

Arecent Food and Drug Administration (FDA) proposal aims to speed the evaluation process for new high-risk medical devices that are intended to address unmet medical needs,1 much like existing expedited approval processes, such as the humanitarian device exemption rule for devices intended to treat rare diseases. Such programs are strongly supported by the medical device industry and some patient advocacy groups, which have criticized the FDA for being too stringent in its evidentiary requirements for investigational devices, leading to delays in the approval of potentially helpful products.2–4 For example, in 2011, the FDA approved a transcatheter aortic valve replacement system that demonstrated significant improvements over conventional treatment options for selected patients with severe aortic stenosis.5,6 However, the United States was the 43rd country to approve the device, roughly 4 years after the European Union.7
Yet expedited approval for high-risk medical devices raises the possibility that these devices will not be as effective as predicted in their limited premarket testing or that they could cause unanticipated harms after approval.8 Of course, well-studied devices may present unexpected safety concerns years after approval,9,10 and even the most rigorous conventional premarket approval process will result in some devices later found to be unsafe or ineffective.11–13 Safety of approved medical devices and the proper scope of premarket testing remain contentious issues after recalls of several widely used devices, including popular models of implantable cardioverter defibrillator leads14,15 and metal-on-metal hip implants.16
Inherent limitations in premarket testing, along with the prospect of lowered evidentiary standards for expedited device reviews, place greater pressures on postapproval monitoring of devices to follow clinical performance and to identify emerging public health problems. Medical device manufacturers routinely perform this sort of vigilance, and the FDA has many tools at its disposal to ensure proper post-market evaluation of approved devices, with several initiatives now under development to expand its postmarket surveillance network.17,18 We sought to analyze current FDA postmarket programs, including their existing structure and prospects for effective medical device surveillance in the United States, particularly as they relate to cardiovascular medicine.
Postapproval Surveillance Strategies
Postapproval device surveillance is a systematic way of gathering and analyzing data to direct consequent policies that minimize device-related morbidity and mortality.17 These activities fall into 3 main categories. One is passive surveillance, in which outside parties report data to the FDA, which then collates the information and determines whether an intervention is necessary. By contrast, there are 2 kinds of more active postapproval surveillance: conduct of prospective organized clinical studies and organization of active medical product registries capable of generating device safety and performance data. In this section, we discuss the FDA's initiatives relating to each of these categories.
Postapproval Passive Surveillance
Passive surveillance systems at the FDA are centered on Medical Device Reporting (MDR), through which manufacturers, physicians, and consumers submit reports of device-associated malfunctions, injuries, or deaths. Although voluntary submissions are an aspect of MDR, manufacturers must file reports with the FDA within 30 days if evidence suggests the device contributed to or caused a malfunction, injury, or death, and within 5 days if more than routine maintenance of the device is needed.19 Hospitals must submit their reports to manufacturers or the FDA within 10 days of an event.19 Failure to comply could lead to civil penalties and freezing device use.17 Patients and healthcare providers are not legally required to submit reports but can do so through the MedWatch program.20 All submitted reports are archived within a single publicly available database called the manufacturer and user facility device experience (MAUDE) database.21 Each year, the FDA receives approximately 100 000 reports.20
A secondary source of passive surveillance is the FDA's Medical Product Safety Network (MedSun). Started in 2003, MedSun consists of approximately 280 hospitals and other facilities that report on medical device use and adverse events.22 Theoretically, MedSun is different from MDR reporting in that these particular facilities are specifically trained in collecting and submitting adverse event reports to the FDA. As such, the quality and relative quantity of reports received from MedSun institutions are typically greater.19 The FDA receives around 5000 annual MedSun reports, and each facility has collective access to all reports submitted through MedSun. In return, the FDA can work with each clinical site to understand and resolve problems associated with medical devices.22
Passive surveillance has helped answer questions about the performance of certain devices. In 2012, Hauser et al23 used MAUDE to assess the presence and modes of deaths related to malfunctioning and failures of Riata and Riata ST leads manufactured by St. Jude Medical. In May 2014, Woerdeman and Cochrane24 used MAUDE to identify failures of hydrocephalus shunt valves. The authors noticed that these valve failures were because of rough handling of delicate shunt valves, and they substantiated their claims with similar reports within MAUDE.
Despite positive outcomes from the passive surveillance system, many of the reports have flaws. Of the 141 065 MDR reports submitted in 2007, roughly half were missing information about device identification and about 11% were lacking critical information on the adverse event itself.19,20 For example, one death associated with use of a vagus nerve stimulator was submitted to MAUDE with the death attributed to sudden unexpected death from epilepsy as confirmed by only a visual autopsy, which can, in reality, only be established with a full autopsy.25 In addition, many of the required reports are submitted late, with 31% of the 5-day manufacturer reports and 39% of 10-day user facility reports submitted late in 2007, despite the possibility of penalties for such violations.19,20 MAUDE can also be subject to double counting from duplicate reports.
Insufficient information on reports can hamper proper assessment of device safety, and even detailed reports leave uncertainty on what is truly device malfunction versus procedural errors or complications unrelated to the device. For example, the MAUDE database currently has 11 reports for an electrophysiological mapping catheter that was recalled in early 2014. The described events—aortic dissection, cardiac tamponade, and entrapment within the mitral valve—are difficult to ascribe specifically to the catheter rather than to technical or patient-related factors. A separate report claimed that during an atrial fibrillation procedure, the catheter's electrode shaft came apart with the wires exposed while inside the patient's body.21 This seems more clearly related to device malfunction but remains difficult to evaluate absent further details.
