

Abstract
Triple-negative breast cancer (TNBC) occurs in 10–15% of patients yet accounts for almost half of all breast cancer deaths. TNBCs lack expression of estrogen and progesterone receptors and HER-2 overexpression and cannot be treated with current targeted therapies. TNBCs often occur in African American and younger women. Although initially responsive to some chemotherapies, TNBCs tend to relapse and metastasize. Thus, it is critical to find new therapeutic targets. A second ER gene product, termed ERβ, in the absence of ERα may be such a target. Using human TNBC specimens with known clinical outcomes to assess ERβ expression, we find that ERβ1 associates with significantly worse 5-year overall survival. Further, a panel of TNBC cell lines exhibit significant levels of ERβ protein. To assess ERβ effects on proliferation, ERβ expression in TNBC cells was silenced using shRNA, resulting in a significant reduction in TNBC proliferation. ERβ-specific antagonists similarly suppressed TNBC growth. Growth-stimulating effects of ERβ may be due in part to downstream actions that promote VEGF, amphiregulin, and Wnt-10b secretion, other factors associated with tumor promotion. In vivo, insulin-like growth factor-2 (IGF-2), along with ERβ1, is significantly expressed in TNBC and stimulates high ERβ mRNA in TNBC cells. This work may help elucidate the interplay of metabolic and growth factors in TNBC.
1. Introduction
Breast cancer (BC) is the most common malignancy in women [1, 2]. About 70% of patients with breast cancer express estrogen receptor-α (ERα). Due to the success of endocrine therapies, the mortality of patients with ERα-positive tumors has declined significantly in the past decade. Similarly, about 15% of patients have tumors that overexpress HER2 receptor and thus are candidates for HER2-targeted treatments. In contrast, TNBC occurs in 10–15% of patients, yet this disease subtype accounts for about half of all breast cancer deaths. TNBCs lack clinical expression of ERα, progesterone receptor, and HER2 overexpression (ERα−/PR−/HER2−). TNBCs have incomplete overlap with basal-like breast tumors, a subgroup of breast cancers defined by gene-expression profiling that express specific cytokeratins, and with some hereditary breast cancers. Though heterogeneous, TNBCs typically occur in younger women and African American women as well as among some patients with BRCA1 gene defects [1, 2]. Population-based data show that African American women have a higher incidence of TNBC and present with more advanced stages than Caucasian women [3]. This cancer subtype also associates with adverse biological features including high mitotic count and very aggressive behavior. Of note, some recent reports indicate that the incidence of ERα-negative BC and TNBC, high-risk breast cancer subtypes, may correlate with the extent of African ancestry [4]. Though initially responsive to chemotherapy, TNBCs tend to relapse and metastasize early and have a prognosis worse than other subtypes. Currently specific therapies for TNBC are unavailable [1–3].
Estrogens promote progression of ERα-positive cancers, effects exerted by binding of estradiol to ERα, a ligand-activated transcription factor [5]. ERα is predominantly a nuclear-localized protein. Immunohistochemical (IHC) detection of nuclear ERα in tumors is a standard clinical assay used to plan patient management [6]. Of special note, recent reports show that a second type of estrogen receptor, termed estrogen receptor-beta (ERβ), is expressed in TNBC cells [7, 8]. ERα and ERβ are encoded by two different genes, yet ERβ has 96% homology with ERα at the DNA-binding domain and 60% homology at the ligand-binding domain (LBD). However, it is important to note that ERβ is not identified in standard assays for ERα. The role of ERβ in breast cancer remains to be elucidated but some studies show ERβ is a biomarker related to a more aggressive clinical course [8] and correlates with Ki-67, a marker of proliferation [7, 9]. Early studies demonstrate higher levels of ERβ in breast tumors of African American as compared to Caucasian women, suggesting that ERβ may play a critical role in TNBC development [10–14].
Based on current data, estradiol regulates gene expression by genomic and nongenomic inputs [15, 16]. Genomic signals involve direct action of nuclear-localized ERα as an estradiol-regulated transcription factor or coregulator. By contrast, nongenomic signaling involves extranuclear events mediated by extranuclear ERs often in cooperation with coactivator or adaptor proteins [17]; these then impact gene expression indirectly by modulating signaling cascades such as MAPK, PI3K/AKT, and mTOR [8, 14–19] to regulate transcription [5, 15, 16]. In target cells, extranuclear ERα forms are derived from the same transcript as nuclear ERα; however minor extranuclear ERα splice variants occur [15, 16]. In TNBC, less is known about the role of ERβ in cancer progression [7, 8, 11]. Several ERβ isoforms occur in breast cancers, including ERβ1, ERβ2, ERβ4, and ERβ5, but only ERβ1 retains an intact LBD to interact with specific ligands, thus ERβ1 is a preferred clinical target [17, 20–22]. ERβ forms occur in tumor cell nuclei but, as ERα forms, may also occur at extranuclear sites [15, 16, 23]. Like ERα, ERβ activates transcription by genomic pathways or nongenomic pathways by interaction with coactivators/coregulators [17] that in turn modulate signaling cascades to impact gene expression and tumor progression [5, 12, 14, 20, 21, 23]. Of note, ERβ target genes appear to be those that regulate cell death and survival, cell movement, and cell development, growth, and proliferation, as well as genes involved in the Wnt/β-catenin and the G1/S cell cycle phase checkpoint pathways [24].
Obesity and the metabolic syndrome are associated with multiple factors that may cause an increased risk for cancer and cancer-related mortality [25]. One example is the insulin family of proteins which have pleiotropic effects on metabolism and growth. A large body of evidence indicates the insulin/insulin-like growth factor (IGF-1, IGF-2) pathway in breast cancer progression [25–29]. Of note, IGF-2 occurs in an unprocessed (pIGF-2) and mature (mIGF-2) form and plays a role in breast cell proliferation and inhibition of apoptosis [28–33]. Under normal conditions, IGF-2 is tightly bound and sequestered [34–38], but overexpression of IGF-2 is associated with breast cancer development and increased tumor formation [39, 40]. Most human cancers overexpress both the IGF-1 receptor (IGF-1R) and insulin receptor (IR) isoforms, leading to the formation of hybrid IGF-1R/IRs. IGF-2 is a known ligand for these receptors as well as the mannose-6-phosphate/IGF-2R and high-affinity binding proteins [25–29, 32]. IGF-2 expression is strongly enhanced in invasive breast cancers and downstream mTOR signaling is stimulated [41] as is TNBC cell migration [29]. Of note, disparities in the expression of IGF-2 and its receptors are reported to occur in breast tissue samples from African American women as compared to Caucasian women and may contribute to differences in clinical outcomes [42]. IGF-2 is detected in both tumor stroma and epithelial breast cancer cells and correlates with both breast epithelial [27, 43–45] and stromal cell proliferation [46]. Westley and May [26] also report that estrogen signaling may cross communicate with IGF pathways, with estrogen promoting increased breast cancer production of IGF-2.
This report details interactions of ERβ with IGF-2 and other growth factor pathways in TNBC [46–52]. Our findings using TNBC models and archival specimens suggest that IGF-2 may regulate ERβ expression which in turn modulates metabolic and growth factor pathways in cancer progression.
2. Materials and Methods
2.1. Breast Cancer Cell Lines
For these studies, we used the following triple-negative breast cancer cell lines (ATCC) which have been previously well characterized as lacking expression of ERα and PR as well as overexpression of HER2 [47, 48]: MDA-MB-231, MDA-MB-435, BT549, HCC38, HCC1143, HCC1937, and HCC1806. As controls, we used MCF-7 (expressing abundant ERα and minimal/no ERβ) and T47D (expresses ERα and more abundant ERβ). Cell cultures were routinely maintained at 37°C in a 5% CO2 incubator using RPMI 1640 media supplemented with penicillin/streptomycin (10,000 units/mL penicillin and 10,000 units/mL streptomycin sulfate) and 10% fetal bovine serum unless other specific media were recommended by the supplier (ATCC). For the MDA-MB-231 cell line, we created stable transfectants with a specific ERβ shRNA producing a knockdown of ERβ. As controls, we used a stable transfectant with a scrambled shRNA and vector control prepared as detailed below (all reagents from Origene).
2.2. Reagents
ERβ ligands for use in these experiments included the following: (a) diarylpropionitrile (DPN), an ERβ agonist (Tocris), (b) 4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP), ERβ antagonist, and (c) 4,4′,4′′-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT), an ERα agonist [49, 50].
2.3. Assays for Cell Proliferation
In experiments to assess proliferative effects of ERβ ligands, cells were grown in phenol red-free, estrogen-free media with 0.1% dextran-coated charcoal-treated- (DCC-) FBS for 48 hours and then treated with selected doses of DPN, PPT, or PHTPP. Cell counts and viability tests (Trypan blue) were done every 24 hours for 3 days. After 72 hours, proliferation was assessed using the BrdU cell proliferation ELISA (Roche). Cell numbers were also assessed initially by cell counts to confirm ELISA data.
2.4. Assays for Growth Factor Secretion
Tumor cells were cultured in estrogen-free media and then treated 20–120 minutes with DPN, followed by harvest of particle-free media and application of established ELISA assays for VEGF, amphiregulin, WNT 10b/12 [51–53], signaling molecules that activate angiogenesis, EGFR, and WNT pathways, respectively, which promote TNBC [1, 52, 53].
