

Abstract
Backgroundy
The microbiome of the home is of great interest because of its possible impact on health. Our goal was to identify some of the factors that determine the richness, evenness and diversity of the home's fungal and bacterial microbiomes.
Methods
Vacuumed settled dust from homes (n=35) in Cincinnati, OH, were analyzed by pyrosequencing to determine the fungal and bacterial relative sequence occurrence. The correlation coefficients between home environmental characteristics, including age of home, Environmental Relative Moldiness Index (ERMI) values, occupant number, relative humidity and temperature, as well as pets (dog and cat) were evaluated for their influence on fungal and bacterial communities. In addition, linear discriminant analysis (LDA) was used for identifying fungal and bacterial genera and species associated with those housing determinants found to be significant.
Results
The fungal richness was found to be positively correlated with age of home (p=0.002), ERMI value (p=0.003), and relative humidity (p=0.015) in the home. However, fungal evenness and diversity were only correlated with the age of home (p=0.001). Diversity and evenness (not richness) of the bacterial microbiome in the homes were associated with dog ownership.
Linear discriminant analysis showed total of 39 putative fungal genera/species with significantly higher LDA scores in high ERMI homes and 47 genera/species with significantly higher LDA scores in homes with high relative humidity. When categorized according to the age of the home, a total of 67 fungal genera/species had LDA scores above the significance threshold. Dog ownership appeared to have the most influence on the bacterial microbiome, since a total of 130 bacterial genera/species had significantly higher LDA scores in homes with dogs.
Conclusions
Some key determinants of the fungal and bacterial microbiome appear to be excess moisture, age of the home and dog ownership.
Keywords: indoor environment, microbial diversity, bacteria, fungi, microbiome
1. Introduction
The factors that determine the make-up of the home's fungal and bacterial microbiomes are of growing interest. However, the home itself has its own microbiome and this microbiome may also be important for adverse human health effects such as immune disorders (Parker and Ollerton, 2013) influencing allergy and asthma incidence. Therefore, the factors that determine the make-up of the home's fungal and bacterial microbiomes are of growing interest.
Although there have been many studies that have measured specific fungal species or groups of fungi in homes (e.g., Pitkäranta et al., 2011), the home's fungal microbiome has been considered in only a few studies (Nonnenmann et al., 2012; Adams et al., 2013a,b; Adams et al., 2014). In addition, many studies have measured specific bacteria or groups of bacteria (e.g. Gram-positive or negative bacteria) in homes (e.g., Adhikari et al., 2014) but the home's (or other indoor environments’) only a few studies have been conducted on bacterial microbiome (Kembel et al., 2012; Qian et al., 2012; Kembel et al., 2014; Adams et al., 2014).
Recently, Adams et al. (2014) showed that outdoor air had a major input into the richness of the home's fungal microbiome but human inputs dominated the home's bacterial microbiome. This is consistent with a recent study of the bacterial microbiome that demonstrated that the bacterial content of the home was linked to the human and animal occupants, and components of the occupants’ bacterial microbiome were readily transferred to newly occupied homes (Lax et al., 2014). However, these studies did not consider the microbiome in water-damaged versus non-water-damaged homes as well as other possible environmental determinants of bacteria considered in the present study.
In this study, we examined the bacterial and fungal microbiome of homes associated with increased-risk of asthma, i.e., homes with higher Environmental Relative Moldiness Index (ERMI) values, compared to homes with low ERMI values (Reponen et al., 2011; Reponen et al., 2012). Our goal was to determine the factors associated with the home that influence the composition of both the bacterial and fungal microbiome. By examining household factors, such as age of home, ERMI, occupant number, relative humidity, temperature, and the presence of pets (dog or cat), we hope to expand our understanding of factors that create and influence the microbiome of homes, especially homes that are associated with a higher risk of asthma.
