). Immunecomplexes were then eluted by incubation with 1% SDS and 100 mM NaHCO3. To reverse cross-links, eluates and input-DNA were incubated overnight at 65 °C. DNA was purified using the Qiagen PCR clean-up kit (Qiagen, Valencia, CA, USA). PCRs were performed with primers flanking the Pink1 promoter (forward, 5′-TGAGAGCACTTGGGAGTGGGGGAGAAGAG -3′ and reverse, 5′ CTGTCGCACCGCCATGGTGGCGCGGTGACC -3′). PCR products were resolved by electrophoresis in agarose gel and visualized with ethidium bromide.
Western blotting
After transfections and treatments, cells were lysed in a buffer containing 2% sodium dodecylsulphate, 2 mM EDTA, 2 mM EGTA, 50 mM Tris pH 7.5, supplemented with phosphatase inhibitors (1 mM Na3VO4, 50 mM NaF) and protease inhibitors (100 μM phenylmethylsulfonyl fluoride, 50 μg/mL anti-papain, 50 μg/mL pepstatin, 50 μg/mL amastatin, 50 μg/mL leupeptin, 50 μg/mL bestatin and 50 μg/mL soybean trypsin inhibitor). Samples were stored on ice for 30 min and boiled for 10 min. Aliquots of cell extracts were subjected to SDS polyacrylamide gel (MiniProtean®, Bio-Rad) and blotted with antibodies overnight at 4 °C. Signal detection was performed with an enhanced chemiluminescence kit (ECL Plus Western blotting detection reagent from GE Healthcare). Hif1α, and DJ1 antibodies were purchased from Abcam. Pink1 and Hexokinase II (Hk2) were from Santa Cruz Biotechnology (Heidelberg, Germany) and β-actin was purchased from Sigma. Biologically independent replicates (three) were always performed, and a representative western blot is shown. The protein abundances were measured by densitometry of the bands on the films using ImageJ 1.48u4 software (National Institutes of Health, USA), and were normalized against the corresponding loading control.
Dj1-Foxo3a co-immunoprecipitation
WT MEFs were lysed in a buffer containing Tris-HCl (50 mM), NaCl (150 mM), EDTA (2 mM) and NP-40 (1%), pH 7.6 for 5 min on ice plus 45 min in an orbital roller at 4 °C. Lysates were centrifuged at 16,000 × g for 20 min, and the supernatant was collected. Immunoprecipitation was performed in 300 μg of cell lysates with Dynabeads (Life Technologies), using Foxo3a antibody (1:100; Cell Signalling, catalog number 2497) overnight at 4 °C in an orbital roller. Proteins were eluted from the beads by heating at 70 °C for 10 min in Laemmli's Buffer (β-mercaptoethanol 5%; SDS 2%; bromophenol blue 0.05%; glycerol 10%; Tris 60 mM). Eluted proteins were loaded directly onto 12% SDS-PAGE. For the input, 100 μg of cell lysates were loaded. Western blot was performed using anti-DJ1 (1/1000, Abcam ab4150) and anti-foxo3a (1/500) antibodies, both for the eluted proteins and the input.
Enzymatic analyses
Cells were collected and suspended in 0.1 M potassium phosphate buffer (pH 7.0). After freeze/thawing three times to ensure cellular disruption, NADH-CoQ1 reductase (complex I) and citrate synthase activities were determined as previously described [27].
Protein Determination
Protein concentrations were determined in the cell suspensions, lysates or in parallel cell culture incubations after solubilization with 0.1 M NaOH. Protein concentrations were determined as described [28] using bovine serum albumin as a standard.
Statistical analysis
All measurements in cell culture were carried out, at least, in triplicate, and the results are expressed as the mean ± SEM values from at least three different culture preparations. Statistical analysis of the results was performed by one-way analysis of variance (ANOVA), followed by Bonferroni test, or by the Student's t test for comparisons between two groups of values. In all cases, p<0.05 was considered significant.