2.5. Knockdown of ERβ Expression
To suppress ERβ expression, we used the HuSH 29 mer shRNA constructs (Origene) designed to target human ERβ (ESR2) and included positive and negative controls. Plasmids were designed and validated specifically to knockdown expression of specific genes by RNA interference and allow for enrichment of transfected cells. Each vector expresses a short hairpin RNA (shRNA) under control of the U6 promoter and puromycin resistance gene to select stably transfected cells. Cells were transfected with negative control, scrambled negative control, and shRNA plasmids for ERβ using MegaTran 1.0 transfection reagent (Origene). After 48 hours, cells were replated at low density in the presence of an effective concentration of puromycin. Culture medium was replaced every 2-3 days, with cells replated every week for 2 weeks. As stable transfectants were obtained, we isolated total RNA to identify colonies with optimal ERβ1 knockdown, as confirmed by qRT-PCR and immunoassays. Expression of transcripts was done as before [14] and protein levels of ERβ variants were determined (data not shown). After ERβ1 knockdown, cell proliferation was determined by established methods in the presence of vehicle or specific ERβ ligands.
2.6. ERβ Expression by Quantitative RT-PCR (QRT-PCR)
To assess ERβ transcript levels in TNBC cells [54], total RNA was isolated by the Aurum total RNA mini kit (Bio-Rad). UV spectroscopy and RNA quality indicator (RQI) values obtained from the Experion automated electrophoresis system (Bio-Rad) were used to determine RNA integrity. Primer pairs used for ERβ (Qiagen SA Biosciences): ERβ forward: 5′-GCTCATCTTTGCTCCAGATCTTG-3′ and ERβ reverse: 5′-GATGCTTTGGTTTGGGTGATTG-3. cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad) with 400 ng total RNA. The iQ SYBR Green Supermix (Bio-Rad) was used for PCR amplification. Each reaction was performed in triplicate and ribosomal protein 36B4 mRNA, a housekeeping gene unaffected by estrogen, was used as the internal control. The cycling conditions consisted of 95.0°C for 30 seconds followed by 39 cycles of 95.0°C for 5 seconds, 57.0°C for 15 seconds, and 72.0°C for 90 seconds in a CFX96 Touch Thermocycler (Bio-Rad). Transcript levels of ERβ were normalized to 36B4. Fold induction or repression was measured relative to controls and calculated after adjusting for 36B4 RNA (endogenous control) using 2−ΔΔCt, where ΔCt = Ct interested gene − Ct 36B4 RNA and ΔΔCt = ΔCt treatment − ΔCt vehicle control. For experiments that evaluated IGF-2 effects on ERβ, cell lines were seeded and cultured to 70–75% confluence in complete media followed by 24 hours in serum/phenol red-free media. Following serum starvation, cultures were treated with 100 ng/mL of human recombinant pIGF-2 or mIGF-2 (GroPep) for 24 hours in serum-free, phenol-red-free media before total RNA isolation.
2.7. Gel Electrophoresis and Immunoblotting
TNBC cells were maintained in estrogen-free conditions 48 hours before experiments. Cells were then incubated with vehicle control or 10 nM DPN for 15 minutes or 24 hours and then harvested and lysed. Total cell proteins were resolved by 4–15% SDS-PAGE, transferred to polyvinylidene difluoride membranes, and probed with antibodies directed against phosphotyrosine-1068-EGFR (Cell Signaling Technology D7A5), total EGFR (Calbiochem, GR15), or HER-3 (Santa Cruz Biotechnology, C17). Phospho-p44/p42 MAPK (Thr202/Tyr204; #9101), total MAPK (#9102), phospho-mTOR (Ser-2448, clone D9C2), and total mTOR (7C10) antibodies were from Cell Signaling Technology. Proteins were detected by using horseradish peroxidase (HRP) conjugated secondary antibodies and Thermo Scientific Pierce ECL Western Blotting Substrate with enhanced chemiluminescent for detection of activity from HRP. Membranes were stripped and reprobed with β-actin as a loading control; anti-beta-actin antibody C4 was from Santa Cruz Biotechnology (sc-47778). Immunoblots shown in figures are a representative of at least three independent experiments.
2.8. Patients and Analyses of Archival TNBC Specimens
Overall 19 TNBC cases were available for this study, with 14 provided by tumor banks associated with the UCLA Early Detection Research Network [55, 56] and the Division of Cancer Research at Charles Drew University School of Medicine and Science (CDU) [57]. Patient specimens were obtained from archival breast cancer studies between 1995 and 2007. This study was approved by our Institutional Review Boards and written informed consent was obtained from all participants. TNBC was confirmed by surgical biopsy/pathology and follow-up data. Of the 14 UCLA-CDU cases, each is linked to deidentified clinical and outcome data. Follow-up time was performed up to 5 years. The specimens include those from 11 non-Hispanic Caucasian and 3 non-Hispanic African American female patients. General characteristics of these patients were reported previously [55–57].
An additional 15 specimens were provided by the NCI-supported Cooperative Human Tissue Network (http://www.chtn.nci.nih.gov/). These archival specimens included 5 cases from female patients with TNBC as well as controls for comparison from female patients with ERα-negative breast cancer (non-TNBC) (n = 5) and ERα-positive breast cancer (n = 5). Overall, all histologies included cases with invasive tumors with some associated with metastases. Among the 15 NCI CHTN tumor cases, tissue samples were taken from neighboring regions of nonmalignant tissue (n = 9). All specimens were collected with the appropriate institutional human subject protection committee approvals and patient consent.
Formalin-fixed and paraffin-embedded tissue samples were stained using standard IHC protocols as before [55–62]. Antibodies used included ERα (clone 1D5, DAKO); ERβ, anti-ERβ (GeneTex); anti-ERβ-1 clone PPG5/10 (AbD Serotec); IGF-2 (Abcam); EGFR; and Ki-67 (DAKO). Appropriate controls were included to assess specificity and validate each antibody including (i) appropriate preimmune control, (ii) dose-dependent titration, (iii) known positive and negative tissues, (iv) use of specific peptide competitors, and (v) other established approaches in our laboratory [58, 62]. For IHC, antibody binding was detected by using the “Envision+” System-HRP (DAB) followed by chromogen detection with diaminobenzidine (DAKO). Sections were counterstained with Harris hematoxylin, followed by dehydration through graded alcohol solutions, and mounted. Images were captured by using an Olympus BX41 microscope hooked to a Pixera Pro 150 ES or Evos xl Core microscope. Care was taken to evaluate staining of specific target structures only. Both intensity of staining and percentage of cells were recorded for both neoplastic and normal cells expressing antigen in both nuclear and extranuclear localizations. Board-certified pathologists quantified expression as before [55–58]. We used an Allred scoring system to quantify expression in tumors [62]. Of note, stromal expression of growth factors or steroid receptors in endometrial cancer are reported to be biomarkers to predict response to hormonal therapy [63]. Hence, we explored stromal as well as tumor expression of selected biomarkers, particularly ERβ1 and IGF-2 (Abcam).
2.9. Statistical Analyses
For in vitro work, experiments were done in triplicate. Student's t-test, ANOVA, or Kruskal-Wallis test, if outcomes were nonnormally distributed, was used to compare intervention groups. Analyses were evaluated using bar and scatter graphs with means, SD, and SE. Time trend curves for agents under different conditions were obtained as appropriate. Repeated measures ANOVA were used to assess time, condition, and time by condition interaction effects. P < 0.05 was considered significant. For analyses of clinical TNBC specimens, we used methods and protocols for assessing novel associations with clinical/pathological variables as well as marker associations as detailed before [56–62]. The log-rank test was used to compare overall survival (OS) between those subjects with positive nuclear ERβ1 expression score versus those with negative scores.
3. Results
3.1. ERβ Expression in Archival TNBC Specimens
Several reports indicate that ERβ expression in node-positive breast cancer is a biomarker for more aggressive disease [8, 21]. For these studies, we used 14 archival TNBC specimens with demographic characteristics including American Joint Committee on Cancer (AJCC) stages provided in Table 1. Using immunohistochemistry, specific ERβ1 staining was observed and scored in nuclear sites, with our observations noted in Table 2. Representative immune staining patterns are shown in Figure 1. Of note, we also observed evidence of diffuse extranuclear ERβ1 staining in most specimens but this staining was not scored or analyzed for this report. In further analyses, we assessed the clinical outcome of 14 patients with a 5-year follow-up whose tumors expressed or did not express ERβ1. In this group of patients with advanced TNBC, overall survival (OS) was significantly worse for TNBC patients with high nuclear ERβ1 (positive) as compared to those with low (negative) ERβ1 (P < 0.001; see Figure 2). Finally, we note that, of the African American patients included in our sample of 14 TNBC patients (11 Caucasian and 3 African American women), all three had tumors that were ERβ1-positive.
Table 1.