Indeed, there may also be bias in the distribution of adverse event reports. More than 90% of reports are from manufacturers presumably acting in accordance with reporting requirements. The remaining reports are voluntarily submitted by users and caregivers,19,26,27 suggesting under-reporting of adverse events by these parties.25,26 Under-reporting by users and caregivers occurs because of a lack of integration of device reporting into clinical operations, inabilities to connect outcomes with device use, failures in understanding a responsibility to report device-related events, and fear of personal litigation.27,28 Many times, malfunctioning products are not returned to the manufacturer, impeding study of potential safety issues. Yet under-reporting by manufacturers is also a problem. Because of the absence of a standardized reporting threshold, manufacturers have discretion in determining whether an adverse event is related to a device,26,29; so manufacturers are disincentivized to report adverse events, and some device-related problems may be inappropriately ascribed to user errors.
Overall, the passive surveillance system rarely leads to the identification of device safety problems. One fundamental drawback of passive surveillance is the lack of a denominator.17,25 With limited ability to track the number of devices actually used and their long-term outcomes, FDA analysts are limited in their ability to detect safety signals from noise.25 The FDA is routinely delayed in reading adverse event reports and rarely penalizes for late MDR submissions.19 When a safety signal is detected, the FDA has been criticized for not addressing the problem in a prompt and consistent manner.25 In addition, the MAUDE database cannot be linked to subsequent actions taken by FDA, such as onsite inspections and warning letters.19
Postapproval Clinical Studies
A postapproval clinical study is designed to provide information on a specific aspect of a device under controlled settings. The FDA can mandate conduct of postapproval studies of medical devices under 2 main provisions: postapproval studies (PAS) or 522 studies. PAS can be made a condition of medical device approval and can be issued alongside a premarket approval or humanitarian device exemption process. For example, since 2008, each new implantable cardioverter defibrillator lead has been required to undergo a PAS that collects data on device performance in ≥1000 patients for 5 years post implantation.28 By contrast, a 522 study can be mandated at any time in a device's market existence and can be applied to high-risk medical devices approved through conventional premarket approval processes or moderate-risk devices cleared through expedited 510(k) processes.30 Failure to fulfill the requirements of a PAS or 522 can be grounds for withdrawal of a device's authorization, but the FDA has yet to exercise this power.31
According to public reports on the FDA website, for the 291 PAS studies mandated from 2005 to 2012, 207 (71%) have a status of complete or adequate progress. Completed the 291 PAS studies are critical for evaluating long-term device performance.32 In March 2014, Vemuri et al33 conducted a retrospective outcomes study of 57 patients with risky cases of juxtarenal aortic aneurysms that were treated using a specific endovascular graft. The study demonstrated how these cases, which were usually untreatable via other available options, were successfully treated with the study graft without increased risk of complications. Another group pooled several PAS studies to study the safety of an openly irrigated ablation catheter. They discovered that a significant proportion of procedural complications was because of a user error, which was reduced when an operator oversight program was initiated.34 A total of 62 (21%) PAS studies showed inadequate progress or have been terminated. For example, the FDA ordered a PAS of Integra LifeScience's Duraseal Exact Spine Sealant in September 2009 to study the incidence of postoperative cerebrospinal fluid leaks. But as of February 2014, the study has not enrolled a sufficient number of patients. If a study is terminated, the manufacturer must communicate early with the FDA to establish a new PAS protocol to meet the postapproval study condition.35 In the past, inadequate completion was attributed to a lack of a standardized PAS tracking in the FDA—which the FDA has since tried to fix—and to a high turnover rate among PAS reviewers.36 These results are presented in Figure 1.
Figure 1.

Status of Food and Drug Administration (FDA)–mandated postapproval studies (PAS) of medical devices. Information current as of June 2014. All 14 studies in 2014 and 22 of the 48 studies in 2013 are still pending. Other annual study totals include 31 in 2005, 45 in 2006, 25 in 2007, 37 in 2008, 29 in 2009, 27 in 2010, 42 in 2011, and 55 in 2012. Definitions of study statuses: completed=manufacturer has fulfilled condition of approval and FDA has closed the study; progress adequate=study is progressing in accordance with the PAS protocol; progress inadequate=study is not progressing in accordance with the PAS protocol (eg, not meeting enrollment and timeline schedules, not adequately following up with patients, and not evaluating all end points); terminated=manufacturer has not fulfilled or cannot fulfill the PAS order for various reasons (eg, device is no longer being sold or has been changed, study questions are obsolete, manufacturer is no longer seeking premarket approval, study is not designed to answer relevant questions), all efforts to fulfill the PAS have been exhausted and the study has been terminated; study pending=the manufacturer's study protocol has been approved and the FDA is awaiting the first progress report; other=the study does not fit within another category (eg, study has changed leadership, device is being redesigned, a separate study is being used).35
Completion rates of 522 studies are similar. Twenty of 336 (6%) 522 studies initiated from 2005 to 2012 were complete or demonstrated adequate progress by June 2014, whereas another 218 (65%) have been consolidated, meaning that the 522 order was later combined with others into a single study (Figure 2).37 These results are displayed in Figure 2. For example, in May 2011, different 522 orders for detection of metal ion levels of various components of Biomet's metal-on-metal acetabular system were consolidated into a single study. Around one-fourth of the studies during this time-line were terminated (78, 23%). For example, Herniamesh's Bioabsorbable Polymer Sling and Surgical Mesh was subject to a 522 study order in April 2012 to assess its effectiveness in surgical repair of female stress urinary incontinence. However, the study was not completed and has been terminated. The remaining 114 (34%) were labeled with an interim other status. When completed, PAS and 522 studies can provide important information about device performance, but it is unclear whether studies themselves tend to not get done properly or whether the knowledge of their progress is just inexact. Moreover, the exact nature of FDA action after completion of the PAS and 522 studies is also not routinely disseminated. Gartenberg et al10 demonstrated how the FDA did not remove the Wingspan intracranial stent system from market in 2012 despite postapproval data suggesting significantly high stroke and mortality risks. Instead, the FDA reformed the indications for the device and ordered a 522 study for the device, which is still pending.10 The most common result from a PAS completed study is a labeling change of the studied device, which has been ordered for 31 studies to date.31
Figure 2.