Results and Discussion
To elucidate whether DJ1 regulates glucose metabolism, we first obtained dj1-/- and dj1+/+ (wild type or WT) mouse primary embryonic fibroblasts (MEF). The glycolytic flux in dj1-/- MEF were ∼1.7 higher when compared with WT (Figure 1A). Expression of a plasmid vector harboring full-length dj1 in dj1-/- MEF rescued this effect (Figure 1A). In contrast, expression of dj1 (L166P), i.e. a mutant form of DJ1 found in PD patients, did not fully reach the same glycolytic rate values to those of WT (Figure 1A). Similar results were obtained when determining the release of lactate to the culture medium (Figure 1B), often used as an index of the glycolytic rate. In good agreement with previous studies reporting mitochondrial dysfunction by DJ1-loss of function [29], dj1-/- MEF showed a marked reduction in complex I activity when compared with WT cells (Figure 1C) and no change in the mitochondrial mass index, citrate synthase (Figure 1C). Acute mitochondrial dysfunction is known to cause rapid, otherwise transient, glycolytic activation in healthy cells [30]. However, under our circumstances, dj1-/- cells show a phenotype compatible with a persistent glycolytic activation, suggesting long-term transcriptional and/or translational re-programming of glucose metabolism. In view that most glycolytic-promoting enzymes are transcriptionally regulated by the hypoxia-inducible factor-1 (Hif1) [31], we analyzed its expression in dj1-/- cells. As shown in Figure 1D, the protein abundances of Hif1α, as well as the Hif1 well-known target hexokinase-2 (Hk2) [31], were higher in dj1-/- cells when compared with WT. A role for DJ1 in regulating Hif1α has been previously studied, providing conflicting results [32-34]. Our data reporting Hif1α stabilization along with its target Hk2, as well as increased glycolytic rate, strongly supports the notion that DJ1 is a negative regulator of Hif1 and glycolysis. In agreement with this, a mitochondrial respiration-glycolytic shift has been suggested in DJ1 knockdown astrocytes [35], although the mechanism for this effect remained unclear.
Figure 1. DJ1 loss of function enhances glycolysis and HIf1α.
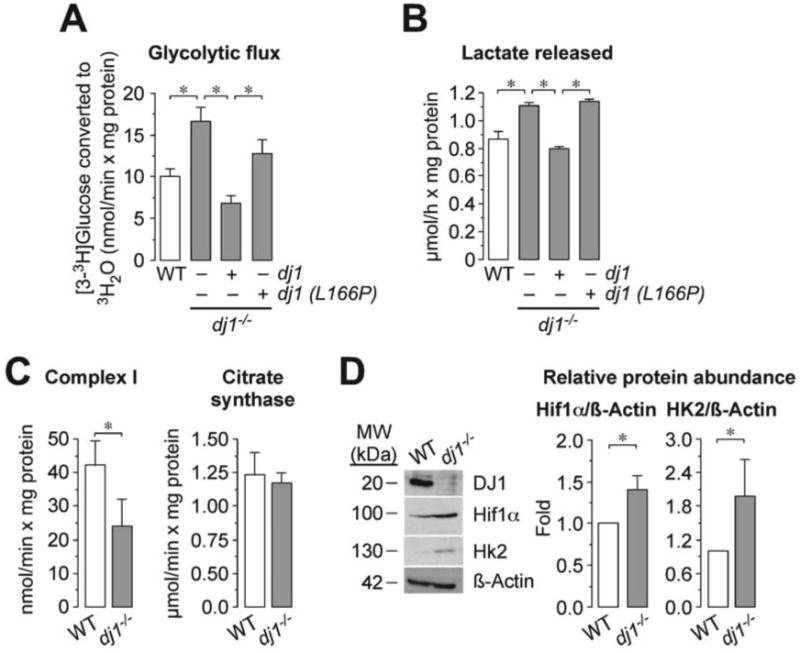
(A) Glycolytic flux was higher in dj1-/- MEF when compared with wild type (WT) MEF obtained from the same offspring. Expression of the full-length wild type dj1, but not the dj1 (L166P) mutant form of dj1, rescued the increased glycolytic flux. (B) Lactate released was higher in dj1-/- MEF when compared with WT. Expression of dj1, but not dj1 (L166P), rescued the increased release in lactate. (C) The activity of mitochondrial complex I was reduced in dj1-/- cells, whereas citrate synthase activity remained unchanged. (D) Hif1α and Hk2 protein abundances are higher in dj1-/- MEF when compared with WT. ß-Actin was used as loading control. A representative western blot is shown out of three. The right-hand side panel shows the relative Hif1α and HK2 protein abundances, as normalized with ß-Actin, averaged from three different blots. Data are expressed as mean±S.E.M. *p<0.05 (ANOVA followed by Bonferroni test; n=3-4 independent experiments).
