

Abstract
The urea cycle is the final pathway for nitrogen metabolism. Urea cycle disorders (UCDs) include a variety of genetic defects, which lead to inefficient urea synthesis. Elevated blood ammonium level is usually dominant in the clinical pattern and the primary manifestations affect the central nervous system. Herein, we report the case of a 17-year-old girl who was diagnosed with UCD at the age of 3. Despite a controlled diet, she was hospitalized several times for acute attacks with recurrent life risk. She came to our attention for a hyperammonemic episode. We proposed an orthotopic liver transplant (OLT) as a treatment; the patient and her family were in complete agreement. On February 28, 2007, she successfully received a transplant. Following the surgery, she has remained well, and she is currently leading a normal life. Usually for UCDs diet plays the primary therapeutic role, while OLT is often considered as a last resort. Our case report and the recent literature data on the quality of life and prognosis of traditionally treated patients vs OLT patients, support OLT as a primary intervention to prevent life-threatening acute episodes and chronic mental impairment.
Keywords: Urea cycle disorders, Hyperammonemia, Diet, Liver transplantation, Quality of life
Core tip: Urea cycle disorders (UCDs) include a variety of genetic defects which lead to inefficient urea-synthesis with hyperammonemia. The liver is the main site of urea cycle’s enzymatic activity. Diet has traditionally been the primary therapy; orthotopic liver transplantation (OLT) is often considered as a last resort. This case report presents a 17-year-old girl with UCD who was successfully treated with OLT. Her recovery and a recent literature review support OLT as a primary intervention for UCDs patients to prevent life-threatening acute episodes and chronic mental impairment, with an approximately 90% survival rate and a better quality of life.
INTRODUCTION
The urea cycle is the final pathway for nitrogen metabolism, which occurs primarily through amino acid catabolism with urea production and subsequent excretion (Figure 1). Urea cycle disorders (UCDs) include a variety of genetic defects in ammonia metabolism in which one of the urea-synthesis passages is defective, leading to hyperammonemia. The genetic defect can involve one of the enzymes involved in the urea cycle or a transporting protein related to the metabolic process. The liver is the most important site of this enzymatic activity.
Figure 1.
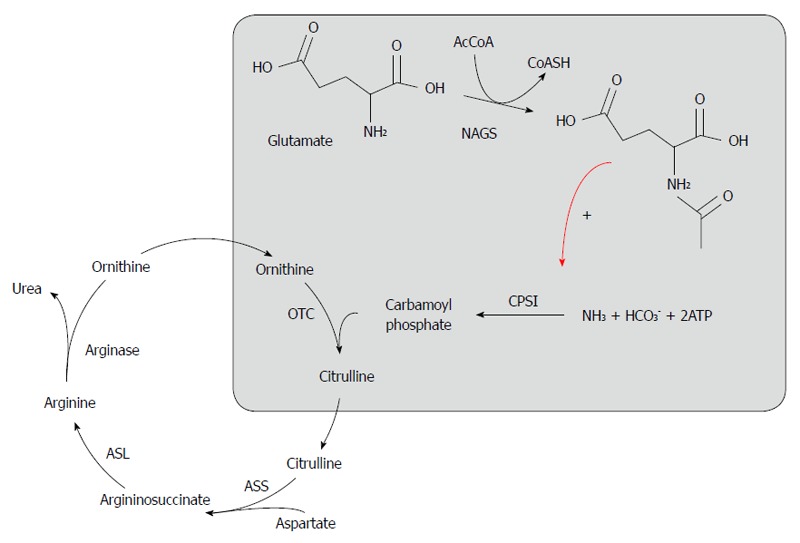
Urea cycle. NAGS: N-acetylglutamate synthetase; NAG: N-acetylglutamate; CPS I: Carbamyl phosphate synthetase 1; OTC: Ornithine transcarbamylase; ASS: Argininosuccinate synthetase; ASL: Argininosuccinate lyase.
Although data from population studies are lacking, the prevalence of this type of genetic defect is approximately 1: 30.000-46.000 live births[1].