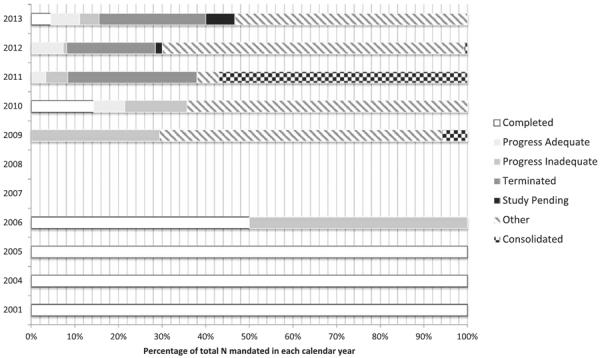
Status of Food and Drug Administration (FDA)-mandated 522 studies of medical devices. Information current as of June 2014. No studies in 2007 and 2008 were present in the database. Only 1 study was mandated in 2001, 2004, and 2005, with all 3 studies having been completed. In 2006, 2 studies were orders with 1 completion and 1 still showing inadequate process. Other annual study totals include 17 in 2009, 14 in 2010, 179 in 2011, 123 in 2012, and 45 in 2013. Definitions of study statuses: completed=manufacturer has fulfilled postmarket surveillance 522 order and FDA has closed the study; progress adequate=study is progressing in accordance with the 522 plan; progress inadequate=study is not progressing in accordance with the 522 plan (eg, not meeting enrollment and timeline schedules, not adequately following up with patients, and not evaluating all end points) or the study plan is overdue; terminated=manufacturer has not fulfilled or cannot fulfill the 522 order for various reasons (eg, device is no longer being sold or has been changed, study questions are obsolete, manufacturer is no longer seeking premarket approval, study is not designed to answer relevant questions), all efforts to fulfill the 522 have been exhausted and the study has been terminated; study pending=the manufacturer's study protocol has been approved and the FDA is awaiting the first progress report; other=the study does not fit within another category (eg, study has changed leadership, device is being redesigned, a separate study is being used).37
Postapproval Registry Surveillance
A device registry is a structured database that actively collects and stores data on individual exposures to specific devices to monitor general device safety prospectively.17 Currently, the FDA registry surveillance program is small. In 2007, the FDA was authorized to create an active surveillance network to monitor pharmaceuticals and medical products, which led to the creation of the Sentinel Initiative.38 Sentinel is modeled to be a distributed data system in which deidentified administrative or claims data are institutionally maintained and sourced by a central hub to monitor the safety of regulated products.17 Although currently still a pharmaceutical data system, Sentinel is beginning to be expanded to include medical devices.
In 2012, the FDA also formalized its plan to pursue a nationwide system that conducts active postmarket surveillance of medical devices.17 The FDA envisions a distributed data system, much like Sentinel, in which registries are maintained by independent entities that can then be accessed centrally. Currently, many organizations maintain registries, such as the American College of Cardiology's National Cardiovascular Data Registry (Table 1). Registries may also be based on community, hospital, or manufacturer settings (Table 2). The FDA is holding workshops and developing partnerships with professional medical societies to facilitate additional registry initiatives.17,28
Table 1.
Selection of National Cardiovascular Registries Used in Published Medical Device Studies in 2013 to 2014
| Registry | Professional Organization | Year | Focus |
|---|---|---|---|
| National Cardiovascular Data Registry | ACC | 1997 | ⋯ |
| ACTION Registry—GWTG | ACC/AHA | 2007 | Acute coronary syndromes |
| CARE Registry | ACC | 2006 | Carotid revascularization, endarterectomy |
| CathPCI Registry | ACC/SCAI | 1998 | Cardiac catherization, percutaneous coronary intervention |
| ICD Registry | ACC/HRS | 2005 | Implantable cardioverter defibrillators and leads pediatric and adult congenital procedures |
| IMPACT Registry | ACC/SCAI | 2007 | Outpatient cardiovascular care |
| PINNACLE Registry | ACC | 2008 | Transcatheter valve procedures |
| TVT Registry | STS/ACC | 2012 | ⋯ |
| STS National Database | |||
| Adult Cardiac Surgery Database | STS | 1989 | Quality improvement and patient safety |
| General Thoracic Surgery Database | |||
| Congenital Heart Surgery Database | |||
| Vascular Registry | SVS | N/A | Carotid stents and embolic protection devices |
| National Surgical Quality Improvement Program | ACS | 1994 | Surgical quality improvement, patient outcomes |
| Translational Research Investigating Underlying Disparities in Acute Myocardial Infarction Patients' Health Status (TRIUMPH) | NHLBI | 2005 | Healthcare disparities in acute myocardial infarction risk |
Registries listed here were identified via a MEDLINE search supplemented with analysis of Food and Drug Administration and individual registry Web sites. Note that this is not an exhaustive list of registries and represents a sample of current national registries for cardiovascular devices. ACC indicates American College of Cardiology; ACS, American College of Surgeons; ACTION, Acute Coronary Treatment and Intervention Outcomes Network; AHA, American Heart Association; CARE, Carotid Artery Revascularization and Endarterectomy; CathPCI, Catheterized Percutaneous Coronary Intervention; GWTG, Get With The Guidelines; HRS, Hearth Rhythm Society; ICD, implantable cardioverter defibrillator; IMPACT, Improving Pediatric and Adult Congenital Treatment; NHLBI, National Heart, Lung, and Blood Institute; PINNACLE, Practice Innovation And Clinical Excellence; SCAI, Society for Cardiovascular Angiography and Interventions; STS, Society of Thoracic Surgeons; SVS, Society for Vascular Surgery, TRIUMPH, Translational Research Investigating Underlying Disparities in Acute Myocardial Infarction Patients' Health Status; and TVT, Transcatheter Valve Therapy.