Next, we aimed to elucidate the mechanism whereby DJ1-loss up-regulated Hif1α leading to increased glycolytic rate. We have recently found that the loss of Pink1 stabilizes Hif1α through reactive oxygen species leading to enhanced glycolytic rate [19]. Since it has been postulated that Pink1 is a downstream DJ1-target [15], we hypothesized whether the effects of the loss of DJ1 could be re-established by ectopically expressing Pink1. As shown in Figure 2A, expression of pink1 in dj1-/- cells fully prevented the increase in the glycolytic flux of DJ1-null cells. Furthermore, the increased Hif1α abundance caused by DJ1-loss was counteracted by the expression of pink1 (Figure 2B). Thus, DJ1 loss of function enhances Hif1α and glycolytic rate through a mechanism that can be replaced by Pink1.
Figure 2. Pink1 rescues DJ1-loss-mediated glycolysis and HIf1α enhancements.
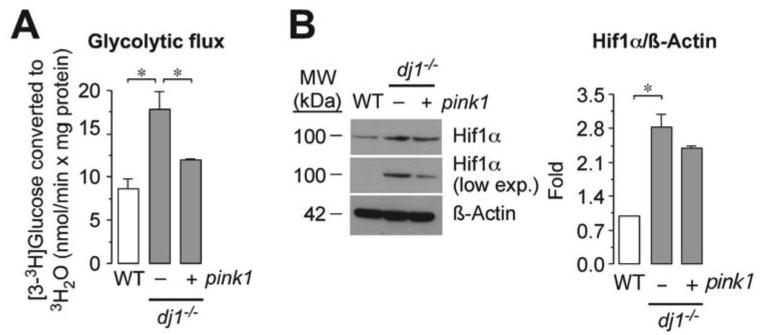
(A) The increased glycolytic flux of dj1-/- MEF is prevented by the expression of pink1. (B) The increased Hif1α abundance of KO MEF was counteracted by the expression of pink1. ß-Actin was used as loading control. A low exposed film (low exp.) is shown to better appreciate the differences in Hif1α abundances. A representative western blot is shown out of three. The right-hand side panel shows the relative Hif1α protein abundance as normalized with ß-Actin, averaged from three different blots. Data are expressed as mean±S.E.M. *p<0.05 (ANOVA followed by Bonferroni test; n=3-4 independent experiments).
Given that (i) Pink1 rescues the loss of function of DJ1 at regulating Hif1α and glycolysis (this work), (ii) Pink1 has been reported to be a tumor suppressor [36] and (iii) Pink1-loss increases Hif1α-mediated cell proliferation [19], we hypothesized whether dj1-/- cells had altered their proliferation rate. As shown in Figure 3A, dj1-/-cells proliferated faster than the wild type, and the expression of pink1 in dj1-/- cells rescued the rate of proliferation to the WT values. To confirm this result further, we studied the rate of bromo-deoxy-uridine (BrdU) incorporation into dj1-/- and WT MEF. We found that the proportion of BrdU-positive cells was ∼3-fold higher in dj1-/- cells than in WT; moreover, pink1 expression abrogated this effect (Figure 3B). Since increased BrdU incorporation is a reflection of increased DNA replication, we next analyzed the cell cycle phases under these conditions. As shown in Figure 3C, loss of DJ1 induced a decrease in cells in the G0/G1 phase and an increase in cells in the S and G2/M phases, a result that is compatible with increased proliferation rate. Moreover, this effect on the cell cycle was abolished by the expression of pink1 (Figure 3C). Uncontrolled activation of DJ1, in cooperation with Ras [1], promotes tumorigenesis, which might argue against a role for DJ1 as a negative regulator of cell proliferation. Whilst such an oncogenic role for DJ1 are difficult to conciliate with our data, it should be mentioned that we have not explored the effect of a dominant-positive expression of DJ1, but rather its loss of function. In view of the rather promiscuity of DJ1 [1-8], one would speculate that the overall effect of DJ1 –or its loss– on cell proliferation would depend on the cellular type and regulatory context. Thus, our results suggest that loss of DJ1 enhances cell proliferation through a molecular mechanism that can be replaced by ectopically expressing Pink1.
Figure 3. DJ1 loss of function enhances cell proliferation through a mechanism that can be replaced by Pink1.