Demographic characteristics.
| Variable | N (%) |
|---|---|
| Race | |
| African American | 3 (21.4) |
| Caucasian | 11 (78.6) |
| AJCC stage | |
| 0-I | 0 |
| II | 7 (50) |
| III-IV | 7 (50) |
| Ki-67 status | |
| Low | 3 (21.4) |
| High | 11 (78.6) |
| Age, mean years (range) | |
| <50 yrs | 8 (32–58) |
| >50 yrs | 6 (53–65) |
Table 2.
ERβ1 biomarker distribution.
| Variable | N (%) |
|---|---|
| Nuclear ERβ1 primary tumor | |
| Positive | 8 (57.1) |
| Negative | 6 (42.8) |
Figure 1.

ERβ1 expression in archival TNBC specimens. Representative examples are shown of IHC staining of tumor and nonmalignant tissue specimens using anti-ERβ1 antibody (AbDSerotec PPG5/10). (a) TNBC specimen shows nuclear (and cytoplasmic) immunostaining of ERβ1 at low magnification. (b) The same TNBC specimen shows nuclear (and cytoplasmic) immunostaining of ERβ1 at higher magnification. (c) Expression of nuclear ERβ1 is also observed in neighboring nonmalignant mammary tissue from the clinical specimen used in panels (a) and (b). (d) Negative ERβ1 detected in a different clinical specimen as shown as comparison. Antibody binding was detected by using the “Envision+” amplification system followed by chromogen detection with diaminobenzidine (DAKO). Sections were counterstained with Harris hematoxylin followed by dehydration through graded alcohol solutions and mounting. See Table 2 for a summary of findings on all TNBC cases examined.
Figure 2.
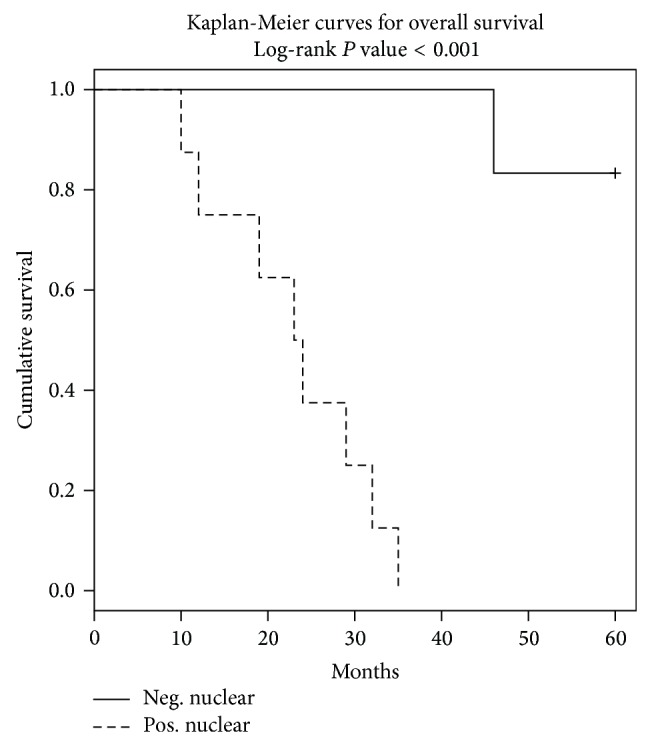
ERβ1 expression reduces overall survival (OS) in TNBC. TNBCs from 3 African American and 11 Caucasian women were scored for nuclear ERβ1 using IHC with validated antibody (see Table 2). Allred scores >2 are denoted as positive. In this group of patients with advanced TNBC, overall survival (OS) was significantly worse for TNBC patients with high nuclear ERβ1 (positive) as compared to those with low (negative) ERβ1 (P < 0.001). We noted that TNBCs from all 3 African American women were ERβ1-positive.
3.2. ERβ Expression in Breast Cancer Cell Lines
We assessed ERβ expression in a panel of established TNBC [47, 48] and control breast cancer cell lines, MCF-7 and T47D. All TNBC cell lines were confirmed to express ERβ. Lysates of reported TNBC cells (MDA-MB-231, MDA-MB-435, BT549, HCC-38, HCC-1143, and HCC-1937) and nuclear ERα-positive controls (MCF-7, T47D) were subjected to gel electrophoresis and immunoblots with anti-ERβ antibody and anti-ERα antibody. Results are shown in Figure 3. T47D cells were also used as a positive control for ERβ expression. These findings indicate significant expression of ERβ TNBC cell lines.
Figure 3.
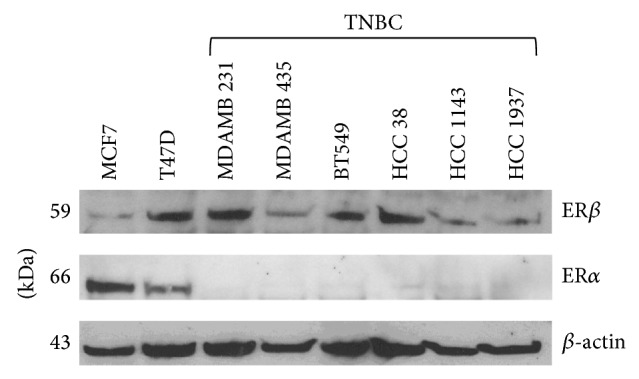
ERβ is expressed in TNBC cells. Lysates of reported TNBC cells (MDA-MB-231, MDA-MB-435, BT549, HCC38, HCC1143, and HCC1937) and nuclear ERα-positive controls (MCF-7, T47D) were subjected to polyacrylamide gel electrophoresis and immunoblots with anti-ERβ antibody (D7N, Zymed/Invitrogen; confirmed with GeneTex ERβ1 antibody (not shown)) and anti-ERα antibody (1D5, DAKO). T47D cells are also a positive control for ERβ expression. β-actin is used as a loading control. Methods were as before [60, 61]. Blot shown is a representative of at least three independent experiments.
3.3. ERβ Regulates Breast Cancer Cell Proliferation
MDA-MB-231 TNBC cells were stably transfected with an empty control vector, nonspecific shRNA scrambled sequence control plasmid, or an ERβ-specific shRNA plasmid to suppress ERβ expression. Evidence of expected molecular alterations in ERβ protein expression in MDA-MB-231 cells is shown in Figure 4(a). Transfected cells were treated with known estrogen receptor agonists and/or antagonists [49, 50] including DPN (ERβ agonist), PPT (ERα agonist), and PHTPP (ERβ antagonist) alone. As shown in Figure 4(b), PHTPP decreased proliferation in ERβ-expressing cells but not in cells where ERβ expression was suppressed. Notably, DPN (ERβ agonist) increased proliferation only in cells expressing ERβ (Figure 4(b)). These results are consistent with the hypothesis that ERβ1 plays a significant role in modulating TNBC proliferation. This finding is consistent with reports that associate ERβ1 expression with Ki-67, a marker of cell proliferation, in ERα-null tumors [17].
Figure 4.

Blockade of ligand-induced proliferation of triple-negative MDA-MB-231 cells by ERβ antagonists and by ERβ shRNA. (a) Estrogen receptor-β knockdown. MDA-MB-231 TNBC cells were transfected with shRNA control (CON), ERβ-targeted shRNA (1, 2, 3), and scrambled shRNA vectors (SCRB) (Origene #TG320347). Stable transfectants were selected using puromycin. Cell lysates were processed for Western blot, with results shown for a negative control (CON), 3 different ERβ knockdown clones (1, 2, and 3), and scrambled shRNA (SCRB). β-actin was used as a loading control. (b) Blockade of ligand-induced proliferation of triple-negative MDA-MB-231 cells by ERβ antagonists and by ERβ shRNA. Cells were stably transfected with empty vector (white bars), nonspecific shRNA scrambled-sequence plasmid (grey bars), or an ERβ shRNA plasmid (black bars). As indicated in the figure, cells were treated with different ligands: DPN (ERβ1 agonist), PPT (ERα agonist), and PHTPP (ERβ antagonist) [49, 50]. Cell proliferation was determined after 72 hours using the BrdU cell proliferation ELISA (Roche) (n > 3). Graph shows percentage of surviving cells relative to untreated controls (vehicle control), defined as 100% for each transfection condition: empty vector, scrambled, and ERβ shRNA. * P = 0.007, ** P = 0.005, and *** P = 0.006. Data represents at least three independent experiments.
3.4. ERβ1 Stimulates Secretion of Growth Factors in TNBC
To investigate potential cross talk between ERβ1 and growth factor signaling pathways [15] which may promote TNBC progression (such as VEGF, amphiregulin, and WNT 10b/12) [1, 15, 52, 53, 64], we cultured TNBC cells in the presence and absence of DPN. Using established ELISA methods as detailed before [51], levels of selected secreted growth factors were assayed in particle-free, extracellular media (Figure 5). DPN promoted secretion of several critical growth factors (P < 0.001).
Figure 5.