2. Methods
2.1. Study Homes
The protocols for collection and analysis of household dust samples previously approved by the Institutional Review Board at the University of Cincinnati were followed during the home sampling. The families recruited for this study (n=35) were a subset of the cohort participating in the Cincinnati Childhood Allergy and Air Pollution Study (CCAAPS) undertaken in Cincinnati, Ohio and Northern Kentucky in 2001 (LeMasters et al., 2006). The ERMI values for these homes were determined previously (Adhikari et al., 2014). Based on the ERMI values, homes were selected for inclusion in a low ERMI (<2) group (n=17) or a high ERMI (≥2) group (n=18).
2.2. On-site Home Visit and Sampling
On-site home visits were performed by two-person teams at child's age eight. Floor-dust samples were obtained for the assessment of bacteria and fungi by vacuuming the floor in the child's primary activity room (Cho et al., 2006). Dust samples for carpeted floor were collected from an area of 2 m2 at a vacuuming rate of 2 min/m2. For non-carpeted floor (hard wood, linoleum, tile, or sheet floor), the sample was collected from the entire room at a rate of 1 min/m2. Large dust particles were removed by sieving (355 μm mesh sieve), and the resulting fine dust was stored at −20°C before analyses. Temperature (°C) and relative humidity (%) were recorded in each home by a portable thermo-hygrometer (Fisher Scientific, Pittsburgh, PA) and data on the number of occupants, age of the homes, and pet ownership were collected through a questionnaire survey.
2.3. DNA Extraction from Environmental Samples
DNA was extracted from 50.0 ± 0.1 mg of the indoor dust samples into 100 μl solutions, using a protocol developed at the U.S. EPA for quantitative analysis of DNA from dust. However, we did not add the external standard (Haugland et al., 2002) because the addition of a large number of external cells could influence the results. Extracted DNA samples were shipped to Research and Testing Laboratories, LLC (Lubbock, TX) for pyrosequencing.
2.4. Pyrosequencing primers
Extracted DNA samples were amplified for pyrosequencing using forward and reverse fusion primers. For bacteria, the forward primer was constructed with (5’-3’) the Roche A linker (CCATCTCATCCCTGCGTGTCTCCGACTCAG), an 8-10 bp barcode (>2 nucleotide differences between primers), and the 28F primer (5’- GAGTTTGATCNTGGCTCAG -3’). The reverse fusion primer was constructed with (5’-3’) a biotin molecule, the Roche B linker (CCTATCCCCTGTGTGCCTTGGCAGTCTCAG), and the universal eubacterial 519R primer (5’- GTNTTACNGCGGCKGCTG -3’). For fungal DNA, the same approach was used, however, the fungal specific ITS1 primer (CTTGGTCATTTAGAGGAAGTAA) replaced 28F in the forward primer and the fungal specific ITS4 (TCCTCCGCTTATTGATATGC) replaced the 519R in the reverse primer.
2.5. Amplification
Amplifications were performed in 25 μl reactions with Qiagen HotStart Taq Master Mix (Qiagen Inc., Valencia, CA), 1μl of each 5μM primer, and 1μl of template on ABI Veriti thermocyclers (Applied Biosytems, Foster City, CA) under the following thermal profile: 95°C for 5 min, then 35 cycles of 94°C for 30 sec, 54°C for 40 sec, 72°C for 1 min, followed by one cycle of 72°C for 10 min and 4°C hold. Amplification products were visualized with eGels (Life Technologies, Inc., Grand Island, NY) and pooled equimolar. Each pool was cleaned with Diffinity RapidTip (Diffinity Genomics, West Henrietta, NY), and size selected using Agencourt AMPure XP (BeckmanCoulter, Brea, CA) following Roche 454 protocols (454 Life Sciences). Size selected pools were quantified and 150 ng of DNA hybridized to Dynabeads M-270 (Life Technologies, Inc., Grand Island, NY) to create single stranded DNA following Roche 454 protocols (454 Life Sciences, Branford, CT). Single stranded DNA was diluted and used in emPCR reactions. Sequencing was accomplished following established manufacturer protocols (454 Life Sciences, Branford, CT) on FLX Sequencers using titanium chemistry.
