

Abstract
Hepatitis C virus (HCV) is a major human pathogen of chronic hepatitis and related liver diseases. Innate immunity is the first line of defense against invading foreign pathogens, and its activation is dependent on the recognition of these pathogens by several key sensors. The interferon (IFN) system plays an essential role in the restriction of HCV infection via the induction of hundreds of IFN-stimulated genes (ISGs) that inhibit viral replication and spread. However, numerous factors that trigger immune dysregulation, including viral factors and host genetic factors, can help HCV to escape host immune response, facilitating viral persistence. In this review, we aim to summarize recent advances in understanding the innate immune response to HCV infection and the mechanisms of ISGs to suppress viral survival, as well as the immune evasion strategies for chronic HCV infection.
Keywords: Hepatitis C virus, Interferon, Interferon-stimulated gene, RIG-I, Toll-like receptor, Virus-host interaction, Chronic hepatitis, Immune evasion, Cell culture system for hepatitis C virus
Core tip: The complex interaction between hepatitis C virus (HCV) and its host determines which side the balance tends to be tipped between antiviral innate response and viral immune evasion. The development of new cell culture systems and small animal models for HCV research permits increased understanding of how the host responds to viral infection and what leads to HCV evasion of innate immunity. Recent discoveries in these areas reveal many pivotal factors imparting the control of viral-induced innate immunity, and facilitate the development of novel drugs and effective vaccines for HCV infection.
INTRODUCTION
Hepatitis C virus (HCV) is a member of the Flaviviridae family that causes liver disease globally. HCV infects approximately 170 million people worldwide, with 3-4 million new infections per year[1]. Following infection, 20%-30% of infected individuals clear the virus spontaneously without any therapy during this phase, while 70%-80% of them become persistently infected[2]. The virus replicates in the liver continuously and establishes intrahepatic persistent infection progressively, putting patients at risk of hepatic fibrosis, cirrhosis and hepatocellular carcinoma[3]. Although direct acting antivirals (DAAs) have been introduced recently, PEGylated interferon-α (PEGIFN-α) plus ribavirin is still the current standard-of-care therapy for HCV in many treatment centers. However, only 40%-50% of patients infected with difficult-to-treat HCV genotypes achieve sustained viral response with PEGIFN-α treatment regimen[4]. A prophylactic anti-HCV vaccine is still lacking[5].
The first immune defense that senses HCV infection is the intrinsic innate immunity within hepatocytes. The pattern recognition receptors (PRRs) within the infected cells sense the virus as non-self and induce antiviral defenses through activation of downstream signaling cascades to clear the virus. Evolutionarily, HCV has acquired strategies to modulate and escape immune recognition by the host, which contributes to HCV persistence[6]. Furthermore, the polymorphisms in IFNL3 (also referred to as IL28B), as the host factor, influence both the outcome of infection and the response to therapy[7-11]. The mechanisms that underlie the different outcomes of viral infection are still not fully understood, but likely reflect a complex interaction between the virus and the host during the immune response. Understanding how HCV activates or evades innate immune responses is essential to develop new antiviral approaches and to design effective vaccine applications to reduce the disease burden of HCV-induced liver disease and cancer. In this review, we describe how HCV regulates the antiviral innate immune response in the hepatocytes and discuss how virus-host interactions affect the outcome of HCV infection.
LIFE CYCLE OF HCV
The HCV genome consists of an approximately 9.6 kb positive single-stranded RNA (+ssRNA)[12]. Its genome encodes a large polyprotein of about 3000 amino acids that is processed by a combination of host signal peptidases and viral proteases into 10 individual proteins, including three structural proteins (core, E1 and E2) and seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). Based on the viral genetic variation of the Core, E1 and NS5B, HCV is classified into seven genotypes (1-7) and up to 67 different subtypes[13]. The different HCV genotypes exhibit different geographical distributions, differential responses to therapy and distinct characteristics in pathogenesis[14].
During infection of hepatocytes by HCV, much progress has been made in understanding how this enveloped RNA virus enters cells. The first described receptors for HCV were the tetraspanin CD81[15] and scavenger receptor class B type I (SR-BI)[16]. Glycosaminoglycans and low-density lipoprotein receptor mediate initial viral binding to hepatocytes before HCV envelope protein interacts with CD81 and SR-BI[14]. The cellular protein apolipoprotein E (apoE) is needed for HCV attachment to cells[17]. Using an iterative complementary DNA library screening approach, two tight junction proteins, claudin-1 (CLDN1)[18] and occludin (OCLN)[19], were identified as co-receptors for the later step of HCV entry. Moreover, the major block to HCV entry of mouse hepatocytes can be overcome by the expression of human CD81 and OCLN in the context of mouse CLDN1 and SR-BI[19]. Recently, the cholesterol absorption receptor, Niemann-Pick C1-like 1, the epidermal growth factor receptor and ephrin receptor typeA2, were reported as essential HCV entry factors that function after viral binding[20,21]. The membrane-spanning syndecan-1 and cell death-inducing DFFA-like effector B were also identified as HCV entry cofactors[22,23]. However, the next step of pH-dependent fusion and uncoating, and release of the viral genome into the cytoplasm is poorly understood[24].
After virion entry into hepatocytes, the internal ribosomal entry site (IRES) sequence located at the 5′ end of the untranslated region of the viral RNA genome is recognized by ribosomes studded in host endoplasmic reticulum (ER) to mediate viral polyprotein translation[25]. The HCV nonstructural proteins, including NS3, NS4A, NS4B, NS5A, and NS5B, comprise the RNA replication machinery within ER-derived structures known as the membranous web[26]. Lipid droplets (LDs) are found in replication sites located at the ER membranous web and play a central role in viral RNA synthesis[27]. HCV particle assembly involves the coordinated action of the ER-resident E1-E2 glycoprotein complex, recruitment of LDs-associated core protein to package viral RNA, and several viral and host factors[28]. HCV uses the lipoprotein production pathway to further assemble infectious particles and to release them from the infected cells. AMP-activated protein kinase plays a key role in regulation of both lipid and glucose metabolism and is implicated in HCV replication[29,30]. Recently, based on the method of selective evolution of ligands by exponential enrichment, we screened a series of specific aptamers targeting viral proteins Core[31], E1E2[32], NS2[33], NS5A[34], respectively. The enzyme-linked oligonucleotide assay, based on core-specific aptamers, can be used to detect serum samples from hepatitis C patients[31]. Furthermore, core-specific aptamers inhibit the production of infectious virus particles by disrupting the localization of core with LDs and NS5A, and perturbing the association of core protein with viral RNA[31]. Like core-specific aptamers, the aptamers targeting NS5A also exhibit suppression of viral production by reversing the interaction of NS5A and core[34]. Moreover, NS5A-specific aptamers repress the RNA replication of HCV[34]. The aptamers targeting E1E2 selectively inhibit HCV entry at early binding step by disrupting the association of E2 and viral receptors[32]. Interestingly, NS2-specific aptamers suppress HCV RNA replication, assembly and release[33]. These aptamers may hold promise for the investigation of the detailed mechanisms of HCV life cycle and development as therapeutic drugs for hepatitis C patients.
MODELS FOR HCV INFECTION
Cell culture models
There is a lack of efficient HCV cell culture systems; therefore, research on the virus has been impeded since the identification of HCV in 1989. The first robust HCV replicon system derived from the Con1 strain (genotype 1b) was established in Huh7 cells in 1999[35]. Until 2005, the first fully permissive cell culture system for HCV became possible using a clinic isolate (genotype 2, termed JFH1) from a Japanese patient with fulminant hepatitis C[36-39]. Similar to the JFH1-derived cell culture system, the efficient infectious culture systems based on H77 (genotype 1a) and Con1 isolates were also described, though both are less robust than JFH-1[40,41]. Continuous culture of JFH1 virus in vitro leads to accumulation of viral genomic mutations responsible for enhancing of virus titer[42]. Several groups reported multiple mutations residing in the p7 and NS2 regions, consistent with their important role in virus assembly[42-45]. Other viral proteins associated with HCV RNA replication, such as NS3 and NS5A, also evolve many titer-enhancing mutations with cell culture adaptation of virus[42,46-48].
Until now, most robust HCV infection has been achieved in Huh7 cells or its derived cells. However, Huh7 cells are not well suited to investigate innate immune response, virus-host interaction and HCV-associated carcinogenesis because they contain some mutant host factors and lack the majority of markers characterizing mature hepatocytes. Hence, it is desirable to develop alternative host cell systems. Indeed, some groups tried to replicate HCV in non-Huh7 cell lines such as HuH-6[49], HepG2[50,51], IMY-N9[51] and LH86[52]. Furthermore, human cell lines derived from the non-liver tissues, including HEK293[53,54], HeLa[54,55], neuroepithelioma cells[56], as well as non-human cells[55,57-61], have been reported to support HCV infection. However, the levels of viral replication in these cells are lower than Huh7 cells, which may reflect insufficient expression of miR-122 and apoE in the non-Huh7 cells[62]. There have been significant advances in the generation of HCV culture systems based on the hepatocyte-like cells induced from pluripotent stem cells[63-65]. Importantly, Schwartz and co-workers demonstrated that induced hepatocyte-like cells mount an antiviral inflammatory response with upregulation of innate immune/inflammatory markers, including CXCL10, CXCL11, TNF-α, IL28B and IL29, by HCV infection[63]. The utilization of the hepatocyte-like cells may be limited because of a series of complicated conditions for induction and culture of cells. To maximally imitate the in vivo infection of virus, we and other groups isolated primary human hepatocytes (PHHs) to establish the closest cell-based in vitro model for HCV[66-68]. After acute HCV infection of PHHs, interferon-β (IFN-β) and interferon-stimulated genes were induced in the cells and the apoptosis of viral-infected cells were triggered by TRAIL-mediated pathway, which indicated that the innate host response is intact in HCV-infected PHHs[66]. Although PHHs can support successful HCV replication, their disadvantages, such extreme lack of donors and highly variable results, may hinder their application. Strikingly, we recently established a PHHs-like hepatoma cell line, termed HLCZ01, and found that it can support both HCVcc (HCV cell culture) and HCV clinical isolates infection and replication[69]. In comparison to Huh7 or its derived cell lines, HLCZ01 mounts an innate immune response to HCV infection[69]. In addition, HLCZ01 also supports infection and propagation of hepatitis B virus produced both in cell culture and clinically[69]. This cell line provides a powerful tool for addressing the virus lifecycle, research of antiviral innate immunity and the development of antivirals and vaccines.