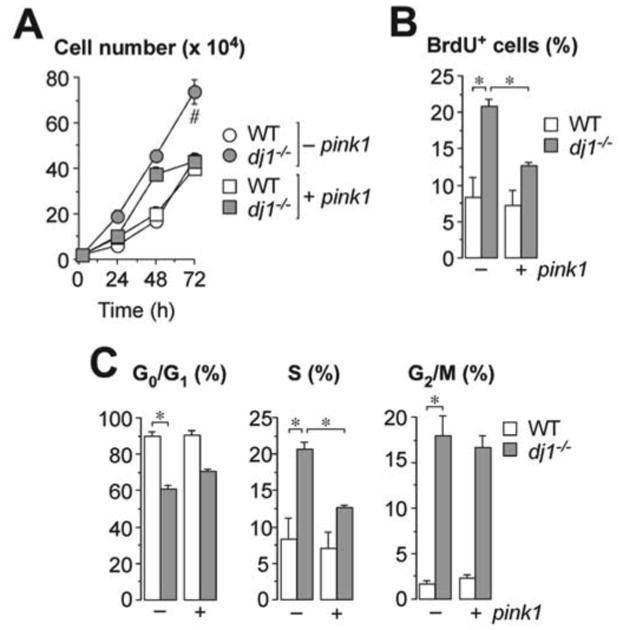
(A) Cell proliferation was higher in dj1-/- MEF than in WT, and the expression of pink1 in dj1-/- MEF restored cell proliferation to the WT values. (B) The proportion of BrdUpositive cells was higher in dj1-/- MEF than in WT, and pink1 expression abrogated this effect. (C) Analysis of the cell cycle phases show that dj1-/- MEF have decreased G0/G1 phase and increased S and G2/M phases, and pink1 expression abolished this effect. Data are expressed as mean±S.E.M. *p<0.05; #p<0.05 versus all other conditions at 72 h (ANOVA followed by Bonferroni test; n=3-4 independent experiments).
In view that the metabolic and cellular phenotype of DJ1-loss could be restored by ectopically expressing Pink1, we hypothesized whether DJ1 is a positive regulator of Pink1. In fact, there is a vast literature suggesting possible interactions amongst DJ1, Pink1 and Parkin [12-15, 37]. It appears to be well established that Pink1 acts upstream of Parkin, promoting its recruitment to mitochondria upon mitochondrial inner membrane potential loss in order to trigger autophagy of damaged mitochondria as a protective mechanism [16, 17]. However, the involvement of DJ1 in this picture has remained largely unknown. Since it has been reported that DJ1 can act as a transcriptional co-activator of several genes [6, 7, 25], we sought to investigate whether DJ1 activated Pink1. As shown in Figure 4A, dj1-/- cells had lower levels of Pink1 protein. This effect could be restored by the ectopic expression, under a dj1-/-background, of dj1, but not of the PD-relevant dj1 (L166P) mutant form of dj1 (Figure 4A). Likewise, pink1 mRNA levels were significantly lower in dj1-/- MEF when compared with the WT, an effect that was normalized by the expression of dj1, but not dj1 (L166P) mutant (Figure 4B). These data support the hypothesis that DJ1 is a transcriptional activator of pink1. To specifically address this, we assessed whether DJ1 could directly bind the pink1 promoter. To do so, we performed a chromatin-anti-DJ1 (α-DJ1) immunoprecipitation (ChIP) assay using DNA purified from either WT or dj1-/-MEF, followed by agarose gel analysis of the PCR-amplified DNA fragments using primers flanking the pink1 promoter. As shown in Figure 4C, α-DJ1 bound to the pink1 promoter from WT, but not from dj1-/- cells. Moreover, expression of wild type –but not the L166P mutant– form of dj1, in a dj1-/- background rescued α-DJ1 binding with the pink1 promoter (Figure 4C). These results strongly suggest that DJ1 is a transcriptional positive regulator of pink1 by directly binding to the pink1 promoter.
Figure 4. DJ1 binds to Foxo3a to transcriptionally activate pink1.