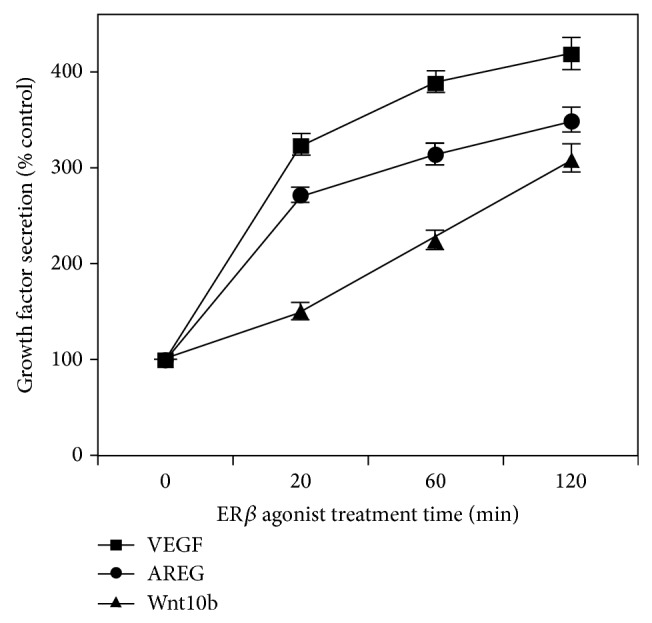
ERβ1 stimulates secretion of VEGF, amphiregulin, and WNT 10b/12. TNBC cells (MDA-MB-231) were cultivated in estrogen-free media and then treated for 20–120 minutes with ERβ agonist (10 nM DPN), followed by harvesting of medium and ELISA assays for VEGF, amphiregulin (AREG), and WNT 10b/12, signaling molecules that are each reported to contribute to the progression of TNBC [52, 53, 64]. Assays were done in triplicate in 3 independent experiments. DPN promoted secretion of several critical growth factors (all at P < 0.001) as compared to appropriate controls.
3.5. Activation of ERβ1 Correlates with an Increase in EGFR Expression and Activation of Downstream Signaling Pathways
Since EGFR is known to be expressed and active in many TNBCs [1, 64], we explored cross communication between ERβ activation and EGFR in TNBC cells. MDA-MB-231 cells treated with DPN for 15 minutes showed increased phosphorylation of Tyr1068-EGFR as well as downstream signaling demonstrated by phosphorylation of p44/p42-MAPK and phospho-Ser2448-mTOR (see Figure 6(a)). Further, TNBC cells treated with DPN for 24 hours had increased EGFR protein levels but levels of HER-3, a related EGFR family member, were not increased (Figure 6(b)). Our findings in Figure 5 that note ERβ-induced promotion of amphiregulin, a known EGFR ligand, as well as EGFR expression (Figure 6(b)) implicate a cascade that could potentially promote downstream EGFR signaling modules such as the Ras/Raf/MEK/ERK1/2 and mTOR pathway for TNBC progression [15, 65]. Hence, ERβ1 elicits increased phosphorylation of EGFR, as well as activation of MAPK and mTOR.
Figure 6.

Estrogen receptor-β agonist DPN promotes expression and activation of EGFR and downstream signaling in MDA-MB-231 cells. (a) Cells were treated with DPN for 15 minutes. Thereafter, cells were lysed and processed for gel electrophoresis and Western immunoblot using antibodies against phosphotyrosine-1068- and total-EGFR, phospho-p44/42- and total-MAPK, and phosphoserine-2448- and total-mTOR. These data are consistent with independent reports on DPN activity [66]. (b) MDA-MB-231 cells were treated with DPN for 24 hours. Then, cells were lysed, processed for gel electrophoresis and Western immunoblot using antibodies to EGFR and HER-3. β-actin was the loading control. Blot shown is representative of at least three independent experiments.
3.6. IGF-2 Stimulates ERβ Transcription
To evaluate potential effects of unprocessed or big (pIGF2) and processed (mIGF2) IGF-2 on ERβ transcription, we used a real-time PCR approach. As compared to T47D cells, which express both ERα and ERβ, higher transcript levels of ERβ were stimulated by mIGF-2 in HCC 1806 and MDA-MB-231 cell lines (Figure 7). These findings indicate a role of IGF-2 in ERβ transcription.
Figure 7.
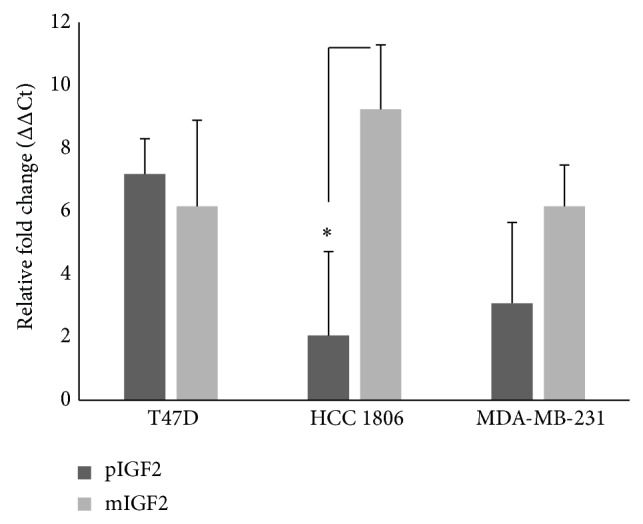
IGF-2 modulates the transcription of ERβ in HCC 1806 and MDA-MB-231 cell lines. Cells were seeded and then cultured for 24 hours in serum-free, phenol red-free media. Following serum starvation, cultures were treated with 100 ng/mL of pIGF-2 or mIGF-2 (GroPep). Following a 24-hour incubation period, total RNA was extracted and qRT-PCR was performed. Data presented are the result of at least six independent experiments performed in triplicate. Statistical significance was determined using Student's t-test (Graphpad). Error bars: SE. * P value = 0.05.
3.7. IGF-2 and ERβ1 Expression in Archival TNBC Specimens
IGF-2 is a secreted protein highly expressed in breast tumor tissue. To assess the association of elevated IGF-2 with a specific tumor subtype, we evaluated IGF-2 expression levels in epithelial and neighboring stromal components of ERα-positive (ER+), ERα-negative (ER−), and TNBC tumors. Following pathology review, we obtained archival tissue from 5 patients in each subtype. The patients selected ranged in age from 35 to 61 years (average age 50) with additional demographic characteristics shown in Table 3. In comparison to ER+ and ER− epithelium, TNBC epithelia expressed significantly higher levels of IGF-2 (P < 0.01; see Figure 8(a)). Additionally, IGF-2 staining in neighboring stromal cells was highest in TNBCs as compared to ER− tumors (P = 0.05) and ER+ tumors (P < 0.01; see Figure 8(b)). These results support a potential apocrine and/or paracrine role of IGF-2 in cancer progression.
Table 3.
Demographic characteristic of archival ER+, ER−, and TNBC breast samples.
| Variable | ER+ | ER− | TNBC |
|---|---|---|---|
| Race | ERα+/PR+/HER2+ | ERα−/PR−/HER2+ | ER−/PR−/HER2− |
| African American | 3 (60%) | 1 (20%) | 2 (40%) |
| Caucasian | 2 (40%) | 4 (80%) | 3 (60%) |
| AJCC stage | |||
| 0-I | 2 (40%) | None | 1 (20%) |
| II | 2 (40%) | None | None |
| III-IV | 1 (20%) | 5 (100%) | 4 (80%) |
| Ki-67 status | |||
| Not recorded | 4 (80%) | 1 (20%) | 1 (20%) |
| Low | 1 (20%) | 1 (20%) | 1 (20%) |
| High | 3 (60%) | 3 (60%) | |
| Metastasis | |||
| Not recorded | 1 (20%) | ||
| Positive | 2 (40%) | 3 (60%) | 2 (40%) |
| Negative | 3 (60%) | 2 (40%) | 2 (40%) |
| Age, mean years (range) | |||
| <50 yrs | 3 (45–49) | 2 (35–49) | 2 (45–49) |
| >50 yrs | 2 (50–70) | 3 (50–70) | 3 (50–70) |
Figure 8.
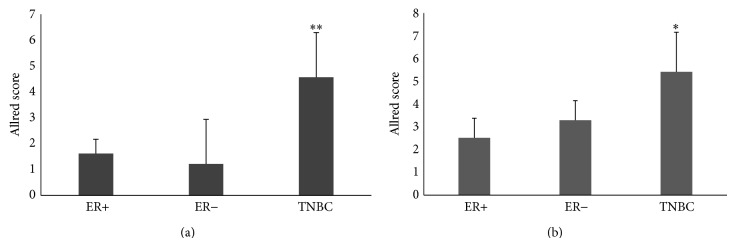
High levels of IGF-2 occur in archival TNBC tumors as compared to ERα+ and ERα− breast cancers. Archival breast tissues from patients with ERα-positive (ER+, n = 5), ERα-negative (ER−, n = 5), and TNBC (n = 5) breast cancer were evaluated for IGF-2 expression. Validated IHC staining methods were used to evaluate IGF-2 expression in tumor epithelium (a) and neighboring stroma (b). Scoring was done using an Allred-based criterion. (a) Significantly higher expression of IGF-2 is found in the epithelium of TNBC tumors as compared to ER+ (** P < 0.01) and ER− breast cancers (** P < 0.01). (b) Compared to ER+ and ER− archival breast tumors, stromal tissue adjacent to TNBC tumors expressed significantly higher levels of IGF-2 than stromal tissue neighboring non-TNBCs (** P < 0.01 and * P < 0.05 for ER+ and ER−, resp.). Error bars: SD.