Animal models
Apart from humans, chimpanzees are the only species that support natural HCV infection. The species-specific host cell factors promoting or restricting HCV replication may have led to this narrow host range[70]. Chimpanzees have played an essential role in the identification of HCV as the etiological agent of non-A non-B hepatitis[71]. In fact, the entire HCV life cycle can be achieved in chimpanzee. Viruses obtained from clinical isolates and in vitro tissue culture have been inoculated into chimpanzees using intravenous or intrahepatic injection[72-75]. The chimpanzee model holds promise for research of both innate and adaptive immunity, and the development or testing of new drugs and vaccines. However, studies based on this model are impeded by limited availability, high costs and ethical concerns. In addition, research on chimpanzees is now banned in most countries[76]. Therefore, alternative animal models are urgently needed. Although the tree shrew, a small squirrel-like mammal, is susceptible to HCV infection[77,78], numerous groups prefer to construct HCV-susceptible mice models.
Humanized mice transplanted with human hepatocytes or expressing essential HCV host factors are now considered as the novel models for analyzing HCV infection and testing therapeutics. The xenotransplantation model, as one of the humanized models, must suffer from an endogenous liver injury and allow stimulated growth of human hepatocytes. The human liver chimeric Alb-uPA[79,80], MUP-uPA[81], HSV-tk[82] and FAH-/-[83] mice were reported to be infected with HCV derived from cell culture and clinic isolates. The AFC8-hu hematopoietic stem cells (HSC)/Hep mice containing both human immune system and liver tissues is a novel model for the study of the immune response against HCV[84]. However, this model is limited because of shortage of human donors to isolate HSCs and hepatocyte progenitors. Another mouse model, comprising genetically humanized mice that were transient adenovirally transfected[85] or showed stable transgenic expression of human CD81 and OCLN[86], could support HCV infection of murine hepatocytes in vivo. Importantly, the genetically humanized mice are immunocompetent models, which provide opportunities to study host immunity during viral infection, in spite of their low efficiency of HCV infection[85]. Although numerous studies have been done to improve the efficacy of the humanized mice, the model remains technically challenging. Currently, there is still a lack of a suitable alternative to the chimpanzee model for to study both innate and adaptive immune responses to HCV infection and to test candidate drugs and vaccines[87].
RECOGNITION OF HCV
RIG-I-like receptors detect HCV
In the host cell, several PRRs sense viruses as foreign invaders through pathogen-associated molecular patterns (PAMPs) recognition to activate the innate immune response. The major key PRRs include RIG-I (retinoic acid inducible gene-I)-like receptors (RLRs), toll-like receptors (TLRs) and other nontraditional PRRs. RLRs consist of RIG-I, melanoma differentiation-associated protein 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2). RIG-I is a cytosolic RNA helicase that contains three major domains: a C-terminal domain, a central DExD/H box RNA helicase domain, and two CARD domains at the N-terminus[88,89]. Mitochondrial antiviral signaling protein (MAVS; also called Cardif, IPS-1 or VISA) is the common adaptor for RLRs. Interestingly, MAVS is located on mitochondria, peroxisomes and mitochondria-associated membranes (MAMs)[90,91] (Figure 1). Activation of MAVS leads to activation of TBK1 or IKKε, which phosphorylates downstream IFN regulatory factor 3 (IRF3) and IRF7. In addition, MAVS also activates NF-κB through the activation of the classical IKK complex.
Figure 1.
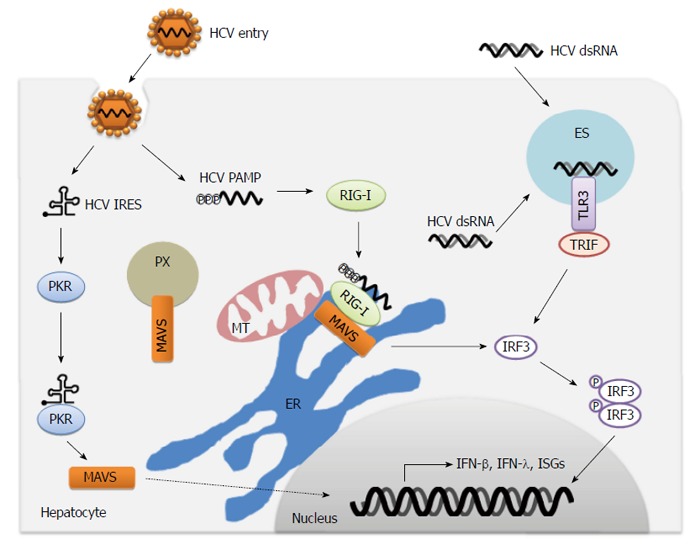
Immune sensing of hepatitis C virus by pattern recognition receptors. During hepatitis C virus (HCV) infection, PKR and RIG-I recognize dsRNA constructed in the HCV IRES and HCV PAMP with 5’-triphosphate plus 3’poly-U/UC region, respectively. In the late period of infection, HCV dsRNA from viral replication intermediates or extracellular dying cells is detected by TLR3. Activation of pattern recognition receptors (PRRs) leads to the transduction of signaling through adaptor protein MAVS or TRIF, and subsequent phosphorylation and dimerization of IRF3 to induce the production of IFN-β, IFN-λ, ISGs and proinflammatory cytokines. MT: Mitochondria; ER: Endoplasmic reticulum; ES: Endosome; PX: Peroxisome; MAVS: Mitochondrial antiviral signaling; PAMP: Pathogen-associated molecular pattern; IFN: Interferon; TRIF: TIR-domain-containing adaptor-inducing interferon-β.
HCV is recognized by RIG-I at the early period of infection[92]. RIG-I senses HCV PAMP that bears 5′ triphosphate and 3′ untranslated region of the HCV genome RNA with poly-U/UC ribonucleotides[93,94] (Figure 1). The recognition of HCV PAMP by RIG-I is dependent on the 34-nucleotide poly-uridine core within the poly U/UC region[95]. Although the 5′ triphosphate and the 3′ poly-U/UC region are at opposite ends of the viral genome, the interactions between the 5′ and 3′ ends of HCV RNA can bring both into proximity for presentation to RIG-I[96]. RIG-I is also crucial for host innate responses to other RNA viruses, including influenza virus, vesicular stomatitis virus, Sendai virus, Japanese encephalititis virus, dengue virus and West Nile virus (WNV)[97-99]. When the HCV RNA binds to RIG-I, it induces a RIG-I conformational change that promotes its oligomerization and translocation from the cytosol into intracellular membranes[100-102]. The E3 ubiquitin ligase tripartite motif-containing protein 25 (TRIM25) induces the lysine 63 (K63)-linked polyubiquitination of the CARD of RIG-I at lysine 172, which is crucial for RIG-I signaling pathway to elicit host antiviral innate immunity[103]. Recent works revealed that two other E3 ubiquitin ligases, Mex3c and Riplet (also called Reul), are also essential for K63-linked polyubiquitination of RIG-I and RIG-I-dependent innate immune responses[104-106]. The chaperone protein 14-3-3e is involved in the association of RIG-I and TRIM25, and facilitates RIG-I translocation to interact with MAVS[102]. Strikingly, unanchored K63-ubiquitin chains potently activate RIG-I by interacting with RIG-I CARD domains[107]. Furthermore, the binding of unanchored K63-ubiquitin chains to RIG-I induces the formation of a large complex comprising four RIG-I and four polyubiquitin chains, which is responsible for activation of antiviral signaling[108]. Siglec-G, a member of lectin family, promotes RIG-I degradation by E3 ubiquitin ligase c-Cbl, which inhibits the innate immune response[109].
MDA5 preferentially senses long dsRNA molecules generated during virus infection[110]. Whether MDA5 serves as a PRR for HCV infection remains to be clarified. Overexpression of MDA5 inhibits HCV infection in vitro and the HCV NS3/4A protein can block the transduction of MDA5 signaling[111]. In addition, suppression of MDA5 by V protein of paramyxovirus enhances HCV replication[112,113]. However, it fails to induce the IFN-β in the RIG-I-/- mouse embryonic fibroblasts (MEFs) by stimulation with HCV RNA[93]. Knockdown of RIG-I in the PHHs and HLCZ01 cells impairs the production of HCV-induced IFN-β[66,114]. Moreover, the MDA5-/- MEFs still produce IFN-β upon stimulation with HCV RNA[93]. These studies suggested that RIG-I is the key sensor for HCV recognition and MDA5 may be involved in regulation of innate immunity triggered by HCV infection.
TLRs sense HCV
TLRs also serve as host PRRs to detect HCV PAMPs. TLR3, TLR7 and TLR9 sense viral nucleic acids, while TLR2 and TLR4 recognize viral proteins[115,116]. In addition, HCV infection can induce the expression of TLR4[117]. As an endosomal sensor of HCV dsRNA, TLR3 is expressed in many cell types within the liver, including hepatocytes and the liver-resident macrophage Kupffer cells[118,119]. TLR3 is type I transmembrane protein and comprises an amino-terminal horse shoe-shaped ectodomain that recognizes viral dsRNA, a transmembrane region, and a cytosolic carboxy-terminal Toll-IL1 receptor homology (TIR) domain that activates downstream signaling[120]. Moreover, the activation of TLR3 signaling is dependent on the acidification of endosomes[121].