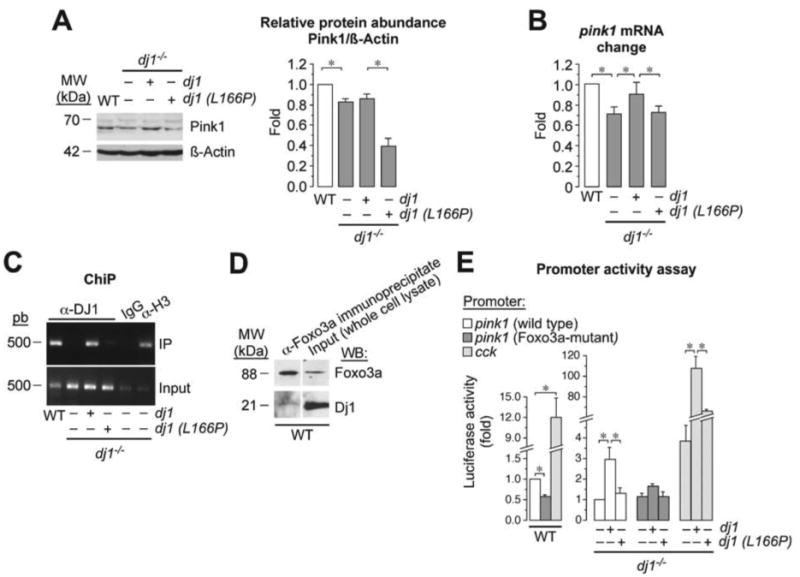
(A) dj1-/- MEF has lower levels of Pink1 protein, as assessed by western blotting, an effect that is normalized by the expression of dj1, but not dj (L166P). A representative western blot is shown out of three. The right-hand side panel shows the relative Pink1 protein abundance as normalized with ß-Actin, averaged from three different blots. (B) Pink1 mRNA levels, as assessed by RT-qPCR, are lower in dj1-/-MEF than in WT, and this effect is restored by expression of dj1, but not dj1 (L166P). mRNA data were calculated from the fold change of each β-actin-normalized transcript abundance in the dj1-/- samples versus that in the WT, which therefore received a value of 1.00. (C) Chromatin-anti-DJ1 (α-DJ1) immunoprecipitation (ChIP) assay using DNA purified from WT or dj1-/- MEF shows that α-DJ1 binds to the pink1 promoter of WT cells, but it does not in dj1-/- cells. α-DJ1 binds to the pink1 promoter of dj1-/- cells if dj1 (but not its mutant L166P form) is over-expressed; immunoglobulin-G (IgG) was used as negative control, and anti-histone-3 (α-H3) as a positive control; input shows equal amplification of DNA in non-immunoprecipitated samples; IP corresponds to the amplified DNA band in the immunoprecipitated samples. (D) Immunoprecipitation of WT cell lysates with an α-Foxo3a antibody, followed by western blotting against α-DJ1, reveals a DJ1 band indicating that physical interaction between Foxo3a and DJ1. Total cell lysates were used as input. (E) Luciferase-reporter promoter activity assays using a plasmid vector harboring a 0.4 kb pink1 promoter region, a mutant form of this 0.4 kb genomic DNA segment harboring three single nucleotide mutations in the Foxo3a binding sequence, or the regulatory elements of the cholecystokinin (cck) gene. Left panel shows decreased luciferase activity in WT cells transfected with the Foxo3amutant pink1 promoter, and increased luciferase activity in these cells when transfected with the cck promoter, when compared with cells transfected with the wild type pink1 promoter. Right panel shows shows that over-expression of dj1 in dj1-/- MEF increases luciferase activity driven by wild type pink1 promoter when compared with dj1-/- MEF not over-expressing dj1; this effect is not observed by over-expression of dj1 (L166P). However, the effect of dj1 over-expression is not observed in cells transfected with the Foxo3a-mutant form of the pink1 promoter. Finally, over-expression of dj1 in dj1-/- MEF increases luciferase activity driven by cck promoter when compared with dj1-/- MEF not over-expressing dj1; this effect is partially rescued by over-expression of dj1 (L166P). Luciferase activity data were normalized to those found in the WT-pink1 promoter samples. Data are expressed as mean±S.E.M. *p<0.05 (ANOVA followed by Bonferroni test; n=3-4 independent experiments).
The only transcription factor described so far to activate pink1 gene is Foxo3a [24]. Accordingly, we next wondered whether DJ1 interacts with Foxo3a to promote the transcriptional activation of pink1. As shown in Figure 4D, immunoprecipitation of WT cell lysates with an α-Foxo3a antibody, followed by western blotting against α-DJ1, revealed that Foxo3a physically interacted with DJ1. These results suggest the possibility that DJ1, by interacting with Foxo3a, would be responsible for the transcriptional activation of pink1 gene. To further confirm this, we performed a luciferase-reporter promoter activity assay using a plasmid vector harboring the 0.4 kb genomic DNA segment immediately preceding the start codon of pink1 gene, which has been previously reported to contain its relevant regulatory elements [24]. We also used a mutant form of this 0.4 kb genomic DNA segment harboring three single nucleotide mutations in the Foxo3a binding sequence [24]. As a positive control, cells were also transfected with a luciferase-reporter construct harboring the regulatory elements of the cholecystokinin (cck) gene, previously reported to be trans-activated by DJ1 [25]. Both WT and dj1-/- MEF were then transfected with these plasmid vectors, and luciferase activity measured as an index of pink1 promoter activity. As shown in Figure 4E, luciferase activity was decreased by ∼45% in cells transfected with the Foxo3a-mutant pink1 promoter, when compared with cells transfected with the wild type pink1 promoter. Moreover, luciferase activity in dj1-/- cells harboring the wild type pink1 promoter increased by ∼twofold by over-expressing wild type dj1, but not the L166P mutant dj1 (Figure 4E). No significant effect was observed in luciferase activity of the dj1-/- cells transfected with the Foxo3a-mutant form of the pink1 promoter (Figure 4E). Finally, promoter activity of cck was ∼12-fold higher than that of pink1 in WT cells, but only ∼4-fold in dj1-/- cells; furthermore, the latter dramatically increased (by ∼100-fold) by over-expression of dj1, an effect that was significantly prevented by the expression of the L166P mutant form of dj1 (Figure 4E).