Since the expression of IGF-2 and ERβ1 is associated with poor patient outcomes, we evaluated their coexpression in normal breast tissue and TNBC tumors (Figure 9). Using immunohistochemistry, archival TNBC tumors and adjacent nonmalignant breast specimens were evaluated for IGF-2 and ERβ1 expression (Figure 9(a)). In the samples evaluated and scored, notably increased expression of IGF-2 (P < 0.01) and ERβ1 (P < 0.005) is found in all TNBC tumors examined (Figure 9(b)).
Figure 9.

IGF-2 and ERβ1 are highly expressed in TNBC tumors. Archival breast tissue from patients with TNBC along with adjacent normal tissue adjacent to TNBC tumors was obtained from the NIH CHTN and UCLA Early Detection Research Network. Immunohistological assays were used to evaluate tissue samples for IGF-2 and ERβ1 expression (see Section 2). (a) Representation of IGF-2 and ERβ1 expression in normal and TNBC breast tissue. IGF-2 (A and B) and ERβ1 (C and D) expression were evaluated and visualized using Evos xl Core microscope. Representative images are presented. (b) Allred scoring of IGF-2 and ERβ1 in archival breast tissue. Samples from neighboring normal tissue (n = 7) and from TNBC tumors (n = 11) were stained for IGF-2 and ERβ1 then scored using Allred-based criterion (see Section 2). IGF-2 (** P < 0.01) and ERβ1 (*** P < 0.005) were notably expressed in TNBC tumor samples. Statistical significance was determined using Student's t-test (Graphpad). Error bars: SE.
4. Discussion
The discovery of ERβ and its expression in TNBC raised hope that targeting ERβ might offer new treatment options for TNBC patients where previously only aggressive chemotherapies were available [1, 2, 17]. However, these predictions have yet to be realized. Some investigators report that ERβ is a favorable prognostic factor [13] or tumor suppressor [10], while others find that ERβ correlates with aggressive phenotypes and worse prognosis [8, 14, 17, 58]. These differences may be due in part to use of nonspecific antibodies to assay ERβ and some studies of archival specimens lack validation of a true TNBC phenotype [17, 20, 21]. To address these problems, we used validated ERβ antibodies [58, 67] and established TNBC specimens (ERα-negative/PR-negative/HER2-overexpression-negative). Our findings support earlier reports on the prognostic potential of ERβ isoforms in TNBC [7, 8, 11, 17, 20, 21, 68, 69], particularly ERβ1. Our data show specific staining for ERβ1 isoform in TNBC specimens, a finding consistent with other recent reports that both ERβ1 and ER coregulator SRAP are predictive biomarkers of tamoxifen-response/benefit in women with ERα-negative breast cancer [17, 70]. It is independently reported that high levels of nuclear ERβ2 are associated with lymph node involvement and serve as an independent biomarker for early tumor relapse in ERα-negative breast cancers, particularly in TNBC subgroups [69]. Since ERβ1 and ERβ2 tend to correlate positively, ERβ2 expression may contribute in part to this finding. This may be critical as ERβ2 can heterodimerize with ERβ1 and modulate gene expression [22, 68]. It will be important in future work to assess the ratio of ERβ1 : ERβ2 expression in a larger sample of TNBC specimens and to correlate receptor protein and transcript levels with treatment outcomes [21]. Expression of ERβ1 is of particular interest because it is the only ERβ isoform that contains an intact LBD, thereby serving as a potential drug target in the clinic [17, 21, 22].
In pursuit of new approaches to treat TNBC, it is important to consider results of some recent studies on TNBC and related ERα-negative disease. Our data appear consistent with independent reports of ERβ1 as a biomarker for improved survival in TNBC patients when treated with tamoxifen [13, 17, 21, 70]. Although previous work suggests that tamoxifen use only reduced the risk of ERα-positive breast cancer, Yan et al. [17] report that both ERβ1 and ER coregulator SRAP are predictive biomarkers of tamoxifen-response/benefit in women with ERα-negative breast cancer. In ERα-negative tumors, ERβ1 expression correlates with Ki-67 proliferation marker suggesting that ERβ1 may have a role in driving proliferation; thus antiestrogen treatment aimed at ERβ1 inhibition could slow tumor progression [17]. With the discovery of ERβ expression alone in TNBC, this presents the possibility that, in this ERα-negative cohort, antiestrogen strategies may potentially mediate activity via ERβ1, the full-length ligand-binding receptor isoform [57, 71, 72].
Although earlier work confirmed that ERα-positive breast cancer cells are more sensitive than ERα-negative breast cells to the growth-inhibitory effects of tamoxifen, moderate antiproliferative responses to tamoxifen and to ICI-164,384 are found in ERα-negative cells [73–75]. These effects may be modulated in part by a second unique binding site identified for hydroxytamoxifen in the coactivator-binding groove of ERβ that may disrupt ERβ-coactivator interactions [76]. Collectively, these reports have important implications since approved breast cancer treatments (tamoxifen, raloxifene) are largely well tolerated and orally administered. Such medications may be alternative treatments for ERβ-positive TNBC patients with generally few options other than cytotoxic chemotherapy [1, 17, 21]. A clinical trial to test this hypothesis in TNBC patients with ERβ-positive status was recently launched [77].
We note that ERβ activity is generally considered antagonistic to that of ERα when both receptors occur together in a cell, but, in isolation, the role of ERβ forms is not well-documented. Molecular studies show that when both ERα and ERβ are present together in tumor cells, each ER restricts the binding site occupancy of the other, with ERα generally being dominant to ERβ. It is clear that ERβ binding and actions in gene regulation are different in the absence of ERα expression in breast tumor cells [78]. Indeed, correlation of ERβ with high proliferative biomarkers is reported in ERα-negative tumors but not in those with ERα expression [13, 17]. Among the first studies on stable transfection of ERβ in MDA-MB-231 cells, it was determined that the proliferation rate of the tumor cells positively correlates with the level of ERβ expression [23]. Results using stable ERβ clones in ERα-negative tumor cells demonstrate increased proliferation as ERβ expression increased. A confirmation of these experiments was published when MDA-MB-435 cells transfected with ERβ led to significant stimulation of tumor progression as well as metastasis in vivo [79]. These reports support ERβ stimulated tumor proliferation in ERα-negative breast cancer cells [23, 79] and complement the data presented in this study. In contrast, in other studies with ERβ transfection in ERα-low or -negative breast cancer cells, cell proliferation was inhibited [80, 81]. Several reasons may explain such contrasting results. For example, transfection of ERα led to the paradoxical finding that ERα was a growth inhibitor in breast cancer, a result that is clearly inconsistent with established clinical findings [82–84]. This unexpected result and similar ERβ transfection data may be due, in part, to excessive levels of ER expression in the model systems used or other complicating factors [23]. Further, conflicting data could be the result of differences in ERβ1 and ERβ2 expression/activity. High nuclear ERβ2 is an independent marker of early relapse in ERα-negative breast cancer and especially TNBC [69]. As noted above, it will be important to assess the impact of ERβ2 [22]. It is possible that ERβ1 and ERβ2 are both necessary partners in promoting aggressive TNBC [85]. Finally, there are conflicting reports on activity of ERβ-specific ligands in TNBC as well as other tumor types [86, 87]. ERβ1 expression correlates with tumor proliferation and progression in lung cancer [58, 87–89] but not in colon cancer [90]. An important new report with direct relevance to TNBC shows that under basal conditions ERβ agonists induced apoptosis in breast cancer cells [65]. However, when extracellular signal-regulated kinases 1 and 2 (ERK1/2) were activated by coincubation with epidermal growth factor (EGF), ERβ1 agonist DPN induced proliferation in breast cancer cells [65]. Such results indicate EGFR-induced signaling activity, known to be frequently overexpressed and active in TNBCs, can modulate ERβ growth-promoting effects [15, 91].
Emerging evidence highlights the increased frequency of obese patients afflicted with TNBC [25, 92–94] and a recent meta-analysis indicates that obese women are at a greater risk of presenting with a TNBC than nonobese women [95]. The insulin family of growth factors are notably critical mediators of metabolic factors [25, 93]. IGF-2 expression is strongly enhanced in invasive breast cancers and stimulates downstream mTOR signaling and TNBC cell migration [29, 41]. An IGF gene signature including those involved in cell growth, survival, metabolism, and biosynthesis is activated in TNBC and ERα-negative breast tumors [96, 97]. Of note, this IGF signature altered mRNA levels of numerous members of both the PI3K and ERK1/2 pathways with enrichment for transcriptional targets of PI3K/Akt/mTOR and EGFR/Ras/MAPK pathways.