TLR3 signals are transduced through the adaptor protein TIR-domain-containing adaptor-inducing interferon-β (TRIF), which activates IRF3 and NF-κB, leading to the production of type I IFN, proinflammatory cytokines and chemokines[122] (Figure 1). TLR3 also triggers the apoptosis of human cancer cells directly [123]. HCV dsRNA replication intermediates (≥ 80-100 bp) accumulated late during HCV replication and represent the HCV PAMP[124]. Actually, HCV infection activates TLR3 signaling late (3-4 d) in infection[118,124], which indicates that the incoming viral genomes or early replication products may not present as TLR3 ligands. In addition to the dsRNA accumulation from viral replication, the HCV ligands for TLR3 are probably obtained from the uptake of extracellular HCV dsRNA (either from dying cells or from the extracellular milieu) by scavenger receptors on nearby uninfected cells[125]. Consequently, TLR3-mediated signaling may act as a secondary innate immune response or monitoring system for uninfected cells after the initial RIG-I detection of HCV.
TLR7 recognizes ssRNAs derived from HCV or other ssRNA viruses in the endosome. TLR7 is expressed mainly in plasmacytoid DCs (pDCs). Phagocytic uptake of HCV RNA triggers an innate immune response via TLR7 signaling in the pDCs. Activated pDCs induce type I IFNs rapidly and the levels of IFNs are more than 100-fold higher than that induced in any other blood cell type[126,127]. Exosomes can transfer HCV RNA from infected cells to pDCs and activate TLR7-dependent innate response in pDCs[128]. Exosomes from both HCV-infected cells and hepatitis C patients transmit HCV infection of human hepatoma cells[129,130]. TLR2 is expressed on the cell surface. The TLR2 pathway is involved in the induction of TNFα after stimulation by synthetic TLR2 ligands in various hepatocyte cell lines[131]. In addition, hepatic expression of TLR2 and TNF-α is associated with hepatic inflammation and liver injury in hepatitis C patients[132,133].
Other sensors recognition of HCV
The protein kinase R (PKR), an antiviral protein, is a serine/threonine kinase that regulates protein synthesis, cell proliferation, apoptosis and signal transduction. PKR is a PRR that detects HCV and activates innate immune signaling[134]. Upon binding to HCV dsRNA, PKR activates its kinase activity for phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α). Activated PKR results in shutting-off of the cap-dependent translation of host mRNAs, while HCV uses an IRES translation mechanism (cap-independent translation) that is insensitive to the phosphorylation of eIF2α[135-138]. PKR binding of HCV dsRNA also activates a kinase-independent signaling that induces a small subset of IFN-stimulated genes (ISGs) and IFN-β through MAVS, TNF receptor-associated factor 3, IRFs and NF-κB transduction before RIG-I activation[134,139,140] (Figure 1). In addition, although the HCV ligand for PKR is the structured RNA at the IRES of the HCV genome[134,138], the detail course of PKR recognition remains unclear.
The Nod (nucleotide oligomerization domain)-like receptors (NLRs) belong to a large family of intracellular PRRs, including NOD1, NOD2, and NLR protein 3 (NLRP3)[141]. NLRs sense the PAMPs from RNA viruses, resulting in activation of the inflammasome[142].The inflammasome comprises a sensor protein, the adaptor protein ASC and the cellular protease caspase 1. The NLRP3 inflammasome is the most characterized inflammasome and is activated by viral dsRNA and ssRNA. NLRP3 triggers the production of proinflammatory cytokines IL-1β in macrophages during HCV infection[143].
IMMUNE EFFECTORS THAT RESTRICT HCV
The immune effectors against invading viral pathogens infection are the products of interferon-stimulated genes (ISGs). More than 300 ISGs are regulated by IFNs through the Jak-Stat signaling pathway. To identify the specific set of ISGs responsible for the inhibition of HCV replication, several screens have been conducted in vitro systems. Identified ISGs that restrict the HCV life cycle are summarized in Table 1. Using an overexpression screening approach, a recent study tested the ability of nearly 400 ISGs to inhibit the replication of several important human and animal viruses, including HCV, yellow fever virus, WNV, chikungunya virus, Venezuelan equine encephalitis virus and human immunodeficiency virus type-1[144]. For HCV, the inhibitory effectors includes IRF family members, RIG-I, MDA5 and other little characterized ISGs, such as DDIT4, IFI44L, MAP3K14, OASL and NT5C3[144,145]. In contrast to the overexpression approach, an RNA interference-based screen was performed by two groups to identify anti-HCV ISGs[146,147]. Zhao et al[146] identified 93 genes that mediate the anti-HCV effect of IFN-α, in which 23 and nine genes are involved in mRNA processing and translation initiation, respectively. Seven ISGs were identified by Metz and co-workers to contribute to the suppression of HCV replication by IFN-α or IFN-γ[147]. Although all ISGs identified in this study were upregulated by either cytokine, PLSCR1 and NOS2 were the main effectors of IFN-γ-mediated anti-HCV activity[147]. In addition to ISG proteins, the IFN-induced microRNAs (miRNAs) may also be capable of inhibiting HCV replication[148]. Similarly, Fusco and co-workers identified ISGs as well as non-transcriptionally induced genes required for the antiviral effect of IFN-α using a genome-wide siRNA screen[149]. In this screen, 120 IFN effector genes were identified that restricted HCV, and 92% of them were non-transcriptionally IFN-induced genes, which suggested an as-yet-unknown action of the IFN pathway[149].
Table 1.
Antiviral interferon-stimulated genes targeting different steps of the hepatitis C virus life cycle
| Gene symbol | Phases of HCV life cycle targeted by ISGs | Infection or replicon for studies | Overexpression or knockdown of ISGs for tests | Ref. |
| IFITM1 | Entry | Infection | oe/kd | [158] |
| Replication | Infection | oe/kd | [159] | |
| Replication | Replicon | kd | [147] | |
| IFITM3 | Translation | Replicon | oe | [161] |
| Replication | Replicon | oe | [160] | |
| Replication | Replicon | oe/kd | [147] | |
| DDIT4 | Translation | Infection/replicon | oe | [144] |
| IFI44L | Translation | Infection/replicon | oe | [144] |
| IRF1 | Translation | Infection/replicon | oe | [144] |
| Replication | Replicon | oe | [162] | |
| IRF2 | Translation | Infection/replicon | oe | [144] |
| IRF7 | Translation | Infection/replicon | oe | [144] |
| MAP3K14 | Translation | Infection/replicon | oe | [144] |
| MDA5 | Translation | Infection/replicon | oe | [144] |
| NT5C3 | Translation | Infection/replicon | oe | [144] |
| RIG-I | Translation | Infection/replicon | oe | [144] |
| OASL | Translation | Infection | oe | [144] |
| Replication | Replicon | oe | [152] | |
| RNaseL | Replication | Replicon | oe/kd | [147] |
| Replication | Infection | oe | [153] | |
| ADAR1 | Replication | Replicon | kd | [163] |
| GBP-1 | Replication | Replicon | oe/kd | [162] |
| IFI27 | Replication | Replicon | oe/kd | [162] |
| IFI6 | Replication | Replicon | oe/kd | [162] |
| IFIT1 | Replication | Infection | oe/kd | [162] |
| IFIT3 | Replication | Replicon | kd | [147] |
| IRF9 | Replication | Replicon | oe | [162] |
| ISG20 | Replication | Replicon | oe | [164] |
| MxA | Replication | Replicon | oe | [162] |
| NOS2 | Replication | Replicon | oe/kd | [147] |
| OAS | Replication | Replicon | oe | [162] |
| PLSCR1 | Replication | Replicon | oe/kd | [147] |
| TRIM14 | Replication | Replicon | oe/kd | [147] |
| Viperin | Replication | Replicon | oe | [164] |
| Replication | Replicon | oe | [165] | |
| Replication | Infection/replicon | oe | [155] | |
| Replication | Infection | kd | [156] | |
| Tetherin | Release | Infection | oe | [166] |
HCV: Hepatitis C virus; ISGs: Interferon-stimulated genes; Infection or replicon: The functions of ISGs were studied by using infectious culture system (infection) or subgenomic replicon (replicon); oe or kd: The functions of ISGs were identified by using overexpression (oe) or knockdown (kd) of indicated ISGs in cells.
Although numerous ISGs have been validated in the process of IFN-mediated anti-HCV activity, the underlying mechanisms of most ISGs are unclear. Several ISGs, including OAS-RNaseL system, Viperin and the IFITM family members are relatively well characterized[150]. During viral infections, viral dsRNA activates one of the three functional human 2′,5′-oligoadenylatesynthetases (OASs) to synthesize 2′-to-5′-linked oligoadenylates (2-5A), which subsequently activate RNaseL to cleave viral RNAs, as well as cellular mRNAs and rRNAs, to induce apoptosis of infected cells[151]. In vitro studies have shown that all three OAS proteins induce RNaseL-dependent antiviral activity against HCV[144,152,153]. Viperin (RSAD2) localizes to the ER and lipid droplets. Viperin interacts with HCV core and NS5A proteins, as well as VAP-A, an important host factor of HCV replication, in the LDs-ER membranes[154-156]. Viperin may cause an alteration of the lipid composition of the membranous web by disturbing NS5A-VAP-A interaction, leading to inhibition of HCV replication[157]. The IFN induced transmembrane proteins (IFITMs), including IFITM1, IFITM2, IFITM3 and IFITM5, contain two anti-parallel transmembrane domains, a leucin-zipper motif and a short cytoplasmic domain. IFITM1 interacts with HCV co-receptors CD81 and occludin to inhibit the process of viral entry[158]. IFITM1 and IFITM3 suppress HCV replication; however, the mechanism of replication inhibition by IFITMs remains to be determined[147,159,160]. In addition, IFITM3 inhibits the translation of both HCV polyprotein and host proteins[161]. IFI27 (also called ISG12a), a mitochondria-anchored protein, also exhibits antiviral activity through inhibition of HCV replication and by triggering apoptosis of HCV-infected cells[114,162]. Although the role of IFI27 in early control of HCV is unclear, IFI27 can mediate TRAIL-dependent apoptosis via induction of Noxa in the late stage of HCV infection[114]. Although single ISG exhibits anti-HCV activity, full restriction of HCV infection requires the combined activity of multiple ISGs and the transduction of other innate immune signaling pathways.