2.6. Putative identifications of bacteria and fungi
Operational taxonomic unit (OTU) clusters for the fungi and bacteria were determined using Kraken as the classifier to assign taxonomy (Wood and Salzberg, 2014). The sequences were sorted using FASTA formatted files and then clustered into OTU clusters with 96.5% identity (3.5% divergence) using USEARCH (Nucleotide, 2011). The file was then queried against a database of high quality sequences derived from NCBI using a distributed .NET algorithm that utilizes BLASTN+ version 0.7.4 (KrakenBLAST; www.krakenblast.com). The BLASTn+ outputs were compiled using a .NET 4.5 standards and C# 5.0 analysis pipeline.
2.7. Data reduction
The data reduction analysis was performed, as previously described (Dowd et al., 2008; Callaway et al., 2009; Callaway et al., 2010). Based upon the above BLASTn+ derived sequence identity (percent of total length query sequence which aligns with a given database sequence) and validation using taxonomic distance methods, the bacteria and fungi were putatively classified at the genus and species taxonomic levels. Sequences representing approximately 82,584 bacterial and 14,438 fungal species were present in the database (Nucleotide, 2011). Sequences with identity scores were compared to known or well-characterized ribosomal DNA sequences. Sequence identities greater than 97% (<3% divergence) were resolved at the species level and between 95 and 97% at the genus level (Nonnenmann et al., 2012). Any match below this percent identity was discarded. In addition, the High Score Pair was at least 75% of the query sequence or it was discarded, regardless of identity (Browning and Browing, 2012).
After resolution based upon these parameters, the percentage of each identity was individually analyzed for the sample by providing relative abundance information based upon relative numbers of 16s and ITS DNA sequences within a given sample for bacteria and fungi, respectively. The putative primary taxonomic identification of the sample sequences was resolved to its closest relative or species level, when possible.
2.8. Statistical Analysis
Four common diversity measures (richness, evenness, Shannon's Diversity Index, and Simpson's Diversity Index) were calculated using species relative abundance values with PCORD v. 6.0 software (MjM Software, Gleneden Beach, OR). Species richness (S) is defined as the number of species (non-zero elements) observed in the sample. Evenness (E) is an estimate of how equitably distributed the species abundances were. Shannon's index (H) measures the information content of a sample and accounts for both abundance and evenness of the species present. The proportion of species i relative to the total number of species (pi) is calculated, and then multiplied by the logarithm of this proportion (log pi). The resulting product is summed across species, and multiplied by -1:
| [1] |
Simpson's Diversity Index (D’) for an infinite population, which is the likelihood that two randomly chosen OTUs will be different species, was defined as:
| [2] |
Preliminary analyses were conducted using SPSS v. 17.0 software (SPSS Inc., Chicago, IL) on the diversity measures to assess normality as determined by Shapiro-Wilk tests. When possible, log10 transformation was utilized to achieve normality. Group means were compared using Student's t-tests on normally distributed variables, whereas Mann-Whitney U tests were used to compare group medians when non-parametric tests were necessary. Correlations (1) between the diversity measures and environmental variables and (2) among the diversity measures themselves were assessed using Pearson and Spearman correlations where appropriate.
The linear discriminant analysis (LDA) effect size (LEfSe) (Segata et al., 2011) algorithm was used for putative bacterial and fungal identification with significant differential relative abundances with respect to ERMI classification of “high” or “low” (based on sample median value) and dog ownership classification of “yes” or “no.” A default threshold value of 2.0 on the logarithmic LDA score was used to identify discriminative features.
3. Results
Table 1 shows the mean and standard deviation for each of the home factors evaluated in this study as well as the occurrence of cats or dogs as pets. The various factors were evaluated for their effect on microbial population richness, which refers to the number of species present in a particular environment; evenness, which refers to the uniformity of the populations in the environment; and diversity, which describes the rarity or commonness of a particular species in a community.