Table 2.
Selection of Hospital-, Community-, and Manufacturer-Based Registries Used in Published Cardiovascular Device Studies From 2013 to 2014
| Registry | Professional Organization | Focus |
|---|---|---|
| INTERMACS | University of Alabama-Birmingham | Durable mechanical circulatory support device therapy to treat advanced heart failure |
| VSGNE database | VSGNE | Vascular surgery operative and perioperative data |
| C3P0 Registry | Children's Hospital of Boston | Outcomes from pediatric catheterization |
| Percutaneous Intervention Registry | Cleveland Clinic | Percutaneous heart valve replacements |
| ClearWay Multicenter Registry | Atrium | ClearWay perfusion catheter |
Registries listed here were identified via a MEDLINE search supplemented with analysis of Food and Drug Administration and individual registry Web sites. Note that this is not an exhaustive list of registries and represents a sample of current cardiovascular registries maintained by individual hospitals, communities, and manufacturers. C3P0 indicates Congenital Cardiac Catheterization Project on Outcomes; INTERMACS, The Interagency Registry for Mechanically Assisted Circulatory Support; and VSGNE, Vascular Study Group of New England.
Among the benefits of a registry-based surveillance system is the opportunity to generate data sets with large sample sizes that also include more diverse sets of patients, which allow for analyses with high generalizability.39 These large data sets can provide more useful safety information that is often lacking in controlled, short-term prospective trials, and studies have subsequently used these data sets to answer important clinical questions.40,41 In addition, registries with linked clinical data (eg, National Cardiovascular Data Registry registries) can also provide information on potential misuse of a device; for example, many studies have found that too many transcatheter aortic valve implantations are being conducted in a transapical fashion than would be expected for a typical patient population.27 Similar studies have suggested lack of adherence to guideline-based indications for intracardiac cardioverter defibrillators and cardiac resynchronization therapy.42,43
If registries are not closely linked to clinical records, the registries may lack critical event-specific data, such as disease severity or comorbidities, that could identify variations among treatments, populations, and device subtypes. A key challenge is that registries may not include follow-up information that could provide more longitudinal analysis. From a study design perspective, this information would be useful for control measures that could minimize the effects of confounding variables within these large data sets.44–46 Until registries are consistently linked to comprehensive clinical information, their data will be susceptible to confounding, which can impair assessment of clinical effectiveness of medical devices. In addition, measures will have to be implemented that ensure data quality, such as basic structural requirements of all registries and audits by overseeing organizations. In their current formats, registries alone are not sufficient to monitor the effectiveness of medical devices and are better suited to assess long-term device safety.
The next step in a registry system is participation. The FDA has not stated how it will encourage organizations to build and maintain local registries. The establishment of a registry can present substantial costs to the managing institution, and mandatory participants may not be prompted to provide complete or unbiased information.47 Within the field of cardiology, smaller healthcare centers may be disincentivized from participating in registries because the data could be used to conclude that only larger institutions be allowed to provide high-risk procedures. Further consideration should include the type of participants because manufacturer-owned registries may lead to biased or untrustworthy data.47 Because registries are maintained locally, it is also up to individual organizations to establish systems that comprehensively collect device information. The FDA will not regulate registry structures,17 so managing organizations must establish internal structures that facilitate reporting to a registry by healthcare providers. Because the FDA cannot mandate registry participation by institutions, the extent of voluntary institutional participation remains to be seen.
Prospects for Device Postmarket Surveillance
The FDA is aware of the shortcomings of device postmarket surveillance systems and is developing many prospective reforms and important innovations to enhance device postmarket surveillance.18 To improve passive adverse event reporting, the FDA plans to work with the November Research Group to replace the MAUDE database with a reporting system similar to the FDA Adverse Event Reporting System that has been used for pharmaceutical products and that will be able to handle larger report volumes, incorporate safety analytics, and potentially employ mobile device or other advanced modalities for reporting.17,48 In addition, the FDA is working to implement software tools that can automatically report adverse events in real time. The FDA created a pilot called the Adverse Drug Events Spontaneous Triggered Event Reporting study, in which adverse event reports are automatically transmitted when they appear in linked medical records.49 The FDA is also interested in modernizing data analysis techniques to identify safety signals rapidly. For example, the Data Extraction and Longitudinal Time Analysis is a proprietary system that evaluates device performance in real time to detect device safety signals rapidly. Data Extraction and Longitudinal Time Analysis has already been effective in identifying safety issues in cardiovascular devices.50 Adverse Drug Events Spontaneous Triggered Event Reporting and Data Extraction and Longitudinal Time Analysis are still years away from broad implementation, but their development suggests a promising future for obtaining improved information from passive post-market surveillance.
In 2010, the FDA initiated MDEpiNet, a public-private partnership designed to study and develop innovative strategies to improve regulation of medical devices.17 MDEpiNet is researching a variety of active surveillance initiatives to collect device data, assess risks, detect safety signals, and act on potential safety issues. One of these initiatives is a nationwide unique device identifier (UDI) system. In September 2013, the FDA issued a final rule requiring medical devices to carry a UDI that places individual labels on devices and device packages to be able to track clinical use and distribution better.51 When integrated with registries, the UDI system will provide a standardized language for device identification by registries. All high-risk devices must bear a UDI by September 2014, moderate-risk devices by September 2016, and low-risk devices by September 2018.51 Despite these compliance deadlines, concrete legislation mandating UDI incorporation is still forthcoming. Accredited issuing agencies will assign device UDIs and will store them in a new database called the Global Unique Device Identification Database. The Global Unique Device Identification Database will allow linkage of the UDI to premarket approval reports or trade and manufacturer information.