The cycle consists of the action of cytoplasmic and mitochondrial enzymes and some transporting proteins, leading to the production of urea molecules from nitrogen atoms. The different metabolic blocks determine the accumulation of nitrogen and intermediate substances, with a variety of corresponding clinical features.
The classification of urea cycle defects is summarized in Table 1.
Table 1.
Main causes of urea cycle defects
| CPS I deficiency | Genetic defect of carbamoyl phosphate synthetase, inheritated as autosomal recessive mutation |
| Block of the first passage of nitrogen in urea cycle | |
| Neonatal onset when there is a severe enzyme deficiency and adult onset when the effects of mutation are mild | |
| OTC deficiency | X-linked disorder, the most common among UCDs |
| Mutation in ornithine transcarbamylase (second passage in urea cycle) | |
| Clinical presentation very similar to CPS I deficiency | |
| ASS deficiency/citrin deficiency (citrullinemia) | Citrullinemia type I: autosomal recessive defect caused by a deficit of argininosuccinate synthetase, with consequent very high citrulline levels in blood |
| Citrullinemia tipe II: caused by a deficit of citrin, a mitochondrial carrier protein of glutamate and aspartate who consequently fail to shuttle to and from the mitochondrion with increase of citrulline levels in blood | |
| ASL deficiency (argininosuccinic aciduria) | Autosomal recessive mutation |
| Loss of argininosuccinate lyase with accumulations in body fluids (also in cerebrospinal fluid) | |
| Seems to lead to mental retardation in all the affected, even when dietary ammonium control is well performed | |
| Arginase deficiency (argininemia) | Arginase I defect |
| Peculiar manifestations such as paraplegia and intractable mental retardation | |
| N-acetylglutamate synthetase deficiency | Very rare and clinically overlapped to CPS I deficiency |
UCDs: Urea cycle disorders; CPS I: Carbamyl phosphate synthetase 1; ASL: Argininosuccinate lyase; OTC: Ornithine transcarbamylase.
In patients with normal hepatic function and hyperammonemia, the diagnosis of UCD should be considered after excluding other causes of elevated blood ammonemia, such as medications (valproate and chemotherapeutic agents), portosystemic shunt, urea producing organisms and hyperalimentation[2]. The diagnosis of UCD is based on clinical, biochemical and molecular data[3,4].
Elevated blood ammonium levels are usually dominant in the clinical pattern and the main manifestations affect the central nervous system[5]. The presentation of UCDs can be quite variable ranging from agitation to coma in acute attacks (common cause of neonatal exitus in these patients) and different degrees of mental impairment chronically (even with correct dietary intake).
Usually, patients with this type of metabolic defect develop a tolerance to high ammonium levels. Therefore, they can be completely asymptomatic even with levels of approximately 200 μg/dL. These episodes are not rare even in mild forms of UCD because many factors (e.g., infections, fever, fasting, surgery) can break down the metabolic equilibrium. Moreover, the long-term compliance of these young patients is usually difficult. The central nervous system damage is at least in part reversible when ammonium levels do not exceed 200-400 μg/dL. Nevertheless, mild neurological damage in the presence of lower ammonium levels cannot be completely ruled out[6,7]. The consequent neurological deterioration is inversely proportional to the frequency of acute episodes and to the ammonia levels. Additional disease-specific symptoms are related to the particular metabolic defect, which are often due to an excess or lack of specific amino acids[8].
Herein, we discuss the case of a girl with UCD caused by carbamyl phosphate synthetase (CPS I) deficit (Table 1) who underwent an orthotopic liver transplantation (OLT) 14 years after her diagnosis.
CASE REPORT
A 17-year-old girl came to our attention in November 2007. She was diagnosed with a CPS I defect in a Pediatric Hospital when she was 3 years old after evidence of lethargy attacks, sight disturbances and growth retardation. Despite a strict dietary regimen, the symptoms control was partial and recurrent acute attacks developed. These attacks were characterized by sudden somnolence, ataxia, coordination defects and temporal cognition distortion (never completely resolved). These often required hospitalization to reduce the ammonia levels with parenteral therapy infusion.