Table 1.
Descriptive statistics of home characteristics.
| Age of Home [years] | 53.8 ± 34.2 |
| ERMI [avg] | 2.89 ± 5.61 |
| Occupants [#] | 4.6 ± 1.6 |
| Relative Humidity [%] | 35.6 ± 8.9 |
| Temperature [°C] | 23.1 ± 3.2 |
| Dog Ownership | |
| Yes | 14 |
| No | 21 |
| Cat Ownership | |
| Yes | 8 |
| No | 27 |
Notes: Values are means ± standard deviation. Pet ownership values represent the number of homes in each category.
The richness of the fungal populations in the homes was positively (p<0.05) correlated with the age of the home (Table 2 and supplementary Fig. S1), the ERMI value (Table 2 and supplementary Fig. S2), and relative humidity (Table 2 and supplementary Fig. S3), but not with the number of occupants or temperature (Table 2) in the home. The fungal evenness and diversity indices (both Shannon and Simpson) were also significantly correlated with the age of the home (Table 2 and supplementary Fig. S1). On the other hand, the bacterial richness, evenness and diversity were not correlated with the age of the home, ERMI, number of occupants, relative humidity, and temperature (Table 2).
Table 2.
Correlation coefficients (r) and their statistical significance (p) between environmental characteristics of home and four diversity metrics for both bacteria and fungi.
| Richness, S | Evenness, E | Shannon Diversity, H | Simpson's Diversity, D’† | ||||||
|---|---|---|---|---|---|---|---|---|---|
| r | p | r | p | r | p | r | p | ||
| Age of Home | Bacteria | 0.215 | 0.216 | 0.123† | 0.482 | 0.214 | 0.217 | 0.175 | 0.315 |
| Fungi | 0.497 | 0.002 | 0.517 | 0.001 | 0.573 | <0.001 | 0.575 | <0.001 | |
| ERMI† | Bacteria | 0.297 | 0.083 | −0.059 | 0.735 | 0.167 | 0.336 | 0.122 | 0.485 |
| Fungi | 0.489 | 0.003 | 0.047 | 0.787 | 0.250 | 0.147 | 0.115 | 0.509 | |
| Occupants | Bacteria | 0.087 | 0.618 | −0.171† | 0.325 | −0.029 | 0.870 | −0.196 | 0.260 |
| Fungi | 0.094 | 0.590 | 0.148 | 0.396 | 0.137 | 0.433 | 0.127 | 0.466 | |
| Relative Humidity | Bacteria | 0.156 | 0.371 | −0.134† | 0.443 | −0.095 | 0.589 | −0.140 | 0.424 |
| Fungi | 0.407 | 0.015 | 0.089 | 0.610 | 0.206 | 0.236 | 0.087 | 0.620 | |
| Temperature | Bacteria | −0.104 | 0.551 | −0.223† | 0.197 | −0.263 | 0.127 | −0.288 | 0.093 |
| Fungi | 0.102 | 0.560 | 0.155 | 0.375 | 0.139 | 0.426 | 0.203 | 0.242 | |
Notes: Values in boldface are considered statistically significant (p<0.05). Pearson correlation on log-transformed data was used unless otherwise noted.
Spearman correlation coefficients are reported for all correlations involving ERMI, Simpson's diversity, and bacteria evenness.
Figure 1 shows that the bacterial populations’ richness (Fig. 1a) was not significantly affected by dog ownership, but evenness (Fig. 1b) and diversity (both Shannon and Simpson- Fig. 1c-d) were significantly higher in homes with dogs. On the other hand, dog ownership had no discernible influence on fungal richness, evenness, and diversity (supplementary Fig. S4). Cat ownership was not associated with bacterial or fungal richness, evenness or diversity (data not given). Overall, there was no correlation between bacterial populations and fungal populations for richness, evenness, or diversity (Table 3). In terms of magnitude, the bacterial population in the house dust had greater richness, evenness, and diversity than the fungal population (Table 3).