Registry building is another area of considerable activity. The FDA envisions the use of voluntary registries as an integral component of a national device surveillance network. Such a registry-based surveillance network could effectively track use of all devices to ascertain exposure and outcome status, better elucidate device-related events, and more efficiently identify safety signals. Yet there is still considerable work to be done, and exactly how this network of voluntary registries will be realized is still uncertain. The FDA will first have to identify the number and type of registries that it hopes to include in this nationwide network, because some databases cover a specific medical device (eg, ClearWay perfusion catheters in a manufacturer-operated registry), whereas others cover a whole class of devices (eg, all coronary interventional catheters in the National Cardiovascular Data Registry's CathPCI registry). Through MDEpiNet, the FDA has developed the National Medical Device Registries Taskforce, which is holding workshops with key stakeholders to develop new registries.18
The FDA will next have to address how it will encourage manufacturers and organizations to build and maintain local registries identified by the Taskforce that can collate device data in an accurate and timely fashion. One suggestion has been to base institutional accreditation of hospitals on participation in national registries, which can also incentivize hospitals to create internal registry reporting requirements for employed healthcare providers. The Centers for Medicare and Medicaid Services has recently linked its reimbursement of transcatheter aortic valve replacements with hospital participation in a prospective, national registry through the Coverage with Evidence Development provision. Linking registry reporting to reimbursement through the Coverage with Evidence Development provision can promote hospital participation in local registries, but these processes still have problems with poor data collection and high implementation costs.52 Placing participation incentives on hospitals may also lead to extra workload for individual physicians and clinical administrators.47 Regardless, the partnership between the FDA and the Centers for Medicare and Medicaid Services may provide the necessary infrastructure to encourage and support hospital and institutional registry initiatives.
Of course, even if registries can be optimally designed, they cannot replace controlled, prospective postmarket studies like the ones required under PAS or 522 orders.4 Device registries are powered by their large sample sizes to answer several questions in a real-world application. But registry data are still limited primarily by selection bias and secondarily by confounding variables—such as disease severity, comorbidities, and medication use—that can make it difficult to assess the effectiveness of a device in an observational study. Postmarket studies mandated by PAS or 522 are useful for controlling against these confounders and for assessing a device's true effectiveness in practice. Studies mandated by 522 and PAS need to be expanded and incorporated with registries to allow the FDA to best assess effectiveness and safety of approved medical devices.
A distributed data system will become increasingly important from a global perspective. Construction of registry surveillance systems will be able to access postapproval data from other countries. European manufacturers have been required to monitor their approved devices under the purview of regional authorities called Notified Bodies.4 Adverse event data are then collated in a centralized repository called The European Databank on Medical Devices.27 Although access to European and other datasets can certainly be helpful, the FDA should remain cautious about prematurely sourcing global registries. European registries still have differing evidence standards among Notified Bodies,27 lack of oversight from local registry authorities,4 and absence of registry interoperability.47 It is still difficult to access European data because of a lack of transparency in premarket device certification and postapproval data sources, such as the European Databank on Medical Devices.53
Researchers in Europe are outlining ways to improve regulations for medical devices. Like the FDA, many European policy advisers are seeking expanded use of registries in collecting device data.4,54 Others have demanded increased transparency of premarket device certifications and of the European Databank on Medical Devices to identify the deficiencies of European device regulation.53 One European analysis called for an FDA-like central body to regulate and oversee the distributed regulatory system of Europe.4 Current proposals being considered by the European Parliament include the establishment of a centralized body to oversee authorization and regulation of medical devices.27 The FDA should actively work with foreign authorities to stabilize and standardize registries for the use in a future globalized distributed data system.
Conclusions
All postmarket surveillance strategies have unique abilities to monitor different aspects of device safety and effectiveness. The FDA is attempting to address current limitations in device surveillance through reform efforts to modernize passive reporting and to construct an active registry-based surveillance network.17,18 The FDA could benefit from taking a centralized leadership role in a distributed data surveillance system in ensuring the standardization, interoperability, and efficiency of different device surveillance strategies, both domestically and globally. However, even optimally designed registries will need to be supplemented by clinical effectiveness data generated from postmarket clinical studies, as well as routine passive surveillance signal detection. Therefore, registries will need to be implemented in a cooperative manner with active clinical study mandates and updated passive surveillance tools to best provide oversight of medical device safety and effectiveness. Policymakers should consider the current status of the postapproval device safety monitoring system when designing programs that accelerate premarket evaluation and review of investigational devices. Although great strides have been made, and additional innovations are planned, numerous issues need to be addressed before the postapproval device surveillance system in the United States will be operating at optimal proficiency.
Acknowledgments
Dr Kramer was supported, in part, by a Paul B. Beeson Career Development Award in Aging Research (K23AG045963), serves as a consultant to the FDA's Circulatory Systems Advisory Panel, and was a member of the American College of Cardiology National Cardiovascular Data Registry Device Surveillance Strategy Work Group. Dr Kesselheim reports grant support from the Greenwall Foundation and an Ignition Award from the Harvard Program in Therapeutic Science.
Footnotes
Disclosures P.V. Rajan reports no conflicts.