In conclusion, here we describe that DJ1 loss of function, often associated with earlyonset PD, enhances the rate of glycolysis and cell proliferation of cultured mouse embryonic fibroblasts. Moreover, we find that this phenotype can be fully replaced by Pink1, a protein whose loss causes similar effects [19] to those we herein describe in DJ1-null cells. Furthermore, we demonstrate that, by binding to Foxo3a, DJ1 is a transcriptional activator of pink1 gene, hence explaining why Pink1 can take the place of these DJ1 functions. These findings may add new insight to the current apparent controversy [12-15] on the role of DJ1 in modulating the function of the Pink1/Parkin pathway. Whether DJ1 loss of function, by altering glucose metabolism, has any deleterious effect on non-dividing cells such as the dopaminergic neurons that degenerate in PD remains elusive. However, it is well known that, in post-mitotic neurons, an increase in the rate of glucose consumption through glycolysis shifts-down the rate of glucose oxidation through the pentose-phosphate pathway (PPP) [22]. Since PPP, by regenerating NADPH, is an essential pathway for the restoration of antioxidant glutathione [38], the increased glycolysis upon DJ1 loss of function might have negative consequences for the neuronal redox status likely contributing to the oxidative stress associated with dopaminergic neurodegeneration in PD [39]. In fact, loss of Pink1 triggers a decrease in PPP activity in neurons [19]. However, whether this effect occurs in DJ1-deficient neurons is an interesting possibility worth pursue investigating to better understand the biochemical hallmarks of PD, and to identify novel therapeutic targets against this devastating neurological disorder.
Acknowledgments
The technical assistances of Ms. Monica Carabias and Ms. Monica Resch are acknowledged.
Funding: This work was funded by the Spanish Ministerio de Economia y Competitividad [SAF2013-41177-R], the Instituto de Salud Carlos III [RD12/0043/0021], the SP3-People-MC-ITN programme of the European Commission [608381], the National Institutes of Drug Abuse/National Institutes of Health (NIH/NIDA) [1R21DA03767801], and the European Regional Development Fund to JPB. AAP was funded by the Instituto de Salud Carlos III [PI12/00685; RD12/0014/0007]. ILB is the recipient of a FPI Fellowship from the Spanish Ministerio de Economia y Competitividad (Spain).
Abbreviations
- BrdU
bromo-deoxy-uridine
- cck
cholecystokinin
- DMEM
Dulbecco's modified Eagle's medium
- FCS
fetal calf serum
- GFP
green fluorescent protein
- Hif1
hypoxia-inducible factor-1
- KO
knockout
- MEF
mouse embryonic fibroblasts
- PD
Parkinson's disease
- PEI
polyethylenimine
- Pink1
PTEN-induced protein kinase-1
- PPP
pentose-phosphate pathway
- WT
wild type
Footnotes
Author Contributions: JPB conceived the idea. JPB and RRA designed research. RRA, ILF, DJB and EF performed research. JPB, RRA and AA analyzed the data. JPB wrote the manuscript.