Our findings show that ERβ transcripts are stimulated severalfold in TNBC cells treated with IGF2 and elevated IGF-2 expression occurs in both archival TNBC and neighboring stromal cells. These data are consistent with independent reports that detected IGF-2 in both tumor stroma and epithelial breast cancer cells and correlated IGF-2 expression with breast epithelial [27, 43–45] and stromal cell proliferation [46]. Westley and May [26] also report cross communication between estrogen and IGF pathways, with estrogen promoting increased production of IGF-2. Extensive cross talk between the IGF signaling pathway and two major growth regulators in breast cancer, namely, ER and EGFR, is detailed in numerous studies [15, 33]. Previous ideas about why patients with TNBC have more aggressive tumors that relapse earlier than patients with other types of breast cancer have focused only on tumor cell-intrinsic properties. Our results support the notion that the host systemic environment and local stromal cell niches may also play a role in tumor behavior [98]. Cross talk between epithelium and stroma is essential for neoplastic transformation in many hormonally regulated tissues [99, 100]. One way this cross talk occurs is through reciprocal signals regulating hormone receptor expression or activity in each cellular compartment. In patients with TNBC, progression of tumor cell populations may depend in part on complex interactions and the bioavailability of IGF-2 and ERβ [98].
5. Conclusions
Results of this study indicate that ERβ1 expression in archival TNBC specimens associates with significantly worse 5-year overall survival, a common characteristic of this disease. Significant expression of ERβ in a panel of TNBC cell lines is consistent with these findings. Of note, IGF-2, known to associate with breast cancer promotion, is expressed in TNBC and neighboring cells in archival clinical specimens and stimulates increased levels of ERβ mRNA in TNBC cells. Using shRNA to silence ERβ expression, we find that ERβ suppression significantly reduced TNBC proliferation. ERβ-specific antagonists similarly reduced TNBC growth. In TNBC, ERβ may stimulate growth through downstream actions that promote VEGF, amphiregulin, and Wnt-10b secretion which in turn activate specific receptor signaling pathways known to be associated with TNBC progression. This work may further our understanding on the interplay of metabolic and growth factors in TNBC, and, hopefully, lead to new therapeutics to manage patients afflicted with this deadly disease.
Acknowledgments
This work was supported by funds from the National Institutes of Health (NIH)/National Cancer Institute U54 CA14393 Partnership to Eliminate Cancer Health Disparities, NIH CA-86366 Early Detection Research Network, NIH/National Center for Advancing Translational Science (NCATS) UCLA CTSI Grant no. UL1TR000124, California Breast Cancer Research Program IDEA Awards 16IB-0042 and 18IB-0034, UCLA Jonsson Cancer Center Foundation Transdisciplinary Research Grant, Stiles Program in Integrative Oncology, Hickey Family Foundation, and the Robert Wood Johnson Foundation Nurse Faculty Scholar Award 69352.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Nalo Hamilton and Diana Márquez-Garbán contributed equally to this work.
References
- 1.Foulkes W. D., Smith I. E., Reis-Filho J. S. Triple-negative breast cancer. The New England Journal of Medicine. 2010;363(20):1938–1948. doi: 10.1056/nejmra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Morris G. J., Naidu S., Topham A. K., et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the national cancer institute's surveillance, epidemiology, and end results database. Cancer. 2007;110(4):876–884. doi: 10.1002/cncr.22836. [DOI] [PubMed] [Google Scholar]
- 3.Amirikia K. C., Mills P., Bush J., Newman L. A. Higher population-based incidence rates of triple-negative breast cancer among young African-American women. Implications for breast cancer screening recommendations. Cancer. 2011;117(12):2747–2753. doi: 10.1002/cncr.25862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stark A., Kleer C. G., Martin I., et al. African ancestry and higher prevalence of triple-negative breast cancer. Cancer. 2010;116(21):4926–4932. doi: 10.1002/cncr.25276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madak-Erdogan Z., Kieser K. J., Sung H. K., Komm B., Katzenellenbogen J. A., Katzenellenbogen B. S. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Molecular Endocrinology. 2008;22(9):2116–2127. doi: 10.1210/me.2008-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists' Collaborative Group. The Lancet. 1998;351(9114):1451–1467. doi: 10.1016/S0140-6736(97)11423-4. [DOI] [PubMed] [Google Scholar]
- 7.Skliris G. P., Leygue E., Curtis-Snell L., Watson P. H., Murphy L. C. Expression of oestrogen receptor-β in oestrogen receptor-α negative human breast tumours. British Journal of Cancer. 2006;95(5):616–626. doi: 10.1038/sj.bjc.6603295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Novelli F., Milella M., Melucci E., et al. A divergent role for estrogen receptor-beta in node-positive and node-negative breast cancer classified according to molecular subtypes: an observational prospective study. Breast Cancer Research. 2008;10(5, article R74) doi: 10.1186/bcr2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jensen E. V., Cheng G., Palmieri C., et al. Estrogen receptors and proliferation markers in primary and recurrent breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(26):15197–15202. doi: 10.1073/pnas.211556298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartman J., Strom A., Gustafsson J. A. Estrogen receptor beta in breast cancer-diagnostic and therapeutic implications. Steroids. 2009;74(8):635–641. doi: 10.1016/j.steroids.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Chen J.-Q., Russo J. ERα-negative and triple negative breast cancer: molecular features and potential therapeutic approaches. Biochimica et Biophysica Acta. 2009;1796(2):162–175. doi: 10.1016/j.bbcan.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phipps A. I., Chlebowski R. T., Prentice R., et al. Reproductive history and oral contraceptive use in relation to risk of triple-negative breast cancer. Journal of the National Cancer Institute. 2011;103(6):470–477. doi: 10.1093/jnci/djr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honma N., Horii R., Iwase T., et al. Clinical importance of estrogen receptor-β evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. Journal of Clinical Oncology. 2008;26(22):3727–3734. doi: 10.1200/jco.2007.14.2968. [DOI] [PubMed] [Google Scholar]
- 14.Poola I., Fuqua S. A. W., de Witty R. L., Abraham J., Marshallack J. J., Liu A. Estrogen receptor α-negative breast cancer tissues express significant levels of estrogen-independent transcription factors, ERβ1 and ERβ5: potential molecular targets for chemoprevention. Clinical Cancer Research. 2005;11(20):7579–7585. doi: 10.1158/1078-0432.ccr-05-0728. [DOI] [PubMed] [Google Scholar]
- 15.Pietras R. J., Márquez-Garbán D. C. Membrane-associated estrogen receptor signaling pathways in human cancers. Clinical Cancer Research. 2007;13(16):4672–4676. doi: 10.1158/1078-0432.CCR-07-1373. [DOI] [PubMed] [Google Scholar]
- 16.Hammes S. R., Levin E. R. Extranuclear steroid receptors: nature and actions. Endocrine Reviews. 2007;28(7):726–741. doi: 10.1210/er.2007-0022. [DOI] [PubMed] [Google Scholar]
- 17.Yan Y., Li X., Blanchard A., et al. Expression of both estrogen receptor-beta 1 (ER-β1) and its co-regulator steroid receptor RNA activator protein (SRAP) are predictive for benefit from tamoxifen therapy in patients with estrogen receptor-alpha (ER-α)-negative early breast cancer (EBC) Annals of Oncology. 2013;24(8):1986–1993. doi: 10.1093/annonc/mdt132. [DOI] [PubMed] [Google Scholar]
- 18.Wessler S., Otto C., Wilck N., Stangl V., Fritzemeier K.-H. Identification of estrogen receptor ligands leading to activation of non-genomic signaling pathways while exhibiting only weak transcriptional activity. The Journal of Steroid Biochemistry and Molecular Biology. 2006;98(1):25–35. doi: 10.1016/j.jsbmb.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Paech K., Webb P., Kuiper G. G. J. M., et al. Differential ligand activation of estrogen receptors ERα and ERrβ at AP1 sites. Science. 1997;277(5331):1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 20.Shaaban A. M., Green A. R., Karthik S., et al. Nuclear and cytoplasmic expression of ERβ1, ERβ2, and ERβ5 identifies distinct prognostic outcome for breast cancer patients. Clinical Cancer Research. 2008;14(16):5228–5235. doi: 10.1158/1078-0432.ccr-07-4528. [DOI] [PubMed] [Google Scholar]
- 21.Murphy L. C., Leygue E. The role of estrogen receptor-β in breast cancer. Seminars in Reproductive Medicine. 2012;30(1):5–13. doi: 10.1055/s-0031-1299592. [DOI] [PubMed] [Google Scholar]