Fig. 1.

Box and whisker plots of the four diversity metrics for bacteria [richness (A) , evenness (B), Shannon's diversity (C), and Simpson's diversity (D)] with homes categorized according to dog ownership. Whiskers on box plots are maximum and minimum values. No significant differences were observed for fungi (see Supplemental Figure IV).
Table 3.
Descriptive statistics and Pearson correlation coefficients (r) between fungal and bacterial diversity metrics.
| Bacteria | Fungi | r | p | |
|---|---|---|---|---|
| Richness | 394 ± 136 | 146 ± 70 | 0.241 | 0.164 |
| Evenness | 0.81 ± 0.05 | 0.55 ± 0.10 | 0.091† | 0.605 |
| Shannon's Diversity | 4.81 ± 0.41 | 2.73 ± 0.71 | 0.131 | 0.452 |
| Simpson's Diversity | 0.97 ± 0.02 | 0.84 ± 0.09 | 0.102† | 0.559 |
Notes: Values are arithmetic means ± standard deviation.
Spearman correlation coefficients are reported for all correlations involving Simpson's diversity and evenness.
As the ERMI value, relative humidity, and age of home determinants appeared to have the most influence on indoor fungal microbiome (Table 2), they were each used to categorize the homes for fungal LDA analysis (Fig. 2). When categorized according to ERMI value, a total of 39 fungi had LDA scores above the significance threshold of ±2.0 which indicated statistically significant enrichment (Fig. 2a and complete list of 33 putative fungi in high ERMI conditions and six in low ERMI conditions in the supplementary Table E1). A total of 47 putative fungi had LDA scores above the significance threshold of ± 2.0 when categorized according to relative humidity status (Fig. 2b and complete list of 44 putative fungi in high relative humidity conditions and three in low relative humidity conditions in the supplementary Table S2). When categorized according to the age of the home, a total of 67 putative fungi had LDA scores above the significance threshold of ±2.0 (Fig. 2c and complete of list of 57 putative fungi in older homes and ten in younger homes in the supplementary Table S3).
Fig. 2.
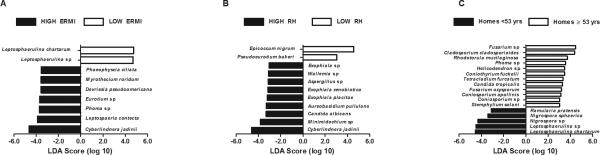
Linear discriminant analysis showing putative fungi associated with high (≥2.0) or low (<2) ERMI homes (A), high (≥36.0%) or low (<36%) relative humidity (B), and older (≥53 years) or younger (<53 years) homes. For the sake of clarity, only putative fungi with LDA scores above 3.5 are shown in panel (A) and above 3.0 in panels (B) and (C).
As dog ownership appeared to have the most influence on the bacterial microbiome in the home (Fig. 1 and supplementary Fig. S5 and Fig. S6), the homes were divided into dog-present or dog-absent homes to categorize the homes for bacterial LDA analysis (Fig. 3). A total of 130 putative bacteria had LDA scores above the significance threshold of ± 2.0 (complete list of 104 putative bacteria in homes with a dog and 26 in homes without a dog in supplementary Table S4).
Fig. 3.

Linear discriminant analysis showing putative bacteria associated with dog ownership. For the sake of clarity, only putative bacteria with LDA scores above 3.0 are shown.
4. Discussion
Fungal growth indoors is known to be enhanced by excess moisture, either as water-damage or humidity (IOM, 2004; WHO, 2009). So it is not surprising that we found excess moisture as important determinant of the home's fungal microbiome. The LDA analysis reinforces this observation. Although the fungal identifications are only putative, the data do demonstrate that about five times more specific fungal genera/species were enriched in higher ERMI homes compared to low ERMI homes and about ten times more fungal genera/species in high versus low relative humidity homes. What is relevant for mold investigations is that the ERMI metric, which is based on only 36 fungi (Vesper et al., 2007), correctly indicated the enrichment of other fungi, not specifically quantified by the ERMI analysis, in homes with excess moisture. Excess moisture may also partially explain why the age of the home is a significant determinant of the home's fungal microbiome.