References
- 1.Nagakubo D, Taira T, Kitaura H, Ikeda M, Tamai K, Iguchi-Ariga SM, Ariga H. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem Biophys Res Commun. 1997;231:509–513. doi: 10.1006/bbrc.1997.6132. [DOI] [PubMed] [Google Scholar]
- 2.Milani P, Ambrosi G, Gammoh O, Blandini F, Cereda C. SOD1 and DJ-1 converge at Nrf2 pathway: a clue for antioxidant therapeutic potential in neurodegeneration. Oxid Med Cell Longev. 2013;2013:836760. doi: 10.1155/2013/836760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yasuda T, Kaji Y, Agatsuma T, Niki T, Arisawa M, Shuto S, Ariga H, Iguchi-Ariga SM. DJ-1 cooperates with PYCR1 in cell protection against oxidative stress. Biochem Biophys Res Commun. 2013;436:289–294. doi: 10.1016/j.bbrc.2013.05.095. [DOI] [PubMed] [Google Scholar]
- 4.Sajjad MU, Green EW, Miller-Fleming L, Hands S, Herrera F, Campesan S, Khoshnan A, Outeiro TF, Giorgini F, Wyttenbach A. DJ-1 modulates aggregation and pathogenesis in models of Huntington's disease. Hum Mol Genet. 2014;23:755–766. doi: 10.1093/hmg/ddt466. [DOI] [PubMed] [Google Scholar]
- 5.Batelli S, Albani D, Rametta R, Polito L, Prato F, Pesaresi M, Negro A, Forloni G. DJ-1 modulates alpha-synuclein aggregation state in a cellular model of oxidative stress: relevance for Parkinson's disease and involvement of HSP70. PLoS One. 2008;3:e1884. doi: 10.1371/journal.pone.0001884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Yamaguchi S, Yamane T, Takahashi-Niki K, Kato I, Niki T, Goldberg MS, Shen J, Ishimoto K, Doi T, Iguchi-Ariga SM, Ariga H. Transcriptional activation of low-density lipoprotein receptor gene by DJ-1 and effect of DJ-1 on cholesterol homeostasis. PLoS One. 2012;7:e38144. doi: 10.1371/journal.pone.0038144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu J, Zhong N, Wang H, Elias JE, Kim CY, Woldman I, Pifl C, Gygi SP, Geula C, Yankner BA. The Parkinson's disease-associated DJ-1 protein is a transcriptional co-activator that protects against neuronal apoptosis. Hum Mol Genet. 2005;14:1231–1241. doi: 10.1093/hmg/ddi134. [DOI] [PubMed] [Google Scholar]
- 8.Trempe JF, Fon EA. Structure and Function of Parkin, PINK1, and DJ-1, the Three Musketeers of Neuroprotection. Front Neurol. 2013;4:38. doi: 10.3389/fneur.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Petrie TG, Liu Y, Liu J, Fujioka H, Zhu X. Parkinson's disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J Neurochem. 2012;121:830–839. doi: 10.1111/j.1471-4159.2012.07734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 11.Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat. 2010;31:763–780. doi: 10.1002/humu.21277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, Xia K, Jiang W, Ronai Z, Zhuang X, Zhang Z. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest. 2009;119:650–660. doi: 10.1172/JCI37617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang B, Xiong H, Sun P, Zhang Y, Wang D, Hu Z, Zhu Z, Ma H, Pan Q, Xia JH, Xia K, Zhang Z. Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson's disease. Hum Mol Genet. 2006;15:1816–1825. doi: 10.1093/hmg/ddl104. [DOI] [PubMed] [Google Scholar]
- 14.Chang C, Wu G, Gao P, Yang L, Liu W, Zuo J. Upregulated Parkin expression protects mitochondrial homeostasis in DJ-1 konckdown cells and cells overexpressing the DJ-1 L166P mutation. Mol Cell Biochem. 2014;387:187–195. doi: 10.1007/s11010-013-1884-3. [DOI] [PubMed] [Google Scholar]
- 15.Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A, Miller D, Maric D, Cedazo-Minguez A, Cookson MR. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet. 2011;20:40–50. doi: 10.1093/hmg/ddq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, Suzuki N, Uchiyama S, Tanaka K, Matsuda N. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem. 2013;288:22019–22032. doi: 10.1074/jbc.M113.467530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, Iguchi-Ariga SM. Neuroprotective function of DJ-1 in Parkinson's disease. Oxid Med Cell Longev. 2013;2013:683920. doi: 10.1155/2013/683920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Requejo-Aguilar R, Lopez-Fabuel I, Fernandez E, Martins LM, Almeida A, Bolaños JP. PINK1 deficiency sustains cell proliferation by reprogramming glucose metabolism through HIF1. Nat Commun. 2014;5:4514. doi: 10.1038/ncomms5514. [DOI] [PubMed] [Google Scholar]