- 22.Leung Y. K., Mak P., Hassan S., Ho S. M. Estrogen receptor (ER)-beta isoforms: a key to understanding ER-beta signaling. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(35):13162–13167. doi: 10.1073/pnas.0605676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tonetti D. A., Rubenstein R., DeLeon M., et al. Stable transfection of an estrogen receptor beta cDNA isoform into MDA-MB-231 breast cancer cells. Journal of Steroid Biochemistry and Molecular Biology. 2003;87(1):47–55. doi: 10.1016/j.jsbmb.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Shanle E. K., Zhao Z., Hawse J., et al. Research resource: global identification of estrogen receptor β target genes in triple negative breast cancer cells. Molecular Endocrinology. 2013;27(10):1762–1775. doi: 10.1210/me.2013-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belardi V., Gallagher E. J., Novosyadlyy R., Leroith D. Insulin and IGFs in obesity-related breast cancer. Journal of Mammary Gland Biology and Neoplasia. 2013;18(3-4):277–289. doi: 10.1007/s10911-013-9303-7. [DOI] [PubMed] [Google Scholar]
- 26.Westley B. R., May F. E. B. Role of insulin-like growth factors in steroid modulated proliferation. The Journal of Steroid Biochemistry and Molecular Biology. 1994;51(1-2):1–9. doi: 10.1016/0960-0760(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 27.Osborne C. K., Clemmons D. R., Arteaga C. L. Regulation of breast cancer growth by insulin-like growth factors. Journal of Steroid Biochemistry and Molecular Biology. 1990;37(6):805–809. doi: 10.1016/0960-0760(90)90423-i. [DOI] [PubMed] [Google Scholar]
- 28.Richardson A. E., Hamilton N., Davis W., Brito C., de León D. Insulin-like growth factor-2 (IGF-2) activates estrogen receptor-α and-β via the IGF-1 and the insulin receptors in breast cancer cells. Growth Factors. 2011;29(2-3):82–93. doi: 10.3109/08977194.2011.565003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mancini M., Gariboldi M. B., Taiana E., et al. Co-targeting the IGF system and HIF-1 inhibits migration and invasion by (triple-negative) breast cancer cells. British Journal of Cancer. 2014;110:2865–2873. doi: 10.1038/bjc.2014.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LeRoith D., Baserga R., Helman L., Roberts C. T., Jr. Insulin-like growth factors and cancer. Annals of Internal Medicine. 1995;122(1):54–59. doi: 10.7326/0003-4819-122-1-199501010-00009. [DOI] [PubMed] [Google Scholar]
- 31.Yang X.-F., Beamer W. G., Huynh H., Pollak M. Reduced growth of human breast cancer xenografts in hosts homozygous for the lit mutation. Cancer Research. 1996;56(7):1509–1511. [PubMed] [Google Scholar]
- 32.Sciacca L., Costantino A., Pandini G., et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999;18(15):2471–2479. doi: 10.1038/sj.onc.1202600. [DOI] [PubMed] [Google Scholar]
- 33.Yee D., Lee A. V. Crosstalk between the insulin-like growth factors and estrogens in breast cancer. Journal of Mammary Gland Biology and Neoplasia. 2000;5(1):107–115. doi: 10.1023/A:1009575518338. [DOI] [PubMed] [Google Scholar]
- 34.Klotz D. M., Hewitt S. C., Ciana P., et al. Requirement of estrogen receptor-α in insulin-like growth factor-1 (IGF-1)-induced uterine responses and in vivo evidence for IGF-1/estrogen receptor cross-talk. Journal of Biological Chemistry. 2002;277(10):8531–8537. doi: 10.1074/jbc.M109592200. [DOI] [PubMed] [Google Scholar]
- 35.Vyas S., Asmerom Y., De León D. D. Resveratrol regulates insulin-like growth factor-II in breast cancer cells. Endocrinology. 2005;146(10):4224–4233. doi: 10.1210/en.2004-1344. [DOI] [PubMed] [Google Scholar]
- 36.Singh S. K., Moretta D., Almaguel F., Wall N. R., de León M., de León D. Differential effect of proIGF-II and IGF-II on resveratrol induced cell death by regulating survivin cellular localization and mitochondrial depolarization in breast cancer cells. Growth Factors. 2007;25(6):363–372. doi: 10.1080/08977190801886905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh S. K., Moretta D., Almaguel F., de León M., de León D. D. Precursor Igf-II (proIGF-II) and mature IGF-II (mIGF-II) induce Bcl-2 and Bcl-XL expression through different signaling pathways in breast cancer cells. Growth Factors. 2008;26(2):92–103. doi: 10.1080/08977190802057258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singer C. F., Mogg M., Koestler W., et al. Insulin-like growth factor (IGF)-I and IGF-II serum concentrations in patients with benign and malignant breast lesions: free IGF-II is correlated with breast cancer size. Clinical Cancer Research. 2004;10(12, part 1):4003–4009. doi: 10.1158/1078-0432.ccr-03-0093. [DOI] [PubMed] [Google Scholar]
- 39.Pacher M., Seewald M. J., Mikula M., et al. Impact of constitutive IGF1/IGF2 stimulation on the transcriptional program of human breast cancer cells. Carcinogenesis. 2007;28(1):49–59. doi: 10.1093/carcin/bgl091. [DOI] [PubMed] [Google Scholar]
- 40.Livingstone C. IGF2 and cancer. Endocrine-Related Cancer. 2013;20(6):R321–R339. doi: 10.1530/erc-13-0231. [DOI] [PubMed] [Google Scholar]
- 41.Gebeshuber C. A., Martinez J. MiR-100 suppresses IGF2 and inhibits breast tumorigenesis by interfering with proliferation and survival signaling. Oncogene. 2013;32(27):3306–3310. doi: 10.1038/onc.2012.372. [DOI] [PubMed] [Google Scholar]
- 42.Kalla Singh S., Tan Q. W., Brito C., De León M., De León D. Insulin-like growth factors I and II receptors in the breast cancer survival disparity among African-American women. Growth Hormone and IGF Research. 2010;20(3):245–254. doi: 10.1016/j.ghir.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mathieu M., Rochefort H., Barenton B., Prebois C., Vignon F. Interactions of cathepsin-D and insulin-like growth factor-II (IGF-II) on the IGF-II/mannose-6-phosphate receptor in human breast cancer cells and possible consequences on mitogenic activity of IGF-II. Molecular Endocrinology. 1990;4(9):1327–1335. doi: 10.1210/mend-4-9-1327. [DOI] [PubMed] [Google Scholar]
- 44.Cullen K. J., Allison A., Martire I., Ellis M., Singer C. Insulin-like growth factor expression in breast cancer epithelium and stroma. Breast Cancer Research and Treatment. 1992;22(1):21–29. doi: 10.1007/BF01833330. [DOI] [PubMed] [Google Scholar]
- 45.de Leon D. D., Wilson D. M., Powers M., Rosenfeld R. G. Effects of insulin-like growth factors (IGFs) and IGF receptor antibodies on the proliferation of human breast cancer cells. Growth Factors. 1992;6(4):327–336. doi: 10.3109/08977199209021544. [DOI] [PubMed] [Google Scholar]
- 46.Giani C., Campani D., Rasmussen A., et al. Insulin-like growth factor II (IGF-II) immunohistochemistry in breast cancer: relationship with the most important morphological and biochemical prognostic parameters. The International Journal of Biological Markers. 2002;17(2):90–95. doi: 10.1177/172460080201700203. [DOI] [PubMed] [Google Scholar]
- 47.Neve R. M., Chin K., Fridlyand J., et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10(6):515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lehmann B. D., Bauer J. A., Chen X., et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. Journal of Clinical Investigation. 2011;121(7):2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrington W. R., Sheng S., Barnett D. H., Petz L. N., Katzenellenbogen J. A., Katzenellenbogen B. S. Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Molecular and Cellular Endocrinology. 2003;206(1-2):13–22. doi: 10.1016/s0303-7207(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 50.Meyers M. J., Sun J., Carlson K. E., Marriner G. A., Katzenellenbogen B. S., Katzenellenbogen J. A. Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. Journal of Medicinal Chemistry. 2001;44(24):4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- 51.Aguilar Z., Akita R. W., Finn R. S., et al. Biologic effects of heregulin/neu differentiation factor on normal and malignant human breast and ovarian epithelial cells. Oncogene. 1999;18(44):6050–6062. doi: 10.1038/sj.onc.1202993. [DOI] [PubMed] [Google Scholar]
- 52.Wend P., Runke S., Wend K., et al. WNT10B/β-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Molecular Medicine. 2013;5(2):264–279. doi: 10.1002/emmm.201201320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walsh S., Flanagan L., Quinn C., et al. mTOR in breast cancer: differential expression in triple-negative and non-triple-negative tumors. Breast. 2012;21(2):178–182. doi: 10.1016/j.breast.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 54.Mandusic V., Dimitrijevic B., Nikolic-Vukosavljevic D., Neskovic-Konstantinovic Z., Kanjer K., Hamann U. Different associations of estrogen receptor β isoforms, ERβ1 and ERβ2, expression levels with tumor size and survival in early- and late-onset breast cancer. Cancer Letters. 2012;321(1):73–79. doi: 10.1016/j.canlet.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 55.Yoon N. K., Maresh E. L., Elshimali Y., et al. Elevated MED28 expression predicts poor outcome in women with breast cancer. BMC Cancer. 2010;10, article 335 doi: 10.1186/1471-2407-10-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoon N. K., Maresh E. L., Shen D., et al. Higher levels of GATA3 predict better survival in women with breast cancer. Human Pathology. 2010;41(12):1794–1801. doi: 10.1016/j.humpath.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Y., Sarkissyan M., Elshimali Y., Vadgama J. V. Triple negative breast tumors in African-American and Hispanic/Latina women are high in CD44+, low in CD24+, and have loss of PTEN. PLoS ONE. 2013;8(10) doi: 10.1371/journal.pone.0078259.e78259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mah V., Marquez D., Alavi M., et al. Expression levels of estrogen receptor beta in conjunction with aromatase predict survival in non-small cell lung cancer. Lung Cancer. 2011;74(2):318–325. doi: 10.1016/j.lungcan.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X., Minin V., Huang Y., Seligson D. B., Horvath S. Statistical methods for analyzing tissue microarray data. Journal of Biopharmaceutical Statistics. 2004;14(3):671–685. doi: 10.1081/BIP-200025657. [DOI] [PubMed] [Google Scholar]