She came to our hospital for an acute attack with lethargy and a blood ammonia concentration of 305 μg/dL. The episode was resolved with a 72-h infusion of a parenteral complex containing sodium benzoate and arginine. Her cerebral magnetic resonance imaging results were normal.
Considering the constant risk of death and the poor quality of life, even with otherwise normal liver parameters, we proposed OLT as a treatment. The patient and her family were in complete agreement.
After a complete work-up, the patient was admitted to the Pediatric list of Bologna Liver Transplantation Center and after two months she underwent OLT. The post-operative course was free from complications with rapid ammonia blood level normalization. After a 7-d recovery period, she was discharged. Currently, her clinical conditions are good; she leads a normal life without dietary restrictions and is free from lethargy episodes and hospital admissions, except for immunosuppressive therapy and routine follow-up visits at the Transplant Center.
DISCUSSION
The treatment for UCDs is a dietary intervention consisting of severe protein restriction[5]. Additionally, citrullin and arginine supplementation (except in cases of argininemia) and sodium benzoate and sodium phenylacetate administration (useful to excrete nitrogen excess) are often used to treat acute hyperammonemia. Nocturnal enteral nutrition and prompt recognition and treatment of intercurrent illnesses are also necessary to prevent catabolism[9]. In cases of acute hyperammonemia attacks, the treatment is based on ammonium removal by dialysis or hemofiltration and on reversing the catabolic state by caloric and arginine supplementation[9]. This strategy, in the actual daily life of teenage patients, could be hindered by the difficulty in maintaining dietary restrictions; daily monitoring helps to control the trigger causes (infections, fasting and other conditions often make the metabolic imbalance unpredictable) of hyperammonemia. In patients whose symptoms cannot be controlled by diet, OLT is a treatment option because the liver is the principal site of urea cycle activity.
In absence of viral reinfection, tumor recurrence and alcohol relapse (typically affecting patients who underwent OLT for more frequent indications), transplants in patients with metabolic disorders (e.g., hemochromatosis, Wilson’s disease, familial amyloidosis, cystic fibrosis) have significantly better outcomes compared with primary liver diseases. The 5 year survival rate is nearly 90% with better results when structural liver disease is absent[10]. Moreover, the consequent metabolic correction is associated with a considerably improved quality of life and elimination of specific death risks.
After OLT, dietary restriction in our patient was no longer needed. Consequently, the other medications for amino acid supplementation and urea excretion were discontinued with no further encephalopathy episodes. Furthermore, our patient has demonstrated increased intellectual performance leading to maturation of personality and behavior comparable with her peers; these improvements are not to be generalized since other authors reported cases of interrupted neurological deterioration without significant intellectual improvements[7,11,12]. A possible explanation could be the timing of OLT, which was performed prior to the development of irreversible brain damage.
The exact number of OLTs for patients with UCDs is unknown; a United States survey in 1998[12] described 16 cases of OLTs for UCD patients. Kyoto University[1] refers to 13 patients who underwent liver transplant for urea cycle defects, and a recent article describes 2 UCD patients who were subjected to OLT in China[13]. A European review[11] reported of 59 transplants with a 93% survival after 3 years. Of these transplant recipients, 6 patients had a CPS I deficit and all of the patients were alive after the 3-year follow-up. In Italy, the Sicilian transplant group[14] reported one case of a living-donor liver transplant (LDLT) for OTC deficiency.
Recently, the United Network for Organ Sharing (UNOS) described 293 patients who underwent liver transplantation for UCDs and organic acidemias with a 5-year graft survival rate of 78% for children < 2 years old at the time of transplant and 88% for children ≥ 2 years old at the time of transplant[15]. Finally, Kasahara et al[16] described 40 patients with UCDs who underwent LDLT, in which the patient and graft survival rate was 96.1% after 15 years.