We have previously shown that older homes are more likely to be water-damaged (higher ERMI values) than newer homes, possibly because the typical infrastructure of a home (roof, pipes, etc) deteriorates with age, resulting in water leaks (Reponen et al., 2013). However, only fungal richness was enhanced in older, high ERMI value and high humidity homes. Fungal community evenness and diversity were only enhanced in older homes. The LDA analysis showed that there were about five times more fungi in the older than in the younger homes. Therefore, it may be that moisture promotes the number of species, i.e., the richness, in the fungal microbiome but, in older homes, there has been sufficient time for the fungal populations to reach population uniformity, i.e., evenness. In addition, the diversity of the fungal microbiome may be greater in older, water-damaged homes compared to younger homes because of the different rates at which fungi colonize homes, after a water-damage event (Yang and Li, 2007); therefore providing more time for colonization by additional fungi, i.e., increased diversity.
On the other hand, the bacterial evenness, richness, and diversity were not significantly determined by the same factors that determined the fungal microbiome. Of the factors tested, bacterial evenness and diversity, but not richness, were significantly increased by the presence of a dog (but not a cat). The LDA analysis showed that homes with dogs contained four times more putative bacterial species compared to homes without a dog.
Fujimura et al. (2010) also found that bacterial diversity was increased in homes with a dog (but not a cat). This diversity might be the result of a dog being allowed outside (whereas, many cats are not). Kembel et al. (2012) reported that the diversity of the outdoor bacterial community was significantly greater than the indoor bacterial community. So if dogs are an important determinant of the home's bacterial microbiome, it may be because dogs are bringing that outdoor bacterial diversity into the home.
The other studies of the bacterial microbiome in homes (Adams et al., 2013a; Adams et al., 2014), and other indoor environments like schools (Qian et al., 2012; Adams et al., 2013b) and a health care facility (Kembel et al., 2012) all demonstrated that the occupants are the most important determinants of the bacterial microbiome in these built environments. Originally, we suspected that water-damage might increase the richness, evenness and diversity of the bacterial community but we found that bacterial microbiomes were about the same, whether the homes were water damaged or not (high vs. low ERMI homes). However, some specific bacteria have been found in higher numbers in water-damaged homes (Rintala et al., 2004; Kettleson et al., 2013). Therefore, although the bacterial microbiome is apparently largely determined by occupants (humans and dog), there may be other factors we did not test which are also determinants of the bacterial microbiome in homes.
In contrast to bacteria, the fungal microbiome is not dominated by occupants. Excess water, as quantified by high relative humidity and high ERMI values, and the age of the home were apparently determinants of the fungal community's composition (richness, evenness and diversity). Previous home microbiome studies did not compare water-damaged and non-water-damaged homes and buildings (Adams et al., 2013a, b; Adams et al., 2014). In those studies, the fungal microbiome was dominated by the fungi from the outdoor air. Yet, it is water-damaged built environments that have been linked to poor respiratory health (IOM, 2004; WHO, 2009; Mendell et al., 2011). Therefore, it appears to be essential to quantify the water-damage in the home (e.g, by moisture measurements or determining the ERMI), when addressing the question of the indoor fungal microbiome and its role in health.
We recognize that our study has limitations beyond just the small number of samples analyzed. The selection of high vs. low ERMI homes was somewhat arbitrary because the samples came from another study; so bias is possible. Also, we only evaluated a few possible determinants that might influence the home's microbiome and certainly there are many other factors that could be important. In addition, pyrosequencing has many inherent limitations, including its lack of standardization of methods, limitations of quantification, reliance on limited databases, and lack of a uniform and comprehensive method of interpreting the results. Despite of these limitations, our study showed that only fungal and bacterial communities are governed by very different determinants.