STRATEGIES FOR INNATE IMMUNITY EVASION OF HCV
Viral factors
Despite the effective recognition of HCV by many sensors to activate innate immune responses, about 80% of patients with acute HCV infection do not effectively clear the virus and develop a chronic infection[167]. HCV may have evolved several mechanisms that are responsible for viral evasion of host innate immune signaling. The key viral factor of the HCV innate immune evasion is the viral NS3/4A protease. NS3/4A, a complex of NS3 and NS4A proteins, is required for the HCV life cycle, including viral RNA replication, polyprotein processing and viral assembly[168]. The NS3/4A complex is anchored to intracellular membranes by the N-terminal α-helix of NS3 and NS4A transmembrane domain that facilitates membrane association and cleavage of membrane-anchored substrates[169,170]. The RIG-I signaling pathway can be blocked by NS3/4A, which cleaves MAVS at Cys508 from intracellular membranes to prevent its dimerization and downstream signaling of innate immunity[92,171-175]. NS3/4A cleaves MAVS at the mitochondrial outer membrane[92,175], and MAVS also localizes to peroxisomes and MAMs[90,91]. Although NS3/4A localizes to all three kinds of membranes during HCV infection, MAM-localized MAVS is cleaved by NS3/4A rather than MAVS anchored on the outer mitochondrial membrane[91] (Figure 2). Activation of the RIG-I pathway by HCV infection may occur via the MAM-localized MAVS to transduce the downstream signaling. In the livers of patients with chronic HCV infection, the cleavage of MAVS by NS3/4A was also observed, which contributes to lower levels of IFN pathway induction[92,176]. Hence, the fact that NS3/4A targets and cleaves MAVS to disrupt RIG-I signaling is an effective strategy for HCV to evade the innate antiviral immunity. HCV NS3/4A also targets the E3 ubiquitin ligase Riplet and inhibits K63-linked polyubiquitination of RIG-I and its association with TRIM25 and TBK1, which provides a new mechanism for HCV evasion of host immunity[105].
Figure 2.
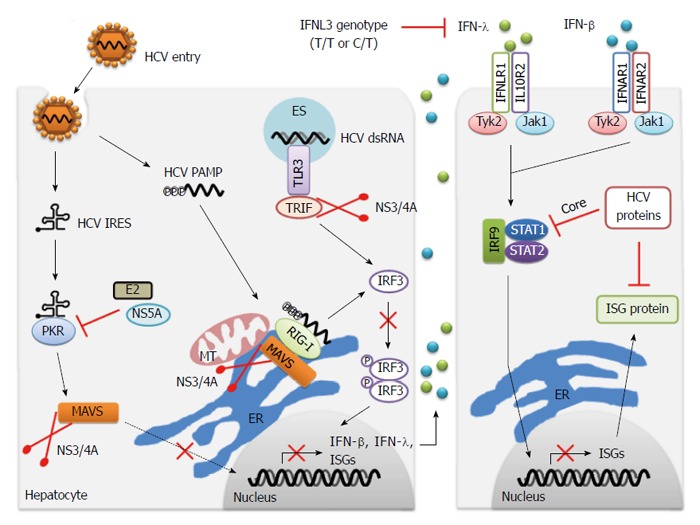
Innate immune evasion of hepatitis C virus infection. Multiple strategies to disrupt both IFN induction and response pathways are responsible for chronic hepatitis C virus (HCV) infection during viral infection. Notably, HCV NS3/4A protease cleaves the adaptor proteins MAVS and TRIF, contributing to the blockage of downstream signaling of PKR, RIG-I and TLR3, and prevention of IFN production. HCV E2 and NS5A proteins suppress the function of PKR by direct binding to this kinase. IFN-β and IFN-λ bind to their distinct receptors and induce the formation of IFN-stimulated gene factor-3 (ISGF3), comprising phosphorylated STAT1, STAT2 and IRF9 through the common JAK/STAT pathway. HCV core and other viral proteins inhibit the activation of STAT signaling, and therefore repress the production of ISGs. The function of specific ISG protein is also suppressed by HCV proteins. Additionally, the unfavorable IFNL3 genotype (T/T or C/T) leads to less expression of IFN-λ, which attenuates the antiviral effect of IFN-λ on HCV infection. MT: Mitochondria; ER: Endoplasmic reticulum; ES: Endosome; PAMP: Pathogen-associated molecular pattern; IRES: Internal ribosomal entry site; TLR: Toll-like receptor; IFN: Interferon; MAVS: Mitochondrial antiviral signaling; ISGs: IFN-stimulated genes; TRIF: TIR-domain-containing adaptor-inducing interferon-β.
TRIF, the TLR3 signaling-adaptor protein, is also cleaved by HCV NS3/4A protease at Cys372, a site containing high sequence homology to the NS4B/5A cleavage site in the HCV polyprotein[177,178] (Figure 2). The infection of hepatoma cells by JFH1 virus in vitro leads to substantial reduction of TRIF protein abundance, which could be partially reversed by NS3/4A inhibitor treatment[118]. Although the relative abundance of TRIF decreased, the specific TRIF proteolytic fragments have not been detected during HCV infection, probably because of technical difficulties in detecting this low abundance protein[118]. Similar to the blockage of RIG-I signaling, NS3/4A-mediated cleavage of TRIF suppresses the transduction of TLR3 signaling, as a mechanism responsible for chronic HCV infection. However, TRIF is also implicated as an adaptor protein for TLR3-independent signaling[179]. This suggests that cleavage of TRIF by NS3/4A may also inhibit other innate immune responses contributing to HCV persistence.
The dsRNA-dependent protein kinase PKR plays multiple roles in cells, including pathogen sensing and response to different stress situations[180]. Binding of HCV dsRNA to PKR activates a kinase-independent signaling that induces some early ISGs and IFN-β through MAVS[134,139,140]. Therefore, cleavage of MAVS by HCV NS3/4A protease probably suppresses the transduction of PKR signaling during HCV infection (Figure 2). Binding to dsRNA also promotes PKR homodimerization and activates the kinase domain to phosphorylate eIF2α. However, the effects of this activation for HCV replication are controversial. During HCV infection, the phosphorylation of eIF2α by PKR inhibits cellular mRNAs translation, which contains host factors important for HCV replication and cellular growth (PKR functions as anti-HCV), while it can also suppress the translation of IFN and ISGs (PKR functions as pro-HCV)[135,138]. Two viral proteins, NS5A and E2, inhibit PKR by directly binding to the kinase[181-183] (Figure 2). NS5A represses PKR through a direct interaction with PKR at a site termed the interferon sensitivity determining region within NS5A[181]. Similarly, E2 can also target and inhibit PKR, and the interaction site in E2 bears a highly conserved amino acid sequence with remarkable sequence homology to PKR autophosphorylation and eIF2α phosphorylation sites[183]. The detailed mechanisms by which NS5A and E2 mediate inhibition of PKR are unknown. It is possible that activation and inhibition of PKR occur in a sequential manner; that is, HCV activates PKR at the early stage of infection and inhibits PKR at a later stage through high levels of synthesized NS5A and E2. Interestingly, HCV E1/E2 protein can downregulate the expression of RIG-I and TLR3 in vitro, which provides a new mechanism of viral evasion of innate immunity for HCV[184].
In addition to disrupting the IFN induction pathway, HCV has evolved strategies to block the IFN response pathway. During HCV infection, HCV viral proteins (such as core protein) are implicated as negative-regulators of the IFN response pathway through blockage of JAK/STAT signaling[185]. In addition, viral proteins also inhibit specific ISGs to evade their restrictions[185] (Figure 2). Interestingly, patients infected with HCV genotype 2 or 3 show the highest response rates to therapy (70%-80%), while only 45%-60% of patients with HCV genotype 1 or 4 infection achieve SVR[186]. Actually, the patients infected with HCV genotypes 1 and 4 have high levels of ISG expression in the liver before IFN therapy, contributing to the resistance of IFN treatment[187-189]. The mechanisms by which HCV persists in the liver, despite high levels of hepatic ISGs expression, are poorly understood and future research focusing on this respect may identify novel strategies by which HCV evades innate immunity.
Host genetic factors
Host genetic factors are also involved in innate immunity evasion by HCV. In fact, the single nucleotide polymorphisms (SNPs) upstream of the IFNL3 locus have been determined as novel predictors for both successful clinical therapy of chronic hepatitis C[7,8,10,11] and spontaneous clearance of HCV infection[9,11]. The IFNL3 locus encodes the antiviral cytokine IFNL3 (also referred to as IL28B), which belongs to type III IFN family. The molecular mechanisms of action of this genetic variation remain unknown. One hypothesis is that these SNPs may alter the mRNA expression of IFNL3 and the expression levels of IFNL3 are probably associated with HCV clearance and therapy effect[7,8,188] (Figure 2). Several groups have verified the hypothesis that the unfavorable SNPs in the IFNL3 locus contribute to lower expression of IFNL3 within the liver, peripheral blood mononuclear cells and whole blood[7,8,189,190,191]. Patients with the unfavorable IFNL3 genotype exhibit the inhibition of innate immune function of NK cells, which suggests that the genetic variation in the IFNL3 locus directly affects antiviral immunity to HCV[192]. Further studies to address how different IFNL3 polymorphisms regulate the innate immune response to HCV infection and the outcome of IFN therapy are encouraged, which will facilitate the development of new therapeutic strategies to clear HCV.
Recently, a newly characterized gene, IFNL4, was identified[193]. IFNL4 is created by a dinucleotide variant ss469415590 (TT or ΔG) in the upstream region of IFNL3 (IL28B) on chromosome 19q13.13[193]. Compared with previously discovered variant rs12979860, ss469415590 is more strongly associated with HCV clearance in individuals of African ancestry[193]. The polymorphism in IFNL4 provides new insights into the genetic regulation of HCV clearance. It is now known that the type III IFNs (IFN-λs) comprise IFNL1 (IL29), IFNL2 (IL28A), IFNL3 (IL28B) and IFNL4. We and other groups have validated the effect of IFN-λs on inhibiting HCV replication in vitro[194,195]. The restricted receptor distribution of IFN-λ makes it likely that IFN-λs could be developed as new drugs for HCV therapy, with fewer side effects than IFN-α[195].