Despite these good results, currently the univocally accepted indications for OLT in patients with UCDs are very severe disease with poor prognosis; progressive liver disease with prospect of liver failure; severe complications that cannot be avoided by other interventions; or when diet is insufficient to maintain normal ammonia blood levels[1,11]. A recent UNOS review highlighted that eight children died while they were waiting for transplantation, thus confirming the lethal nature of this disease. The optimal timing for transplantation remains an important question because younger patients have a higher risk of graft loss due to increased liver transplant complications. Recent expert consensus conference guidelines recommend OLT between 3 and 12 mo of age if the child weighs more than 5 kg[17].
An important aspect highlighted by Hadzic and Vara is that children with these rare conditions are followed by specialists in metabolic units that often have no direct contact with the transplant center. Considering the positive results of transplantation for metabolic diseases, there should be closer contact between these sectors[18].
Moreover, the periodic lack of compliance in these children and adolescents, facing this long-term life restriction, that can cause recurrent hyperammonemia episodes is another factor favoring OLT.
Of course, all patients require an individual evaluation for risk and benefit options. However, when considering the significant improvements in transplant techniques, the overall results and the positive and generally better outcomes following OLT in patients with metabolic defects compared to primary liver diseases, we support the idea of liver transplant as an option for every single patient. Furthermore, liver transplant should no longer be a last resort. In particular, we agree with some authors, such as Meyburg et al[19], who have strongly promoted OLT as the first and only definitive cure for UCDs given that liver transplant is associated with the best survival rate in the whole field of solid organ transplantation. OLT should be performed as soon as possible to prevent irreversible brain damage.
The OLT indications are not different for the different UCDs types, except NAGS deficiency for whom the definitive therapy is N-carbamyl-L-glutamate, a NAG analogue that is taken up enterally and replaces NAG for the activation of CPS1[17]. For all the other UCD forms, liver transplantation should be considered in all patients before irreversible neurological damage and/or repeated crises occur[17].
In the literature, OLT has been reported for patients with all UCD types, except NAGS deficiency. Wakiya et al[20] described twelve children with OTC deficiency who underwent living donor liver transplantation with satisfactory outcomes. Kimura et al[21] reported two cases of adult-onset type II citrullinemia successfully treated with OLT. Nagamani et al[22] report OLT as a long-term correction for patients with ASL deficiency. Silva et al[23] described two cases of children with argininemia successfully treated with OLT. Morioka et al[1] reported 13 patients with UCDs who underwent OLT at Kyoto University and reviewed 38 OLTs for patients with UCDs reported in the worldwide English literature of whom 28 patients had an OTC deficiency; 4 patients had a CPSI deficiency; 4 patients had citrullinemia type I; 27 patients had citrullinemia type II; 1 patient had an arginase deficiency.
Regarding our patient, in our opinion, the transplant should have been performed long before she came to our hospital, which would have prevented more than 10 years of mental impairment and life-threatening episodes during her childhood. The improved quality of life that OLT offers, despite the immunosuppressive therapy, to these young people and the great benefit for their social development should be taken into account.
Therefore, our present experience, together with the results obtained worldwide in patients with this type of metabolic disease, supports OLT as soon as possible. The advantages for these children are an approximate 90% survival rate with normal mental development and a better quality of life.
COMMENTS
Case characteristics
A 17-year-old girl diagnosed with urea cycle disorder (carbamyl phosphate synthetase deficiency diagnosed when she was 3) presented with an acute attack characterized by lethargy.
Clinical diagnosis
Drowsiness, ataxia, coordination defects and temporal cognition distortion.
Differential diagnosis
Urea cycle disorders (UCDs), other metabolic disorders, encephalitis, hepatic encephalopathy, cerebral mass, drug intoxication.
Laboratory diagnosis
The patient’s blood ammonia concentration was 305 µg/dL; the liver function tests were within the normal limits.
Imaging diagnosis
The cerebral magnetic resonance imaging results were normal.
Pathological diagnosis
No pathological investigations were performed.