References
- 1.U.S. Food and Drug Administration . Center for Devices and Radiological Health. 2014. Expedited Access for Premarket Approval Medical Devices Intended for Unmet Medical Need for Life Threatening or Irreversibly Debilitating Diseases or Conditions: Draft Guidance for Industry and Food and Drug Administration Staff. [Google Scholar]
- 2.PricewaterhouseCoopers . Improving America's health V: a survey of the working relationship between the life sciences industry and FDA. PricewaterhouseCoopers; London: 2011. [Google Scholar]
- 3.Makower J, Meer A, Denend L. FDA Impact on U.S. Medical Technology Innovation: A Survey of Over 200 Medical Technology Companies. National Venture Capital Association; Arlington, VI: 2010. [Google Scholar]
- 4.Cohen D, Billingsley M. Europeans are left to their own devices. BMJ. 2011;342:d2748. doi: 10.1136/bmj.d2748. [DOI] [PubMed] [Google Scholar]
- 5.Mack MJ, Brennan JM, Brindis R, Carroll J, Edwards F, Grover F, Shahian D, Tuzcu EM, Peterson ED, Rumsfeld JS, Hewitt K, Shewan C, Michaels J, Christensen B, Christian A, O'Brien S, Holmes D. STS/ACC TVT Registry. Outcomes following transcatheter aortic valve replacement in the United States. JAMA. 2013;310:2069–2077. doi: 10.1001/jama.2013.282043. doi: 10.1001/jama.2013.282043. [DOI] [PubMed] [Google Scholar]
- 6.Leon MB, Smith CR, Mack M, Miller DC, Moses JW, Svensson LG, Tuzcu EM, Webb JG, Fontana GP, Makkar RR, Brown DL, Block PC, Guyton RA, Pichard AD, Bavaria JE, Herrmann HC, Douglas PS, Petersen JL, Akin JJ, Anderson WN, Wang D, Pocock S. PARTNER Trial Investigators. Transcatheter aortic-valve implantation for aortic stenosis in patients who cannot undergo surgery. N Engl J Med. 2010;363:1597–1607. doi: 10.1056/NEJMoa1008232. doi: 10.1056/NEJMoa1008232. [DOI] [PubMed] [Google Scholar]
- 7.Contrera P, Cushing M. AANA Journal Course: Update for nurse anesthetists–part-4—transcatheter aortic valve replacement. AANA J. 2013;81:399–408. [PubMed] [Google Scholar]
- 8.Hwang TJ, Carpenter D, Kesselheim AS. Assessment of US pathway for approving medical devices for rare conditions. BMJ. 2014;348:g217. doi: 10.1136/bmj.g217. [DOI] [PubMed] [Google Scholar]
- 9.Maisel WH. Unanswered questions–drug-eluting stents and the risk of late thrombosis. N Engl J Med. 2007;356:981–984. doi: 10.1056/NEJMp068305. doi: 10.1056/NEJMp068305. [DOI] [PubMed] [Google Scholar]
- 10.Gartenberg AJ, Peleg A, Dhruva SS, Redberg RF. Presumed safe no more: lessons from the Wingspan saga on regulation of devices. BMJ. 2014;348:g93. doi: 10.1136/bmj.g93. [DOI] [PubMed] [Google Scholar]
- 11.Dhruva SS, Bero LA, Redberg RF. Strength of study evidence examined by the FDA in premarket approval of cardiovascular devices. JAMA. 2009;302:2679–2685. doi: 10.1001/jama.2009.1899. doi: 10.1001/jama.2009.1899. [DOI] [PubMed] [Google Scholar]
- 12.Kramer DB, Mallis E, Zuckerman BD, Zimmerman BA, Maisel WH. Premarket clinical evaluation of novel cardiovascular devices: quality analysis of premarket clinical studies submitted to the Food and Drug Administration 2000–2007. Am J Ther. 2010;17:2–7. doi: 10.1097/MJT.0b013e3181ca8105. doi: 10.1097/MJT.0b013e3181ca8105. [DOI] [PubMed] [Google Scholar]
- 13.Chen CE, Dhruva SS, Redberg RF. Inclusion of comparative effectiveness data in high-risk cardiovascular device studies at the time of premarket approval. JAMA. 2012;308:1740–1742. doi: 10.1001/jama.2012.14491. doi: 10.1001/jama.2012.14491. [DOI] [PubMed] [Google Scholar]
- 14.Ellenbogen KA, Wood MA, Swerdlow CD. The Sprint Fidelis lead fracture story: what do we really know and where do we go from here? Heart Rhythm. 2008;5:1380–1381. doi: 10.1016/j.hrthm.2008.08.001. doi: 10.1016/j.hrthm.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Henrikson CA. The Riata story-where are we now? Heart Rhythm. 2012;9:750–751. doi: 10.1016/j.hrthm.2012.01.013. doi: 10.1016/j.hrthm.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Ardaugh BM, Graves SE, Redberg RF. The 510(k) ancestry of a metal-on-metal hip implant. N Engl J Med. 2013;368:97–100. doi: 10.1056/NEJMp1211581. doi: 10.1056/NEJMp1211581. [DOI] [PubMed] [Google Scholar]
- 17.U.S. Food and Drug Administration . Center for Devices and Radiological Health. 2012. Strengthening our National System for Medical Device Postmarket Surveillance. [Google Scholar]
- 18.U.S. Food and Drug Administration . Center for Devices and Radiological Health. 2013. Strengthening our National System for Medical Device Postmarket Surveillance: Update and Next Steps. [Google Scholar]
- 19.Department of Health and Human Services . Office of the Inspector General. 2009. Adverse Event Reporting for Medical Devices. [Google Scholar]
- 20.Institute of Medicine . Medical Devices and the Public's Health: The FDA 510(k) Clearance Process at 35 Years. The National Academies Press; Washington, DC: 2011. [Google Scholar]