CONCLUSION
The host innate immunity is critical for HCV sensing and subsequent viral clearance, while the dysregulation of innate immune signaling can lead to chronic viral infection. HCV triggers this immune dysregulation by evading the host innate immune response via numerous strategies, including viral factors and host genetic factors, which contributes to eventual viral persistence. Multiple types of cells residing in the liver microenvironment are involved in regulation of HCV-induced innate immune response. However, the detailed mechanisms of how hepatocytes and other cell types regulate each other, and how the immune modulation of cells determines the consequence of HCV infection, is still unclear. The intracellular sensors, such as RIG-I, TLR3 and PKR, are essential for HCV recognition; however the cross talk of these receptors in the detection of HCV during viral infection remains to be clarified. Although the detection of HCV by RIG-I and TLR3 is well characterized, it is unknown how and where the HCV PAMPs are presented to PKR and NLRs for innate immune signaling. The potential mechanisms used by HCV to evade PRRs-dependent recognition should be identified. Genetic studies have provided insights into IFNL3 polymorphisms, indicating that variations of IFNL3 predict an effective immune response for both natural and IFN-induced HCV clearance. However, the IFNL3 genotype is not the best predictor of therapeutic responses to HCV infection[167]. Thus, further studies to define new host factors that determine the outcome of viral infection could help to effectively guide the individual treatment of HCV infection, together with known predictors.
Although the IFN-based therapy is still the current standard-of-care therapy for HCV in many treatment centers, significant scientific advances have enabled the development of new classes of antivirals for the treatment of HCV. In the near future, the DAAs targeting HCV NS3 protease, NS5A or NS5B polymerase may well cure virtually all hepatitis C patients with an all-oral, interferon-free regimen[196]. However, viral breakthrough and drug resistance during these therapeutic courses will need to be considered and monitored carefully[197]. Furthermore, the efficacy of these drugs for other genotypes of HCV, not genotype 1, still needs to be determined. A full understanding of how the host antiviral innate immunity responds to HCV infection, and what drives HCV evasion of this protective activity, will be important to the development of new therapeutic approaches and novel vaccines with high-efficacy for HCV. In addition, the establishment of new, efficient cell culture systems and more convenient small animal models is highly encouraged, which will facilitate basic research on viral-host interactions, innate immunity and HCV pathogenesis, leading to the development of drugs and vaccines for chronic hepatitis C.
Footnotes
Conflict-of-interest: The authors declare no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: November 28, 2014
First decision: January 8, 2015
Article in press: February 13, 2015
P- Reviewer: El-mezayen HA, Honge BL, Oshiumi H, Papastergiou V S- Editor: Qi Y L- Editor: Stewart G E- Editor: Wang CH
References
- 1.Lavanchy D. The global burden of hepatitis C. Liver Int. 2009;29 Suppl 1:74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 2.Hoofnagle JH. Course and outcome of hepatitis C. Hepatology. 2002;36:S21–S29. doi: 10.1053/jhep.2002.36227. [DOI] [PubMed] [Google Scholar]
- 3.Heathcote EJ. Prevention of hepatitis C virus-related hepatocellular carcinoma. Gastroenterology. 2004;127:S294–S302. doi: 10.1053/j.gastro.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 4.Soriano V, Peters MG, Zeuzem S. New therapies for hepatitis C virus infection. Clin Infect Dis. 2009;48:313–320. doi: 10.1086/595848. [DOI] [PubMed] [Google Scholar]
- 5.Liang TJ. Current progress in development of hepatitis C virus vaccines. Nat Med. 2013;19:869–878. doi: 10.1038/nm.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rose R, Markov PV, Lam TT, Pybus OG. Viral evolution explains the associations among hepatitis C virus genotype, clinical outcomes, and human genetic variation. Infect Genet Evol. 2013;20:418–421. doi: 10.1016/j.meegid.2013.09.029. [DOI] [PubMed] [Google Scholar]
- 7.Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML, Bassendine M, Spengler U, Dore GJ, Powell E, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, Nakagawa M, Korenaga M, Hino K, Hige S, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 9.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, Kidd J, Kidd K, Khakoo SI, Alexander G, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 11.Rauch A, Kutalik Z, Descombes P, Cai T, Di Iulio J, Mueller T, Bochud M, Battegay M, Bernasconi E, Borovicka J, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138:1338–1345, 1345.e1-7. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 12.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 13.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 16.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol. 2012;86:7256–7267. doi: 10.1128/JVI.07222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 19.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sainz B, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med. 2012;18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi Q, Jiang J, Luo G. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J Virol. 2013;87:6866–6875. doi: 10.1128/JVI.03475-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu X, Lee EM, Hammack C, Robotham JM, Basu M, Lang J, Brinton MA, Tang H. Cell death-inducing DFFA-like effector b is required for hepatitis C virus entry into hepatocytes. J Virol. 2014;88:8433–8444. doi: 10.1128/JVI.00081-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeisel MB, Felmlee DJ, Baumert TF. Hepatitis C virus entry. Curr Top Microbiol Immunol. 2013;369:87–112. doi: 10.1007/978-3-642-27340-7_4. [DOI] [PubMed] [Google Scholar]
- 25.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 26.Rice CM. New insights into HCV replication: potential antiviral targets. Top Antivir Med. 2011;19:117–120. [PMC free article] [PubMed] [Google Scholar]
- 27.Lohmann V. Hepatitis C virus RNA replication. Curr Top Microbiol Immunol. 2013;369:167–198. doi: 10.1007/978-3-642-27340-7_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindenbach BD. Virion assembly and release. Curr Top Microbiol Immunol. 2013;369:199–218. doi: 10.1007/978-3-642-27340-7_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mankouri J, Tedbury PR, Gretton S, Hughes ME, Griffin SD, Dallas ML, Green KA, Hardie DG, Peers C, Harris M. Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP-activated protein kinase. Proc Natl Acad Sci USA. 2010;107:11549–11554. doi: 10.1073/pnas.0912426107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang D, Xue B, Wang X, Yu X, Liu N, Gao Y, Liu C, Zhu H. 2-octynoic acid inhibits hepatitis C virus infection through activation of AMP-activated protein kinase. PLoS One. 2013;8:e64932. doi: 10.1371/journal.pone.0064932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi S, Yu X, Gao Y, Xue B, Wu X, Wang X, Yang D, Zhu H. Inhibition of hepatitis C virus production by aptamers against the core protein. J Virol. 2014;88:1990–1999. doi: 10.1128/JVI.03312-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang D, Meng X, Yu Q, Xu L, Long Y, Liu B, Fang X, Zhu H. Inhibition of hepatitis C virus infection by DNA aptamer against envelope protein. Antimicrob Agents Chemother. 2013;57:4937–4944. doi: 10.1128/AAC.00897-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao Y, Yu X, Xue B, Zhou F, Wang X, Yang D, Liu N, Xu L, Fang X, Zhu H. Inhibition of hepatitis C virus infection by DNA aptamer against NS2 protein. PLoS One. 2014;9:e90333. doi: 10.1371/journal.pone.0090333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu X, Gao Y, Xue B, Wang X, Yang D, Qin Y, Yu R, Liu N, Xu L, Fang X, et al. Inhibition of hepatitis C virus infection by NS5A-specific aptamer. Antiviral Res. 2014;106:116–124. doi: 10.1016/j.antiviral.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 35.Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 36.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 38.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology. 2003;125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 40.Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc Natl Acad Sci USA. 2006;103:2310–2315. doi: 10.1073/pnas.0510727103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pietschmann T, Zayas M, Meuleman P, Long G, Appel N, Koutsoudakis G, Kallis S, Leroux-Roels G, Lohmann V, Bartenschlager R. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog. 2009;5:e1000475. doi: 10.1371/journal.ppat.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaul A, Woerz I, Meuleman P, Leroux-Roels G, Bartenschlager R. Cell culture adaptation of hepatitis C virus and in vivo viability of an adapted variant. J Virol. 2007;81:13168–13179. doi: 10.1128/JVI.01362-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J Virol. 2006;80:11082–11093. doi: 10.1128/JVI.01307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, Purcell RH, Emerson SU. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc Natl Acad Sci USA. 2008;105:4370–4375. doi: 10.1073/pnas.0800422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delgrange D, Pillez A, Castelain S, Cocquerel L, Rouillé Y, Dubuisson J, Wakita T, Duverlie G, Wychowski C. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol. 2007;88:2495–2503. doi: 10.1099/vir.0.82872-0. [DOI] [PubMed] [Google Scholar]