Treatment
The acute episode was resolved with a 72-h infusion of a parenteral complex containing sodium benzoate and arginine; shortly afterwards, after a complete work-up, the patient underwent orthotopic liver transplantation.
Term explanation
The urea cycle is the primary nitrogen disposal pathway in humans. The cycle requires the function of six enzymes to catalyze the conversion of ammonia and bicarbonate into urea. Ammonia is toxic, but urea is relatively inert, is soluble in water and is readily excreted by the kidney into the urine. The liver is the most important site of this enzymatic activity. Urea cycle disorders are metabolic inborn errors caused by a deficiency in one of the enzymes of the cycle. Ammonia (and other toxic intermediates) accumulation leads to predominant neurologic sequelae, ranging from mild cognitive deficits to deep coma and death.
Experiences and lessons
This case report, together with the recent literature data, supports liver transplantation as a primary intervention in patients with UCDs to prevent life-threatening acute episodes and chronic mental impairment.
Peer-review
This case report focuses on the issue of liver transplantation for patients with urea cycle disorders, which is relevant and not thoroughly discussed in the transplant literature.
Footnotes
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 12, 2014
First decision: August 27, 2014
Article in press: October 21, 2014
P- Reviewer: Fourtounas C, Salvadori M S- Editor: Qi Y L- Editor: A E- Editor: Ma S
References
- 1.Morioka D, Kasahara M, Takada Y, Shirouzu Y, Taira K, Sakamoto S, Uryuhara K, Egawa H, Shimada H, Tanaka K. Current role of liver transplantation for the treatment of urea cycle disorders: a review of the worldwide English literature and 13 cases at Kyoto University. Liver Transpl. 2005;11:1332–1342. doi: 10.1002/lt.20587. [DOI] [PubMed] [Google Scholar]
- 2.Bates TR, Lewis BD, Burnett JR, So K, Mitchell A, Delriviere L, Jeffrey GP. Late-onset carbamoyl phosphate synthetase 1 deficiency in an adult cured by liver transplantation. Liver Transpl. 2011;17:1481–1484. doi: 10.1002/lt.22407. [DOI] [PubMed] [Google Scholar]
- 3.Brusilow SW, Horwich AL. Urea enzymes. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds , et al., editors. The Molecular and Metabolic Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001. pp. 1909–1965. [Google Scholar]
- 4.Braissant O. Current concepts in the pathogenesis of urea cycle disorders. Mol Genet Metab. 2010;100 Suppl 1:S3–S12. doi: 10.1016/j.ymgme.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Endo F, Matsuura T, Yanagita K, Matsuda I. Clinical manifestations of inborn errors of the urea cycle and related metabolic disorders during childhood. J Nutr. 2004;134:1605S–1609S; discussion 1630S-1632S, 1667S-1672S. doi: 10.1093/jn/134.6.1605S. [DOI] [PubMed] [Google Scholar]
- 6.Enns GM. Neurologic damage and neurocognitive dysfunction in urea cycle disorders. Semin Pediatr Neurol. 2008;15:132–139. doi: 10.1016/j.spen.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 7.McBride KL, Miller G, Carter S, Karpen S, Goss J, Lee B. Developmental outcomes with early orthotopic liver transplantation for infants with neonatal-onset urea cycle defects and a female patient with late-onset ornithine transcarbamylase deficiency. Pediatrics. 2004;114:e523–e526. doi: 10.1542/peds.2004-0198. [DOI] [PubMed] [Google Scholar]