5. Conclusion
Some key determinants of the fungal microbiome in homes might be excess moisture and age of the home but for the bacterial microbiome, the only determinant, of those tested, was dog ownership.
Supplementary Material
Highlights.
The fungal microbiome was determined by the age of the home and water conditions.
The bacterial microbiome was influenced by dog ownership but not cat.
Bacterial / fungal richness, evenness, or diversity in house dust did not correlate.
Acknowledgements
This study was supported by Grant No. OHLHH0199-09 from the Healthy Homes Technical Studies Program of the U.S. Department of Housing and Urban Development (HUD). We also acknowledge a partial support from the HUD Grant No. OHLHH0162-07, the National Institute for Occupational Safety and Health Pilot Research Project Training Program of University of Cincinnati Education and Research Center Grant No. T42OH008432-07, and the National Institute of Environmental Health Sciences (NIEHS) Grant No. T32ES010957-11 awarded to the University of Cincinnati. The CCAAPS birth cohort study was supported by NIEHS Grant ES11170. Technical assistance from Mr. Chris Schaffer during dust sampling is graciously acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The U.S. Environmental Protection Agency (EPA) through its Office of Research and Development collaborated in the research described here. Although this work was reviewed by EPA and approved for publication it may not necessarily reflect official EPA policy. Mention of trade names or commercial products does not constitute endorsement or recommendation by the EPA for use.
Conflict of Interest The authors declare no conflict of interests as to the interpretation and presentation of this manuscript. Since MSQPCR technology is patented by the US EPA, the Agency has a financial interest in its commercial use.
References
- Adams RI, et al. The diversity and distribution of fungi on residential surfaces. PLoS One. 2013a;8:e78866. doi: 10.1371/journal.pone.0078866. doi: 10.1371/journal.pone.0078866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RI, et al. Dispersal in microbes: fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. ISME J. 2013b;7:1262–73. doi: 10.1038/ismej.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RI, et al. Airborne bacterial communities in residences: similarities and differences with fungi. PLoS One. 2014;9:e91283. doi: 10.1371/journal.pone.0091283. doi: 10.1371/journal.pone.0091283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari A, et al. Dustborne and airborne Gram-positive and Gram-negative bacteria in high versus low ERMI homes. Sci. Total Environ. 482. 2014;483:92–99. doi: 10.1016/j.scitotenv.2014.02.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, et al. Detecting identity by descent and estimating genotype error rates in sequence data. Am. J. Hum. Genet. 2013;93:840–51. doi: 10.1016/j.ajhg.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway TR, et al. Evaluation of the bacterial diversity in cecal contents of laying hens fed various molting diets by using bacterial tag-encoded FLX amplicon pyrosequencing. Poultry Sci. 2009;88:298–302. doi: 10.3382/ps.2008-00222. [DOI] [PubMed] [Google Scholar]
- Callaway TR, et al. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. J. Animal Sci. 2010;88:3977–83. doi: 10.2527/jas.2010-2900. [DOI] [PubMed] [Google Scholar]
- Cho SH, et al. The effect of home characteristics on dust antigen concentrations and loads in homes. Sci. Total Environ. 2006;371:31–43. doi: 10.1016/j.scitotenv.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd SE, et al. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 2008;8:125. doi: 10.1186/1471-2180-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura KE, et al. Man's best friend? The effect of pet ownership on house dust microbial communities. J. Allergy Clin. Immunol. 2010;126:410–2. doi: 10.1016/j.jaci.2010.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugland RA, et al. Evaluation of rapid DNA extraction methods for the quantitative detection of fungal cells using real time PCR analysis. J. Microbiol. Meth. 2002;50:319–23. doi: 10.1016/s0167-7012(02)00037-4. [DOI] [PubMed] [Google Scholar]