- 46.Yi M, Ma Y, Yates J, Lemon SM. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J Virol. 2007;81:629–638. doi: 10.1128/JVI.01890-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scheel TK, Gottwein JM, Carlsen TH, Li YP, Jensen TB, Spengler U, Weis N, Bukh J. Efficient culture adaptation of hepatitis C virus recombinants with genotype-specific core-NS2 by using previously identified mutations. J Virol. 2011;85:2891–2906. doi: 10.1128/JVI.01605-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gottwein JM, Scheel TK, Jensen TB, Lademann JB, Prentoe JC, Knudsen ML, Hoegh AM, Bukh J. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology. 2009;49:364–377. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 49.Windisch MP, Frese M, Kaul A, Trippler M, Lohmann V, Bartenschlager R. Dissecting the interferon-induced inhibition of hepatitis C virus replication by using a novel host cell line. J Virol. 2005;79:13778–13793. doi: 10.1128/JVI.79.21.13778-13793.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Narbus CM, Israelow B, Sourisseau M, Michta ML, Hopcraft SE, Zeiner GM, Evans MJ. HepG2 cells expressing microRNA miR-122 support the entire hepatitis C virus life cycle. J Virol. 2011;85:12087–12092. doi: 10.1128/JVI.05843-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Date T, Kato T, Miyamoto M, Zhao Z, Yasui K, Mizokami M, Wakita T. Genotype 2a hepatitis C virus subgenomic replicon can replicate in HepG2 and IMY-N9 cells. J Biol Chem. 2004;279:22371–22376. doi: 10.1074/jbc.M311120200. [DOI] [PubMed] [Google Scholar]
- 52.Zhu H, Dong H, Eksioglu E, Hemming A, Cao M, Crawford JM, Nelson DR, Liu C. Hepatitis C virus triggers apoptosis of a newly developed hepatoma cell line through antiviral defense system. Gastroenterology. 2007;133:1649–1659. doi: 10.1053/j.gastro.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 53.Ali S, Pellerin C, Lamarre D, Kukolj G. Hepatitis C virus subgenomic replicons in the human embryonic kidney 293 cell line. J Virol. 2004;78:491–501. doi: 10.1128/JVI.78.1.491-501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kato T, Date T, Miyamoto M, Zhao Z, Mizokami M, Wakita T. Nonhepatic cell lines HeLa and 293 support efficient replication of the hepatitis C virus genotype 2a subgenomic replicon. J Virol. 2005;79:592–596. doi: 10.1128/JVI.79.1.592-596.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu Q, Guo JT, Seeger C. Replication of hepatitis C virus subgenomes in nonhepatic epithelial and mouse hepatoma cells. J Virol. 2003;77:9204–9210. doi: 10.1128/JVI.77.17.9204-9210.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fletcher NF, Yang JP, Farquhar MJ, Hu K, Davis C, He Q, Dowd K, Ray SC, Krieger SE, Neyts J, et al. Hepatitis C virus infection of neuroepithelioma cell lines. Gastroenterology. 2010;139:1365–1374. doi: 10.1053/j.gastro.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang KS, Cai Z, Zhang C, Sen GC, Williams BR, Luo G. Replication of hepatitis C virus (HCV) RNA in mouse embryonic fibroblasts: protein kinase R (PKR)-dependent and PKR-independent mechanisms for controlling HCV RNA replication and mediating interferon activities. J Virol. 2006;80:7364–7374. doi: 10.1128/JVI.00586-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Long G, Hiet MS, Windisch MP, Lee JY, Lohmann V, Bartenschlager R. Mouse hepatic cells support assembly of infectious hepatitis C virus particles. Gastroenterology. 2011;141:1057–1066. doi: 10.1053/j.gastro.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 59.Frentzen A, Anggakusuma E, Hueging K, Knocke S, Ginkel C, Brown RJ, Heim M, Dill MT, Kröger A, Kalinke U, et al. Cell entry, efficient RNA replication, and production of infectious hepatitis C virus progeny in mouse liver-derived cells. Hepatology. 2014;59:78–88. doi: 10.1002/hep.26626. [DOI] [PubMed] [Google Scholar]
- 60.Uprichard SL, Chung J, Chisari FV, Wakita T. Replication of a hepatitis C virus replicon clone in mouse cells. Virol J. 2006;3:89. doi: 10.1186/1743-422X-3-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vogt A, Scull MA, Friling T, Horwitz JA, Donovan BM, Dorner M, Gerold G, Labitt RN, Rice CM, Ploss A. Recapitulation of the hepatitis C virus life-cycle in engineered murine cell lines. Virology. 2013;444:1–11. doi: 10.1016/j.virol.2013.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lohmann V, Bartenschlager R. On the history of hepatitis C virus cell culture systems. J Med Chem. 2014;57:1627–1642. doi: 10.1021/jm401401n. [DOI] [PubMed] [Google Scholar]
- 63.Schwartz RE, Trehan K, Andrus L, Sheahan TP, Ploss A, Duncan SA, Rice CM, Bhatia SN. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc Natl Acad Sci USA. 2012;109:2544–2548. doi: 10.1073/pnas.1121400109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu X, Robotham JM, Lee E, Dalton S, Kneteman NM, Gilbert DM, Tang H. Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS Pathog. 2012;8:e1002617. doi: 10.1371/journal.ppat.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roelandt P, Obeid S, Paeshuyse J, Vanhove J, Van Lommel A, Nahmias Y, Nevens F, Neyts J, Verfaillie CM. Human pluripotent stem cell-derived hepatocytes support complete replication of hepatitis C virus. J Hepatol. 2012;57:246–251. doi: 10.1016/j.jhep.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 66.Yang D, Liu N, Zuo C, Lei S, Wu X, Zhou F, Liu C, Zhu H. Innate host response in primary human hepatocytes with hepatitis C virus infection. PLoS One. 2011;6:e27552. doi: 10.1371/journal.pone.0027552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Podevin P, Carpentier A, Pène V, Aoudjehane L, Carrière M, Zaïdi S, Hernandez C, Calle V, Méritet JF, Scatton O, et al. Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology. 2010;139:1355–1364. doi: 10.1053/j.gastro.2010.06.058. [DOI] [PubMed] [Google Scholar]
- 68.Steinmann E, Pietschmann T. Cell culture systems for hepatitis C virus. Curr Top Microbiol Immunol. 2013;369:17–48. doi: 10.1007/978-3-642-27340-7_2. [DOI] [PubMed] [Google Scholar]
- 69.Yang D, Zuo C, Wang X, Meng X, Xue B, Liu N, Yu R, Qin Y, Gao Y, Wang Q, et al. Complete replication of hepatitis B virus and hepatitis C virus in a newly developed hepatoma cell line. Proc Natl Acad Sci USA. 2014;111:E1264–E1273. doi: 10.1073/pnas.1320071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ploss A, Rice CM. Towards a small animal model for hepatitis C. EMBO Rep. 2009;10:1220–1227. doi: 10.1038/embor.2009.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Houghton M. The long and winding road leading to the identification of the hepatitis C virus. J Hepatol. 2009;51:939–948. doi: 10.1016/j.jhep.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 72.Alter HJ, Purcell RH, Holland PV, Popper H. Transmissible agent in non-A, non-B hepatitis. Lancet. 1978;1:459–463. doi: 10.1016/s0140-6736(78)90131-9. [DOI] [PubMed] [Google Scholar]
- 73.Tabor E, Gerety RJ, Drucker JA, Seeff LB, Hoofnagle JH, Jackson DR, April M, Barker LF, Pineda-Tamondong G. Transmission of non-A, non-B hepatitis from man to chimpanzee. Lancet. 1978;1:463–466. doi: 10.1016/s0140-6736(78)90132-0. [DOI] [PubMed] [Google Scholar]
- 74.Lindenbach BD, Meuleman P, Ploss A, Vanwolleghem T, Syder AJ, McKeating JA, Lanford RE, Feinstone SM, Major ME, Leroux-Roels G, et al. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc Natl Acad Sci USA. 2006;103:3805–3809. doi: 10.1073/pnas.0511218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kolykhalov AA, Agapov EV, Blight KJ, Mihalik K, Feinstone SM, Rice CM. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science. 1997;277:570–574. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]
- 76.Institute of Medicine. Chimpanzees in Biomedical and Behavioral Research: Assessing the Necessity. Washington, DC: National Academies Press; 2011. [PubMed] [Google Scholar]
- 77.Tian ZF, Shen H, Fu XH, Chen YC, Blum HE, Baumert TF, Zhao XP. Interaction of hepatitis C virus envelope glycoprotein E2 with the large extracellular loop of tupaia CD81. World J Gastroenterol. 2009;15:240–244. doi: 10.3748/wjg.15.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tong Y, Zhu Y, Xia X, Liu Y, Feng Y, Hua X, Chen Z, Ding H, Gao L, Wang Y, et al. Tupaia CD81, SR-BI, claudin-1, and occludin support hepatitis C virus infection. J Virol. 2011;85:2793–2802. doi: 10.1128/JVI.01818-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A, Addison WR, Fischer KP, Churchill TA, Lakey JR, et al. Hepatitis C virus replication in mice with chimeric human livers. Nat Med. 2001;7:927–933. doi: 10.1038/90968. [DOI] [PubMed] [Google Scholar]
- 80.Meuleman P, Libbrecht L, De Vos R, de Hemptinne B, Gevaert K, Vandekerckhove J, Roskams T, Leroux-Roels G. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology. 2005;41:847–856. doi: 10.1002/hep.20657. [DOI] [PubMed] [Google Scholar]
- 81.Tesfaye A, Stift J, Maric D, Cui Q, Dienes HP, Feinstone SM. Chimeric mouse model for the infection of hepatitis B and C viruses. PLoS One. 2013;8:e77298. doi: 10.1371/journal.pone.0077298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kosaka K, Hiraga N, Imamura M, Yoshimi S, Murakami E, Nakahara T, Honda Y, Ono A, Kawaoka T, Tsuge M, et al. A novel TK-NOG based humanized mouse model for the study of HBV and HCV infections. Biochem Biophys Res Commun. 2013;441:230–235. doi: 10.1016/j.bbrc.2013.10.040. [DOI] [PubMed] [Google Scholar]
- 83.Bissig KD, Wieland SF, Tran P, Isogawa M, Le TT, Chisari FV, Verma IM. Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J Clin Invest. 2010;120:924–930. doi: 10.1172/JCI40094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Washburn ML, Bility MT, Zhang L, Kovalev GI, Buntzman A, Frelinger JA, Barry W, Ploss A, Rice CM, Su L. A humanized mouse model to study hepatitis C virus infection, immune response, and liver disease. Gastroenterology. 2011;140:1334–1344. doi: 10.1053/j.gastro.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dorner M, Horwitz JA, Robbins JB, Barry WT, Feng Q, Mu K, Jones CT, Schoggins JW, Catanese MT, Burton DR, et al. A genetically humanized mouse model for hepatitis C virus infection. Nature. 2011;474:208–211. doi: 10.1038/nature10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T, Vogt A, Catanese MT, Satoh T, Kawai T, et al. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature. 2013;501:237–241. doi: 10.1038/nature12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Billerbeck E, de Jong Y, Dorner M, de la Fuente C, Ploss A. Animal models for hepatitis C. Curr Top Microbiol Immunol. 2013;369:49–86. doi: 10.1007/978-3-642-27340-7_3. [DOI] [PubMed] [Google Scholar]
- 88.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 89.Rehwinkel J, Reis e Sousa C. RIGorous detection: exposing virus through RNA sensing. Science. 2010;327:284–286. doi: 10.1126/science.1185068. [DOI] [PubMed] [Google Scholar]