- 21.U.S. Food and Drug Administration . MAUDE Adverse Event Report: Biosense Webster, Inc. (Juarez) Pentaray Nav Eco high-density mapping catheter, intracardiac mapping, high-density array [Internet] U.S. Food and Drug Administration; Silver Spring, MD: [Accessed November 18 2014]. http://www.accessdata. fda.gov/scripts/cdrh/cfdocs/cfmaude/detail.cfm?mdrfoi__id=3785579. [Google Scholar]
- 22.Engleman D, Rich S, Powell T, Flack M. Medical Product Safety Network (MedSun) Collaborates with Medical Product Users to Create Specialty Subnetworks. In: Henriksen K, Battles JB, Keyes MA, et al., editors. Advances in Patient Safety: New Directions and Alternative Approaches (Vol. 1: Assessment) Agency for Healthcare Research and Quality; Rockville, MD: 2008. Available from http://www.ncbi.nlm.nih.gov/books/NBK43633/ [PubMed] [Google Scholar]
- 23.Hauser RG, Abdelhadi R, McGriff D, Retel LK. Deaths caused by the failure of Riata and Riata ST implantable cardioverter-defibrillator leads. Heart Rhythm. 2012;9:1227–1235. doi: 10.1016/j.hrthm.2012.03.048. doi: 10.1016/j.hrthm.2012.03.048. [DOI] [PubMed] [Google Scholar]
- 24.Woerdeman PA, Cochrane DD. Disruption of silicone valve housing in a Codman Hakim Precision valve with integrated Siphonguard. J Neurosurg Pediatr. 2014;13:532–535. doi: 10.3171/2014.2.PEDS13436. doi: 10.3171/2014.2.PEDS13436. [DOI] [PubMed] [Google Scholar]
- 25.Lenzer J, Brownlee S. Why the FDA can't protect the public. BMJ. 2010;341:c4753. doi: 10.1136/bmj.c4753. [DOI] [PubMed] [Google Scholar]
- 26.Greenfield LJ. Perspective from an academic on postmarket surveillance. In: Brown SL, Bright RA, Tavris DR, editors. Medical Device Epidemiology and Surveillance. John Wiley & Sons, Ltd; Hoboken, NJ: 2007. [Google Scholar]
- 27.Sorenson C, Drummond M. Improving medical device regulation: the United States and Europe in perspective. Milbank Q. 2014;92:114–150. doi: 10.1111/1468-0009.12043. doi: 10.1111/1468-0009.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blake K. Postmarket surveillance of medical devices: current capabilities and future opportunities. J Interv Card Electrophysiol. 2013;36:119–127. doi: 10.1007/s10840-013-9778-6. doi: 10.1007/s10840-013-9778-6. [DOI] [PubMed] [Google Scholar]
- 29.Bren L. Company caught in coverup of medical device malfunctions. FDA Consum. 2003;37:43. [PubMed] [Google Scholar]
- 30.U.S. Food and Drug Administration . Transcript for Public Workshop – Design and Methodology for Postmarket Surveillance Studies under Section 522 of the Federal Food, Drug and Cosmetic Act, March 7, 2012 [Internet] U.S. Food and Drug Administration; Silver Spring, MD: last updated Apr 2012. http://www.fda.gov/medicaldevices/newsevents/workshopsconferences/ucm301847.htm. [Google Scholar]
- 31.Reynolds IS, Rising JP, Coukell AJ, Paulson KH, Redberg RF. Assessing the Safety and Effectiveness of Devices After US Food and Drug Administration Approval: FDA-Mandated Postapproval Studies. JAMA Intern Med. 2014;174:1773–1779. doi: 10.1001/jamainternmed.2014.4194. doi: 10.1001/jamainternmed.2014.4194. [DOI] [PubMed] [Google Scholar]
- 32.Vardi M, Perez J, Griffin PJ, Burke DA, Yeh RW, Cutlip DE. Usefulness of postmarket studies to evaluate long-term safety of coronary eluting stents (from the ENDEAVOR and PROTECT Programs) Am J Cardiol. 2014;114:528–533. doi: 10.1016/j.amjcard.2014.05.028. doi: 10.1016/j.amjcard.2014.05.028. [DOI] [PubMed] [Google Scholar]
- 33.Vemuri C, Oderich GS, Lee JT, Farber MA, Fajardo A, Woo EY, Cayne N, Sanchez LA. Postapproval outcomes of juxtarenal aortic aneurysms treated with the Zenith fenestrated endovascular graft. J Vasc Surg. 2014;60:295–300. doi: 10.1016/j.jvs.2014.01.071. doi: 10.1016/j.jvs.2014.01.071. [DOI] [PubMed] [Google Scholar]
- 34.Waldo AL, Wilber DJ, Marchlinski FE, Stevenson WG, Aker B, Boo LM, Jackman WM. Safety of the open-irrigated ablation catheter for radiofrequency ablation: safety analysis from six clinical studies. Pacing Clin Electrophysiol. 2012;35:1081–1089. doi: 10.1111/j.1540-8159.2012.03480.x. doi: 10.1111/j.1540-8159.2012.03480.x. [DOI] [PubMed] [Google Scholar]
- 35.U.S. Food and Drug Administration . Guidance for Industry and FDA Staff: Procedures for Handling Post-Approval Studies Imposed by PMA Order [Internet] U.S. Food and Drug Administration; Silver Spring, MD: [Accessed June 2009]. http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm071013.pdf. [Google Scholar]
- 36.U.S. Food and Drug Administration Transcript for Circulatory System Devices Panel Meeting, April 22, 2005 [Internet] http://www.fda.gov/ohrms/dockets/ac/05/transcripts/2005-4108t1.htm.
- 37.U.S. Food and Drug Administration . Guidance for Industry and Food and Drug Administration Staff: Procedures for Handling Section 522 Postmarket Surveillance Studies [Internet] U.S. Food and Drug Administration; Silver Spring, MD: [Accessed August 2011]. http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM268141.pdf. [Google Scholar]
- 38.U.S. Department of Health and Human Services [Accessed July 2010];The Sentinel Initiative: Access to Electronic Healthcare Data for More Than 25 Million Lives Achieving FDAAA Section 905 Goal One. http://www.fda.gov/downloads/Safety/FDAsSentinelInitiative/UCM233360.pdf.