- 8.Darwish AA, McKiernan P, Chardot C. Paediatric liver transplantation for metabolic disorders. Part 1: Liver-based metabolic disorders without liver lesions. Clin Res Hepatol Gastroenterol. 2011;35:194–203. doi: 10.1016/j.clinre.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Teufel U, Weitz J, Flechtenmacher C, Prietsch V, Schmidt J, Hoffmann GF, Kölker S, Engelmann G. High urgency liver transplantation in ornithine transcarbamylase deficiency presenting with acute liver failure. Pediatr Transplant. 2011;15:E110–E115. doi: 10.1111/j.1399-3046.2009.01171.x. [DOI] [PubMed] [Google Scholar]
- 10.Treem WR. Liver transplantation for non-hepatotoxic inborn errors of metabolism. Curr Gastroenterol Rep. 2006;8:215–223. doi: 10.1007/s11894-006-0078-5. [DOI] [PubMed] [Google Scholar]
- 11.Leonard JV, McKiernan PJ. The role of liver transplantation in urea cycle disorders. Mol Genet Metab. 2004;81 Suppl 1:S74–S78. doi: 10.1016/j.ymgme.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 12.Whitington PF, Alonso EM, Boyle JT, Molleston JP, Rosenthal P, Emond JC, Millis JM. Liver transplantation for the treatment of urea cycle disorders. J Inherit Metab Dis. 1998;21 Suppl 1:112–118. doi: 10.1023/a:1005317909946. [DOI] [PubMed] [Google Scholar]
- 13.Shen ZY, Wang ZF, Zhu ZJ, Zang YJ, Zheng H, Deng YL, Pan C, Chen XG. Pediatric liver transplantation in 31 consecutive children. Chin Med J (Engl) 2008;121:2001–2003. [PubMed] [Google Scholar]
- 14.Miller EW, Bian SX, Chang CJ. A fluorescent sensor for imaging reversible redox cycles in living cells. J Am Chem Soc. 2007;129:3458–3459. doi: 10.1021/ja0668973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perito ER, Rhee S, Roberts JP, Rosenthal P. Pediatric liver transplantation for urea cycle disorders and organic acidemias: United Network for Organ Sharing data for 2002-2012. Liver Transpl. 2014;20:89–99. doi: 10.1002/lt.23765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasahara M, Sakamoto S, Horikawa R, Koji U, Mizuta K, Shinkai M, Takahito Y, Taguchi T, Inomata Y, Uemoto S, et al. Living donor liver transplantation for pediatric patients with metabolic disorders: the Japanese multicenter registry. Pediatr Transplant. 2014;18:6–15. doi: 10.1111/petr.12196. [DOI] [PubMed] [Google Scholar]
- 17.Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS, Santer R, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. doi: 10.1186/1750-1172-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hadžić N, Vara R. The times they are a-changin’. Liver Transpl. 2014;20:1–3. doi: 10.1002/lt.23802. [DOI] [PubMed] [Google Scholar]
- 19.Meyburg J, Hoffmann GF. Liver, liver cell and stem cell transplantation for the treatment of urea cycle defects. Mol Genet Metab. 2010;100 Suppl 1:S77–S83. doi: 10.1016/j.ymgme.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 20.Wakiya T, Sanada Y, Mizuta K, Umehara M, Urahasi T, Egami S, Hishikawa S, Fujiwara T, Sakuma Y, Hyodo M, et al. Living donor liver transplantation for ornithine transcarbamylase deficiency. Pediatr Transplant. 2011;15:390–395. doi: 10.1111/j.1399-3046.2011.01494.x. [DOI] [PubMed] [Google Scholar]
- 21.Kimura N, Kubo N, Narumi S, Toyoki Y, Ishido K, Kudo D, Umehara M, Yakoshi Y, Hakamada K. Liver transplantation versus conservative treatment for adult-onset type II citrullinemia: our experience and a review of the literature. Transplant Proc. 2013;45:3432–3437. doi: 10.1016/j.transproceed.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 22.Nagamani SC, Erez A, Lee B. Argininosuccinate lyase deficiency. Genet Med. 2012;14:501–507. doi: 10.1038/gim.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silva ES, Cardoso ML, Vilarinho L, Medina M, Barbot C, Martins E. Liver transplantation prevents progressive neurological impairment in argininemia. JIMD Rep. 2013;11:25–30. doi: 10.1007/8904_2013_218. [DOI] [PMC free article] [PubMed] [Google Scholar]