- IOM (Institute of Medicine) Damp Indoor Spaces and Health. National Academic Press; Washington, D.C.: 2004. [Google Scholar]
- Kembel SW, et al. Architectural design influences the diversity and structure of the built environment microbiome. ISME J. 2012;8:1469–79. doi: 10.1038/ismej.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kembel SW, et al. Architectural design drives the biogeography of indoor bacterial communities. PLoS One. 2014;9:e87093. doi: 10.1371/journal.pone.0087093. doi: 10.1371/journal.pone.0087093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettleson E, et al. Stenotrophomonas, Mycobacterium, and Streptomyces in home dust and air: Associations with moldiness and other home/family characteristics. Indoor Air. 2013;23:387–96. doi: 10.1111/ina.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lax S, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2014;345(6200):1048–52. doi: 10.1126/science.1254529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMasters GK, et al. High prevalence of aeroallergen sensitization among infants of atopic parents. J. Pediatr. 2006;149:505–11. doi: 10.1016/j.jpeds.2006.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell MJ, et al. Respiratory and allergic health effects of dampness, mold, and dampness-related agents: a review of the epidemiologic evidence. Environ. Health Perspect. 2011;19(6):748–56. doi: 10.1289/ehp.1002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonnenmann M, et al. Utilizing pyrosequencing and quantitative PCR to characterize fungal populations in house dust samples. J. Environ. Monit. 2012;14:2038–43. doi: 10.1039/c2em30229b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucleotide [12 Oct. 2013];The National Institutes of Health. 2011 < http://www.ncbi.nlm.nih.gov/nucleotide/>.
- Parker W, et al. Evolutionary biology and anthropology suggest biome reconstitution as a necessary approach toward dealing with immune disorders. Evol. Med. Public Health. 2013;13:89–103. doi: 10.1093/emph/eot008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkäranta M, et al. Molecular profiling of fungal communities in moisture damaged buildings before and after remediation--a comparison of culture-dependent and culture-independent methods.B.M.C. Microbiol. 2011;11:235. doi: 10.1186/1471-2180-11-235. doi: 10.1186/1471-2180-11-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, et al. Size-resolved emission rates of airborne bacteria and fungi in an occupied classroom. Indoor Air. 2012;22:339–51. doi: 10.1111/j.1600-0668.2012.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reponen T, et al. High Environmental Relative Moldiness Index during infancy as predictor of age seven asthma. Ann. Allergy Asthma Immun. 2011;107:120–6. doi: 10.1016/j.anai.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reponen T, et al. Infant origins of childhood asthma associated with specific molds. J. Allergy Clin. Immunol. 2012;130:639–44. doi: 10.1016/j.jaci.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reponen T, et al. Family and home characteristics correlate with mold in homes. Environ. Res. 2013;124:67–70. doi: 10.1016/j.envres.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintala H, et al. Detection of streptomycetes in house dust--comparison of culture and PCR methods. Indoor Air. 2004;14:112–9. doi: 10.1111/j.1600-0668.2003.00219.x. [DOI] [PubMed] [Google Scholar]
- Segata N, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. doi:10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesper SJ, et al. Development of an environmental relative moldiness index for homes in the U.S. J. Occu. Environ. Med. 2007;49:829–33. doi: 10.1097/JOM.0b013e3181255e98. [DOI] [PubMed] [Google Scholar]
- Wood DE, et al. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15(3):R46. doi: 10.1186/gb-2014-15-3-r46. 3 doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (World Health Organization) Europe . WHO guidelines for indoor air quality: Dampness and mould. Copenhagen, Denmark.: 2009. [PubMed] [Google Scholar]
- Yang CS, et al. Ecology of fungi in the indoor environment. In: Yang CS, Heinsohn P, editors. Sampling and Analysis of Indoor Microorganisms. John Wiley & Sons, Inc.; Hoboken, NJ: 2007. pp. 191–214. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.