- 39.Black N, Tan S. Use of national clinical databases for informing and for evaluating health care policies. Health Policy. 2013;109:131–136. doi: 10.1016/j.healthpol.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 40.Fröbert O, Lagerqvist B, Olivecrona GK, Omerovic E, Gudnason T, Maeng M, Aasa M, Angerås O, Calais F, Danielewicz M, Erlinge D, Hellsten L, Jensen U, Johansson AC, Kåregren A, Nilsson J, Robertson L, Sandhall L, Sjögren I, Ostlund O, Harnek J, James SK. TASTE Trial. Thrombus aspiration during ST-segment elevation myocardial infarction. N Engl J Med. 2013;369:1587–1597. doi: 10.1056/NEJMoa1308789. [DOI] [PubMed] [Google Scholar]
- 41.Rao SV, Hess CN, Barham B, Aberle LH, Anstrom KJ, Patel TB, Jorgensen JP, Mazzaferri EL, Jr, Jolly SS, Jacobs A, Newby LK, Gibson CM, Kong DF, Mehran R, Waksman R, Gilchrist IC, McCourt BJ, Messenger JC, Peterson ED, Harrington RA, Krucoff MW. A registry-based randomized trial comparing radial and femoral approaches in women undergoing percutaneous coronary intervention: the SAFE-PCI for Women (Study of Access Site for Enhancement of PCI for Women) trial. JACC Cardiovasc Interv. 2014;7:857–867. doi: 10.1016/j.jcin.2014.04.007. doi: 10.1016/j.jcin.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 42.Fein AS, Wang Y, Curtis JP, Masoudi FA, Varosy PD, Reynolds MR. National Cardiovascular Data Registry. Prevalence and predictors of off-label use of cardiac resynchronization therapy in patients enrolled in the National Cardiovascular Data Registry Implantable Cardiac-Defibrillator Registry. J Am Coll Cardiol. 2010;56:766–773. doi: 10.1016/j.jacc.2010.05.025. doi: 10.1016/j.jacc.2010.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Khatib SM, Hellkamp A, Curtis J, Mark D, Peterson E, Sanders GD, Heidenreich PA, Hernandez AF, Curtis H, Hammill S. Non-evidence-based ICD implantations in the United States. JAMA. 2011;305:43–49. doi: 10.1001/jama.2010.1915. doi: 10.1001/jama.2010.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneeweiss S, Avorn J. A review of uses of health care utilization databases for epidemiologic research on therapeutics. J Clin Epidemiol. 2005;58:323–337. doi: 10.1016/j.jclinepi.2004.10.012. doi: 10.1016/j.jclinepi.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 45.Normand SL, Hatfield L, Drozda J, Resnic FS. Postmarket surveillance for medical devices: America's new strategy. BMJ. 2012;345:e6848. doi: 10.1136/bmj.e6848. [DOI] [PubMed] [Google Scholar]
- 46.Setoguchi S, Zhu Y, Jalbert JJ, Williams LA, Chen CY. Validity of deterministic record linkage using multiple indirect personal identifiers: linking a large registry to claims data. Circ Cardiovasc Qual Outcomes. 2014;7:475–480. doi: 10.1161/CIRCOUTCOMES.113.000294. doi: 10.1161/CIRCOUTCOMES.113.000294. [DOI] [PubMed] [Google Scholar]
- 47.Niederländer C, Wahlster P, Kriza C, Kolominsky-Rabas P. Registries of implantable medical devices in Europe. Health Policy. 2013;113:20–37. doi: 10.1016/j.healthpol.2013.08.008. doi: 10.1016/j.healthpol.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 48.November Research Group [Accessed July 2013];FDA Selects November Research Group's PRIMO Software Platform as Next Generation Product Vigilance Solution [Internet] Business Wire. http://www.businesswire.com/news/home/20130917005257/en/FDA-Selects-November-Research-Group's-PRIMO-Software#.U63OAhZFImt.
- 49.Brajovic S, Piazza-Hepp T, Swartz L, Dal Pan G. Quality assessment of spontaneous triggered adverse event reports received by the Food and Drug Administration. Pharmacoepidemiol Drug Saf. 2012;21:565–570. doi: 10.1002/pds.3223. discussion 571. doi: 10.1002/pds.3223. [DOI] [PubMed] [Google Scholar]
- 50.Vidi VD, Matheny ME, Donnelly S, Resnic FS. An evaluation of a distributed medical device safety surveillance system: the DELTA network study. Contemp Clin Trials. 2011;32:309–317. doi: 10.1016/j.cct.2011.02.001. doi: 10.1016/j.cct.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Department of Health and Human Services [Accessed July 2012];Unique Device Identification System. Federal Register. https://www.federalregister.gov/articles/2012/07/10/2012-16621/unique-device-identification-system.
- 52.Neumann PJ, Chambers J. [Accessed April 2013];Medicare's reset on `Coverage With Evidence Development' [Internet] Health Affairs blog. http://healthaffairs.org/blog/2013/04/01/medicares-reset-on-coverage-with-evidence-development/
- 53.Eikermann M, Gluud C, Perleth M, Wild C, Sauerland S, Gutierrez-Ibarluzea I, Antoine SL, Demotes-Mainard J, Neugebauer EA. Signatories of Our Open Letter to the European Union. Commentary: Europe needs a central, transparent, and evidence based regulation process for devices. BMJ. 2013;346:f2771. doi: 10.1136/bmj.f2771. [DOI] [PubMed] [Google Scholar]
- 54.Campbell B, Stainthorpe AC, Longson CM. How can we get high quality routine data to monitor the safety of devices and procedures? BMJ. 2013;346:f2782. doi: 10.1136/bmj.f2782. [DOI] [PubMed] [Google Scholar]