- 90.Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Horner SM, Liu HM, Park HS, Briley J, Gale M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci USA. 2011;108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, Carney DS, Wang T, Ishida H, Yoneyama M, et al. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci USA. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Uzri D, Gehrke L. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J Virol. 2009;83:4174–4184. doi: 10.1128/JVI.02449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schnell G, Loo YM, Marcotrigiano J, Gale M. Uridine composition of the poly-U/UC tract of HCV RNA defines non-self recognition by RIG-I. PLoS Pathog. 2012;8:e1002839. doi: 10.1371/journal.ppat.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.You S, Rice CM. 3’ RNA elements in hepatitis C virus replication: kissing partners and long poly(U) J Virol. 2008;82:184–195. doi: 10.1128/JVI.01796-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M. Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J Virol. 2008;82:609–616. doi: 10.1128/JVI.01305-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 99.Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, García-Sastre A, Katze MG, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci USA. 2007;104:582–587. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jiang F, Ramanathan A, Miller MT, Tang GQ, Gale M, Patel SS, Marcotrigiano J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479:423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu HM, Loo YM, Horner SM, Zornetzer GA, Katze MG, Gale M. The mitochondrial targeting chaperone 14-3-3ε regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe. 2012;11:528–537. doi: 10.1016/j.chom.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 104.Kuniyoshi K, Takeuchi O, Pandey S, Satoh T, Iwasaki H, Akira S, Kawai T. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc Natl Acad Sci USA. 2014;111:5646–5651. doi: 10.1073/pnas.1401674111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oshiumi H, Miyashita M, Matsumoto M, Seya T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013;9:e1003533. doi: 10.1371/journal.ppat.1003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Oshiumi H, Miyashita M, Inoue N, Okabe M, Matsumoto M, Seya T. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe. 2010;8:496–509. doi: 10.1016/j.chom.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 107.Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, Chen ZJ. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 2012;36:959–973. doi: 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen W, Han C, Xie B, Hu X, Yu Q, Shi L, Wang Q, Li D, Wang J, Zheng P, et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell. 2013;152:467–478. doi: 10.1016/j.cell.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 110.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Akira S, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 112.Andrus L, Marukian S, Jones CT, Catanese MT, Sheahan TP, Schoggins JW, Barry WT, Dustin LB, Trehan K, Ploss A, et al. Expression of paramyxovirus V proteins promotes replication and spread of hepatitis C virus in cultures of primary human fetal liver cells. Hepatology. 2011;54:1901–1912. doi: 10.1002/hep.24557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ramachandran A, Horvath CM. Dissociation of paramyxovirus interferon evasion activities: universal and virus-specific requirements for conserved V protein amino acids in MDA5 interference. J Virol. 2010;84:11152–11163. doi: 10.1128/JVI.01375-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang D, Meng X, Xue B, Liu N, Wang X, Zhu H. MiR-942 mediates hepatitis C virus-induced apoptosis via regulation of ISG12a. PLoS One. 2014;9:e94501. doi: 10.1371/journal.pone.0094501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chang S, Dolganiuc A, Szabo G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J Leukoc Biol. 2007;82:479–487. doi: 10.1189/jlb.0207128. [DOI] [PubMed] [Google Scholar]
- 116.Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, Szabo G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology. 2004;127:1513–1524. doi: 10.1053/j.gastro.2004.08.067. [DOI] [PubMed] [Google Scholar]
- 117.Machida K, Cheng KT, Sung VM, Levine AM, Foung S, Lai MM. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J Virol. 2006;80:866–874. doi: 10.1128/JVI.80.2.866-874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang N, Liang Y, Devaraj S, Wang J, Lemon SM, Li K. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J Virol. 2009;83:9824–9834. doi: 10.1128/JVI.01125-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 120.Kawasaki T, Kawai T, Akira S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev. 2011;243:61–73. doi: 10.1111/j.1600-065X.2011.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Funami K, Matsumoto M, Oshiumi H, Akazawa T, Yamamoto A, Seya T. The cytoplasmic ‘linker region’ in Toll-like receptor 3 controls receptor localization and signaling. Int Immunol. 2004;16:1143–1154. doi: 10.1093/intimm/dxh115. [DOI] [PubMed] [Google Scholar]
- 122.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. 2006;176:4894–4901. doi: 10.4049/jimmunol.176.8.4894. [DOI] [PubMed] [Google Scholar]
- 124.Li K, Li NL, Wei D, Pfeffer SR, Fan M, Pfeffer LM. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology. 2012;55:666–675. doi: 10.1002/hep.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dansako H, Yamane D, Welsch C, McGivern DR, Hu F, Kato N, Lemon SM. Class A scavenger receptor 1 (MSR1) restricts hepatitis C virus replication by mediating toll-like receptor 3 recognition of viral RNAs produced in neighboring cells. PLoS Pathog. 2013;9:e1003345. doi: 10.1371/journal.ppat.1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 127.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 128.Dreux M, Garaigorta U, Boyd B, Décembre E, Chung J, Whitten-Bauer C, Wieland S, Chisari FV. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe. 2012;12:558–570. doi: 10.1016/j.chom.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan Q, de Ruiter PE, Willemsen R, Demmers JA, Stalin Raj V, Jenster G, et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc Natl Acad Sci USA. 2013;110:13109–13113. doi: 10.1073/pnas.1221899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014;10:e1004424. doi: 10.1371/journal.ppat.1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Preiss S, Thompson A, Chen X, Rodgers S, Markovska V, Desmond P, Visvanathan K, Li K, Locarnini S, Revill P. Characterization of the innate immune signalling pathways in hepatocyte cell lines. J Viral Hepat. 2008;15:888–900. doi: 10.1111/j.1365-2893.2008.01001.x. [DOI] [PubMed] [Google Scholar]
- 132.Berzsenyi MD, Roberts SK, Preiss S, Woollard DJ, Beard MR, Skinner NA, Bowden DS, Visvanathan K. Hepatic TLR2 & amp; TLR4 expression correlates with hepatic inflammation and TNF-α in HCV & amp; HCV/HIV infection. J Viral Hepat. 2011;18:852–860. doi: 10.1111/j.1365-2893.2010.01390.x. [DOI] [PubMed] [Google Scholar]
- 133.Riordan SM, Skinner NA, Kurtovic J, Locarnini S, McIver CJ, Williams R, Visvanathan K. Toll-like receptor expression in chronic hepatitis C: correlation with pro-inflammatory cytokine levels and liver injury. Inflamm Res. 2006;55:279–285. doi: 10.1007/s00011-006-0082-0. [DOI] [PubMed] [Google Scholar]
- 134.Arnaud N, Dabo S, Akazawa D, Fukasawa M, Shinkai-Ouchi F, Hugon J, Wakita T, Meurs EF. Hepatitis C virus reveals a novel early control in acute immune response. PLoS Pathog. 2011;7:e1002289. doi: 10.1371/journal.ppat.1002289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Arnaud N, Dabo S, Maillard P, Budkowska A, Kalliampakou KI, Mavromara P, Garcin D, Hugon J, Gatignol A, Akazawa D, et al. Hepatitis C virus controls interferon production through PKR activation. PLoS One. 2010;5:e10575. doi: 10.1371/journal.pone.0010575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe. 2009;6:513–522. doi: 10.1016/j.chom.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Koev G, Duncan RF, Lai MM. Hepatitis C virus IRES-dependent translation is insensitive to an eIF2alpha-independent mechanism of inhibition by interferon in hepatocyte cell lines. Virology. 2002;297:195–202. doi: 10.1006/viro.2002.1455. [DOI] [PubMed] [Google Scholar]
- 138.Shimoike T, McKenna SA, Lindhout DA, Puglisi JD. Translational insensitivity to potent activation of PKR by HCV IRES RNA. Antiviral Res. 2009;83:228–237. doi: 10.1016/j.antiviral.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 139.Kumar A, Yang YL, Flati V, Der S, Kadereit S, Deb A, Haque J, Reis L, Weissmann C, Williams BR. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-kappaB. EMBO J. 1997;16:406–416. doi: 10.1093/emboj/16.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.McAllister CS, Samuel CE. The RNA-activated protein kinase enhances the induction of interferon-beta and apoptosis mediated by cytoplasmic RNA sensors. J Biol Chem. 2009;284:1644–1651. doi: 10.1074/jbc.M807888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, Delker DA, Jo J, Bertoletti A, Hagedorn CH, et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013;9:e1003330. doi: 10.1371/journal.ppat.1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhao H, Lin W, Kumthip K, Cheng D, Fusco DN, Hofmann O, Jilg N, Tai AW, Goto K, Zhang L, et al. A functional genomic screen reveals novel host genes that mediate interferon-alpha’s effects against hepatitis C virus. J Hepatol. 2012;56:326–333. doi: 10.1016/j.jhep.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Metz P, Dazert E, Ruggieri A, Mazur J, Kaderali L, Kaul A, Zeuge U, Windisch MP, Trippler M, Lohmann V, et al. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology. 2012;56:2082–2093. doi: 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- 148.Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fusco DN, Brisac C, John SP, Huang YW, Chin CR, Xie T, Zhao H, Jilg N, Zhang L, Chevaliez S, et al. A genetic screen identifies interferon-α effector genes required to suppress hepatitis C virus replication. Gastroenterology. 2013;144:1438–1449, 1449.e1-9. doi: 10.1053/j.gastro.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li K, Lemon SM. Innate immune responses in hepatitis C virus infection. Semin Immunopathol. 2013;35:53–72. doi: 10.1007/s00281-012-0332-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Silverman RH. Viral encounters with 2’,5’-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol. 2007;81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ishibashi M, Wakita T, Esumi M. 2’,5’-Oligoadenylate synthetase-like gene highly induced by hepatitis C virus infection in human liver is inhibitory to viral replication in vitro. Biochem Biophys Res Commun. 2010;392:397–402. doi: 10.1016/j.bbrc.2010.01.034. [DOI] [PubMed] [Google Scholar]