- 20.Flicek P, Amode MR, Barrell D, Beal K, Brent S, Chen Y, Clapham P, Coates G, Fairley S, Fitzgerald S, Gordon L, Hendrix M, Hourlier T, Johnson N, Kahari A, Keefe D, Keenan S, Kinsella R, Kokocinski F, Kulesha E, Larsson P, Longden I, McLaren W, Overduin B, Pritchard B, Riat HS, Rios D, Ritchie GR, Ruffier M, Schuster M, Sobral D, Spudich G, Tang YA, Trevanion S, Vandrovcova J, Vilella AJ, White S, Wilder SP, Zadissa A, Zamora J, Aken BL, Birney E, Cunningham F, Dunham I, Durbin R, Fernandez-Suarez XM, Herrero J, Hubbard TJ, Parker A, Proctor G, Vogel J, Searle SM. Ensembl 2011. Nucleic Acids Res. 2010;39:D800–806. doi: 10.1093/nar/gkq1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuende J, Moreno S, Bolanos JP, Almeida A. Retinoic acid downregulates Rae1 leading to APC(Cdh1) activation and neuroblastoma SH-SY5Y differentiation. Oncogene. 2008;27:3339–3344. doi: 10.1038/sj.onc.1210987. [DOI] [PubMed] [Google Scholar]
- 22.Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. 2009;11:747–752. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- 23.Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci U S A. 2005;102:5703–5708. doi: 10.1073/pnas.0500617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mei Y, Zhang Y, Yamamoto K, Xie W, Mak TW, You H. FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc Natl Acad Sci U S A. 2009;106:5153–5158. doi: 10.1073/pnas.0901104106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamane T, Suzui S, Kitaura H, Takahashi-Niki K, Iguchi-Ariga SMM, Ariga H. Transcriptional Activation of the Cholecystokinin Gene by DJ-1 through Interaction of DJ-1 with RREB1 and the Effect of DJ-1 on the Cholecystokinin Level in Mice. PlosOne. 2013;8:e78374. doi: 10.1371/journal.pone.0078374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc. 2006;1:729–748. doi: 10.1038/nprot.2006.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolaños JP, Peuchen S, Heales SJR, Land JM, Clark JB. Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J Neurochem. 1994;63:910–916. doi: 10.1046/j.1471-4159.1994.63030910.x. [DOI] [PubMed] [Google Scholar]
- 28.Lowry OH, Rosebrough NJ, Lewis-Farr A, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 29.Hauser DN, Hastings TG. Mitochondrial dysfunction and oxidative stress in Parkinson's disease and monogenic parkinsonism. Neurobiol Dis. 2013;51:35–42. doi: 10.1016/j.nbd.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Almeida A, Moncada S, Bolaños JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. [DOI] [PubMed] [Google Scholar]
- 31.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheng C, Heng X, Zhang G, Xiong R, Li H, Zhang S, Chen S. DJ-1 deficiency perturbs microtubule dynamics and impairs striatal neurite outgrowth. Neurobiol Aging. 2013;34:489–498. doi: 10.1016/j.neurobiolaging.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Foti R, Zucchelli S, Biagioli M, Roncaglia P, Vilotti S, Calligaris R, Krmac H, Girardini JE, Del Sal G, Gustincich S. Parkinson disease-associated DJ-1 is required for the expression of the glial cell line-derived neurotrophic factor receptor RET in human neuroblastoma cells. J Biol Chem. 2010;285:18565–18574. doi: 10.1074/jbc.M109.088294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vasseur S, Afzal S, Tardivel-Lacombe J, Park DS, Iovanna JL, Mak TW. DJ-1/PARK7 is an important mediator of hypoxia-induced cellular responses. Proc Natl Acad Sci U S A. 2009;106:1111–1116. doi: 10.1073/pnas.0812745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larsen NJ, Ambrosi G, Mullett SJ, Berman SB, Hinkle DA. DJ-1 knock-down impairs astrocyte mitochondrial function. Neuroscience. 2011;196:251–264. doi: 10.1016/j.neuroscience.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Unoki M, Nakamura Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene. 2001;20:4457–4465. doi: 10.1038/sj.onc.1204608. [DOI] [PubMed] [Google Scholar]
- 37.Wilhelmus MM, Nijland PG, Drukarch B, de Vries HE, van Horssen J. Involvement and interplay of Parkin, PINK1, and DJ1 in neurodegenerative and neuroinflammatory disorders. Free Radic Biol Med. 2012;53:983–992. doi: 10.1016/j.freeradbiomed.2012.05.040. [DOI] [PubMed] [Google Scholar]
- 38.Bolaños JP, Almeida A. The pentose-phosphate pathway in neuronal survival against nitrosative stress. IUBMB Life. 2010;62:14–18. doi: 10.1002/iub.280. [DOI] [PubMed] [Google Scholar]
- 39.Gaki GS, Papavassiliou AG. Oxidative stress-induced signaling pathways implicated in the pathogenesis of Parkinson's disease. Neuromolecular Med. 2014;16:217–230. doi: 10.1007/s12017-014-8294-x. [DOI] [PubMed] [Google Scholar]