- 153.Kwon YC, Kang JI, Hwang SB, Ahn BY. The ribonuclease L-dependent antiviral roles of human 2’,5’-oligoadenylate synthetase family members against hepatitis C virus. FEBS Lett. 2013;587:156–164. doi: 10.1016/j.febslet.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 154.Hinson ER, Cresswell P. The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic alpha-helix. Proc Natl Acad Sci USA. 2009;106:20452–20457. doi: 10.1073/pnas.0911679106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang S, Wu X, Pan T, Song W, Wang Y, Zhang F, Yuan Z. Viperin inhibits hepatitis C virus replication by interfering with binding of NS5A to host protein hVAP-33. J Gen Virol. 2012;93:83–92. doi: 10.1099/vir.0.033860-0. [DOI] [PubMed] [Google Scholar]
- 156.Helbig KJ, Eyre NS, Yip E, Narayana S, Li K, Fiches G, McCartney EM, Jangra RK, Lemon SM, Beard MR. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology. 2011;54:1506–1517. doi: 10.1002/hep.24542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Metz P, Reuter A, Bender S, Bartenschlager R. Interferon-stimulated genes and their role in controlling hepatitis C virus. J Hepatol. 2013;59:1331–1341. doi: 10.1016/j.jhep.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 158.Wilkins C, Woodward J, Lau DT, Barnes A, Joyce M, McFarlane N, McKeating JA, Tyrrell DL, Gale M. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology. 2013;57:461–469. doi: 10.1002/hep.26066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Raychoudhuri A, Shrivastava S, Steele R, Kim H, Ray R, Ray RB. ISG56 and IFITM1 proteins inhibit hepatitis C virus replication. J Virol. 2011;85:12881–12889. doi: 10.1128/JVI.05633-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhu H, Liu C. Interleukin-1 inhibits hepatitis C virus subgenomic RNA replication by activation of extracellular regulated kinase pathway. J Virol. 2003;77:5493–5498. doi: 10.1128/JVI.77.9.5493-5498.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yao L, Dong H, Zhu H, Nelson D, Liu C, Lambiase L, Li X. Identification of the IFITM3 gene as an inhibitor of hepatitis C viral translation in a stable STAT1 cell line. J Viral Hepat. 2011;18:e523–e529. doi: 10.1111/j.1365-2893.2011.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Itsui Y, Sakamoto N, Kurosaki M, Kanazawa N, Tanabe Y, Koyama T, Takeda Y, Nakagawa M, Kakinuma S, Sekine Y, et al. Expressional screening of interferon-stimulated genes for antiviral activity against hepatitis C virus replication. J Viral Hepat. 2006;13:690–700. doi: 10.1111/j.1365-2893.2006.00732.x. [DOI] [PubMed] [Google Scholar]
- 163.Taylor DR, Puig M, Darnell ME, Mihalik K, Feinstone SM. New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J Virol. 2005;79:6291–6298. doi: 10.1128/JVI.79.10.6291-6298.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jiang D, Guo H, Xu C, Chang J, Gu B, Wang L, Block TM, Guo JT. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J Virol. 2008;82:1665–1678. doi: 10.1128/JVI.02113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Helbig KJ, Lau DT, Semendric L, Harley HA, Beard MR. Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology. 2005;42:702–710. doi: 10.1002/hep.20844. [DOI] [PubMed] [Google Scholar]
- 166.Dafa-Berger A, Kuzmina A, Fassler M, Yitzhak-Asraf H, Shemer-Avni Y, Taube R. Modulation of hepatitis C virus release by the interferon-induced protein BST-2/tetherin. Virology. 2012;428:98–111. doi: 10.1016/j.virol.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 167.Horner SM, Gale M. Regulation of hepatic innate immunity by hepatitis C virus. Nat Med. 2013;19:879–888. doi: 10.1038/nm.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Morikawa K, Lange CM, Gouttenoire J, Meylan E, Brass V, Penin F, Moradpour D. Nonstructural protein 3-4A: the Swiss army knife of hepatitis C virus. J Viral Hepat. 2011;18:305–315. doi: 10.1111/j.1365-2893.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- 169.Brass V, Berke JM, Montserret R, Blum HE, Penin F, Moradpour D. Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc Natl Acad Sci USA. 2008;105:14545–14550. doi: 10.1073/pnas.0807298105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Horner SM, Park HS, Gale M. Control of innate immune signaling and membrane targeting by the Hepatitis C virus NS3/4A protease are governed by the NS3 helix α0. J Virol. 2012;86:3112–3120. doi: 10.1128/JVI.06727-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Foy E, Li K, Wang C, Sumpter R, Ikeda M, Lemon SM, Gale M. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 172.Foy E, Li K, Sumpter R, Loo YM, Johnson CL, Wang C, Fish PM, Yoneyama M, Fujita T, Lemon SM, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci USA. 2005;102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 174.Baril M, Racine ME, Penin F, Lamarre D. MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J Virol. 2009;83:1299–1311. doi: 10.1128/JVI.01659-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bellecave P, Sarasin-Filipowicz M, Donzé O, Kennel A, Gouttenoire J, Meylan E, Terracciano L, Tschopp J, Sarrazin C, Berg T, et al. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology. 2010;51:1127–1136. doi: 10.1002/hep.23426. [DOI] [PubMed] [Google Scholar]
- 177.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Lemon SM. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ferreon JC, Ferreon AC, Li K, Lemon SM. Molecular determinants of TRIF proteolysis mediated by the hepatitis C virus NS3/4A protease. J Biol Chem. 2005;280:20483–20492. doi: 10.1074/jbc.M500422200. [DOI] [PubMed] [Google Scholar]
- 179.Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, Ghaffari AA, Qin J, Cheng G, Liu YJ. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity. 2011;34:866–878. doi: 10.1016/j.immuni.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dabo S, Meurs EF. dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses. 2012;4:2598–2635. doi: 10.3390/v4112598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gale MJ, Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE, Polyak SJ, Gretch DR, Katze MG. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 182.Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 183.Noguchi T, Satoh S, Noshi T, Hatada E, Fukuda R, Kawai A, Ikeda S, Hijikata M, Shimotohno K. Effects of mutation in hepatitis C virus nonstructural protein 5A on interferon resistance mediated by inhibition of PKR kinase activity in mammalian cells. Microbiol Immunol. 2001;45:829–840. doi: 10.1111/j.1348-0421.2001.tb01322.x. [DOI] [PubMed] [Google Scholar]
- 184.Eksioglu EA, Zhu H, Bayouth L, Bess J, Liu HY, Nelson DR, Liu C. Characterization of HCV interactions with Toll-like receptors and RIG-I in liver cells. PLoS One. 2011;6:e21186. doi: 10.1371/journal.pone.0021186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Horner SM, Gale M. Intracellular innate immune cascades and interferon defenses that control hepatitis C virus. J Interferon Cytokine Res. 2009;29:489–498. doi: 10.1089/jir.2009.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Pang PS, Planet PJ, Glenn JS. The evolution of the major hepatitis C genotypes correlates with clinical response to interferon therapy. PLoS One. 2009;4:e6579. doi: 10.1371/journal.pone.0006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, Heim MH. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci USA. 2008;105:7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dill MT, Duong FH, Vogt JE, Bibert S, Bochud PY, Terracciano L, Papassotiropoulos A, Roth V, Heim MH. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology. 2011;140:1021–1031. doi: 10.1053/j.gastro.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 189.Chen L, Borozan I, Sun J, Guindi M, Fischer S, Feld J, Anand N, Heathcote J, Edwards AM, McGilvray ID. Cell-type specific gene expression signature in liver underlies response to interferon therapy in chronic hepatitis C infection. Gastroenterology. 2010;138:1123–1133.e1-3. doi: 10.1053/j.gastro.2009.10.046. [DOI] [PubMed] [Google Scholar]
- 190.Fukuhara T, Taketomi A, Motomura T, Okano S, Ninomiya A, Abe T, Uchiyama H, Soejima Y, Shirabe K, Matsuura Y, et al. Variants in IL28B in liver recipients and donors correlate with response to peg-interferon and ribavirin therapy for recurrent hepatitis C. Gastroenterology. 2010;139:1577–1585, 1585.e1-3. doi: 10.1053/j.gastro.2010.07.058. [DOI] [PubMed] [Google Scholar]
- 191.Langhans B, Kupfer B, Braunschweiger I, Arndt S, Schulte W, Nischalke HD, Nattermann J, Oldenburg J, Sauerbruch T, Spengler U. Interferon-lambda serum levels in hepatitis C. J Hepatol. 2011;54:859–865. doi: 10.1016/j.jhep.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 192.Naggie S, Osinusi A, Katsounas A, Lempicki R, Herrmann E, Thompson AJ, Clark PJ, Patel K, Muir AJ, McHutchison JG, et al. Dysregulation of innate immunity in hepatitis C virus genotype 1 IL28B-unfavorable genotype patients: impaired viral kinetics and therapeutic response. Hepatology. 2012;56:444–454. doi: 10.1002/hep.25647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, Hergott D, Porter-Gill P, Mumy A, Kohaar I, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45:164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhu H, Butera M, Nelson DR, Liu C. Novel type I interferon IL-28A suppresses hepatitis C viral RNA replication. Virol J. 2005;2:80. doi: 10.1186/1743-422X-2-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kelly C, Klenerman P, Barnes E. Interferon lambdas: the next cytokine storm. Gut. 2011;60:1284–1293. doi: 10.1136/gut.2010.222976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lange CM, Jacobson IM, Rice CM, Zeuzem S. Emerging therapies for the treatment of hepatitis C. EMBO Mol Med. 2014;6:4–15. doi: 10.1002/emmm.201303131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lange CM, Zeuzem S. Perspectives and challenges of interferon-free therapy for chronic hepatitis C. J Hepatol. 2013;58:583–592. doi: 10.1016/j.jhep.2012.10.019. [DOI] [PubMed] [Google Scholar]