- 60.Márquez-Garbán D. C., Chen H.-W., Fishbein M. C., Goodglick L., Pietras R. J. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids. 2007;72(2):135–143. doi: 10.1016/j.steroids.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weinberg O. K., Marquez-Garban D. C., Fishbein M. C., et al. Aromatase inhibitors in human lung cancer therapy. Cancer Research. 2005;65(24):11287–11291. doi: 10.1158/0008-5472.CAN-05-2737. [DOI] [PubMed] [Google Scholar]
- 62.Harvey J. M., Clark G. M., Osborne C. K., Allred D. C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. Journal of Clinical Oncology. 1999;17(5):1474–1481. doi: 10.1200/JCO.1999.17.5.1474. [DOI] [PubMed] [Google Scholar]
- 63.Janzen D. M., Rosales M. A., Paik D. Y., et al. Progesterone receptor signaling in the microenvironment of endometrial cancer influences its response to hormonal therapy. Cancer Research. 2013;73(15):4697–4710. doi: 10.1158/0008-5472.can-13-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bilir B., Kucuk O., Moreno C. S. Wnt signaling blockage inhibits cell proliferation and migration, and induces apoptosis in triple-negative breast cancer cells. Journal of Translational Medicine. 2013;11,article 280 doi: 10.1186/1479-5876-11-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cotrim C. Z., Fabris V., Doria M. L., et al. Estrogen receptor beta growth-inhibitory effects are repressed through activation of MAPK and PI3K signalling in mammary epithelial and breast cancer cells. Oncogene. 2013;32(19):2390–2402. doi: 10.1038/onc.2012.261. [DOI] [PubMed] [Google Scholar]
- 66.Klinge C. M., Blankenship K. A., Risinger K. E., et al. Resveratrol and estradiol rapidly activate MAP kinase signaling through estrogen receptors α and β in endothelial cells. The Journal of Biological Chemistry. 2005;280(9):7460–7468. doi: 10.1074/jbc.m411565200. [DOI] [PubMed] [Google Scholar]
- 67.Wu X., Subramaniam M., Negron V., et al. Development, characterization, and applications of a novel estrogen receptor beta monoclonal antibody. Journal of Cellular Biochemistry. 2012;113(2):711–723. doi: 10.1002/jcb.23443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leung Y. K., Lam H. M., Wu S., et al. Estrogen receptor beta2 and beta5 are associated with poor prognosis in prostate cancer, and promote cancer cell migration and invasion. Endocrine-Related Cancer. 2010;17(3):675–689. doi: 10.1677/erc-09-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chantzi N. I., Tiniakos D. G., Palaiologou M., et al. Estrogen receptor beta 2 is associated with poor prognosis in estrogen receptor alpha-negative breast carcinoma. Journal of Cancer Research and Clinical Oncology. 2013;139(9):1489–1498. doi: 10.1007/s00432-013-1467-4. [DOI] [PubMed] [Google Scholar]
- 70.Mann S., Laucirica R., Carlson N., et al. Estrogen receptor beta expression in invasive breast cancer. Human Pathology. 2001;32(1):113–118. doi: 10.1053/hupa.2001.21506. [DOI] [PubMed] [Google Scholar]
- 71.Phillips K.-A., Milne R. L., Rookus M. A., et al. Tamoxifen and risk of contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. Journal of Clinical Oncology. 2013;31(25):3091–3099. doi: 10.1200/jco.2012.47.8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Litwiniuk M. M., Roznowski K., Filas V., et al. Expression of estrogen receptor beta in the breast carcinoma of BRCA1 mutation carriers. BMC Cancer. 2008;8, article 100 doi: 10.1186/1471-2407-8-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stewart J., King R., Hayward J., Rubens R. Estrogen and progesterone receptors: correlation of response rates, site and timing of receptor analysis. Breast Cancer Research and Treatment. 1982;2(3):243–250. doi: 10.1007/bf01806937. [DOI] [PubMed] [Google Scholar]
- 74.von Maillot K., Gentsch H. H., Gunselmann W. Steroid receptors and response to endocrine treatment and chemotherapy of advanced breast cancer. Journal of Cancer Research and Clinical Oncology. 1980;98(3):301–313. doi: 10.1007/bf00410792. [DOI] [PubMed] [Google Scholar]
- 75.Reddel R. R., Murphy L. C., Hall R. E., Sutherland R. L. Differential sensitivity of human breast cancer cell lines to the growth-inhibitory effects of tamoxifen. Cancer Research. 1985;45(4):1525–1531. [PubMed] [Google Scholar]
- 76.Wang Y., Chirgadze N. Y., Briggs S. L., Khan S., Jensen E. V., Burris T. P. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor β . Proceedings of the National Academy of Sciences of the United States of America. 2006;103(26):9908–9911. doi: 10.1073/pnas.0510596103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kiely B. E., Phillips K. A., Francis P. A., et al. ANZ1001 SORBET: study of oestrogen receptor beta and efficacy of tamoxifen, a single arm, phase II study of the efficacy of tamoxifen in triple-negative but estrogen receptor beta-positive metastatic breast cancer. Journal of Clinical Oncology. 2011;29(supplement) TPS1136. [Google Scholar]
- 78.Charn T. H., Liu E. T.-B., Chang E. C., Lee Y. K., Katzenellenbogen J. A., Katzenellenbogen B. S. Genome-wide dynamics of chromatin binding of estrogen receptors alpha and beta: mutual restriction and competitive site selection. Molecular Endocrinology. 2010;24(1):47–59. doi: 10.1210/me.2009-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hou Y. F., Yuan S. T., Li H. C., et al. ERβ exerts multiple stimulative effects on human breast carcinoma cells. Oncogene. 2004;23(34):5799–5806. doi: 10.1038/sj.onc.1207765. [DOI] [PubMed] [Google Scholar]
- 80.Thomas C., Rajapaksa G., Nikolos F., et al. ERβ1 represses basal-like breast cancer epithelial to mesenchymal transition by destabilizing EGFR. Breast Cancer Research. 2012;14, article R148 doi: 10.1186/bcr3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shanle E. K., Zhao Z., Hawse J., et al. Research resource: global identification of estrogen receptor β target genes in triple negative breast cancer cells. Molecular Endocrinology. 2013;27(10):1762–1775. doi: 10.1210/me.2013-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jeng M.-H., Jiang S.-Y., Jordan V. C. Paradoxical regulation of estrogen-dependent growth factor gene expression in estrogen receptor (ER)-negative human breast cancer cells stably expressing ER. Cancer Letters. 1994;82(2):123–128. doi: 10.1016/0304-3835(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 83.Jiang S.-Y., Jordan V. C. Growth regulation of estrogen receptor-negative breast cancer cells transfected with complementary DNAs for estrogen receptor. Journal of the National Cancer Institute. 1992;84(8):580–591. doi: 10.1093/jnci/84.8.580. [DOI] [PubMed] [Google Scholar]
- 84.Lazennec G., Katzenellenbogen B. S. Expression of human estrogen receptor using an efficient adenoviral gene delivery system is able to restore hormone-dependent features to estrogen receptor-negative breast carcinoma cells. Molecular and Cellular Endocrinology. 1999;149(1-2):93–105. doi: 10.1016/s0303-7207(98)00254-8. [DOI] [PubMed] [Google Scholar]
- 85.Navaratnam S., Skliris G., Qing G., et al. Differential role of estrogen receptor beta in early versus metastatic non-small cell lung cancer. Hormones and Cancer. 2012;3(3):93–100. doi: 10.1007/s12672-012-0105-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nair H. B., Kirma N. B., Ganapathy M., Vadlamudi R. K., Tekmal R. R. Estrogen receptor-β activation in combination with letrozole blocks the growth of breast cancer tumors resistant to letrozole therapy. Steroids. 2011;76(8):792–796. doi: 10.1016/j.steroids.2011.02.038. [DOI] [PubMed] [Google Scholar]
- 87.Hershberger P. A., Stabile L. P., Kanterewicz B., et al. Estrogen receptor beta (ERβ) subtype-specific ligands increase transcription, p44/p42 mitogen activated protein kinase (MAPK) activation and growth in human non-small cell lung cancer cells. The Journal of Steroid Biochemistry and Molecular Biology. 2009;116(1-2):102–109. doi: 10.1016/j.jsbmb.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mah V., Seligson D. B., Li A., et al. Aromatase expression predicts survival in women with early-stage non-small cell lung cancer. Cancer Research. 2007;67(21):10484–10490. doi: 10.1158/0008-5472.can-07-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stabile L. P., Davis A. L., Gubish C. T., et al. Human non-small cell lung tumors and cells derived from normal lung express both estrogen receptor alpha and beta and show biological responses to estrogen. Cancer Research. 2002;62(7):2141–2150. [PubMed] [Google Scholar]
- 90.Rudolph A., Toth C., Hoffmeister M., et al. Expression of oestrogen receptor Β and prognosis of colorectal cancer. British Journal of Cancer. 2012;107(5):831–839. doi: 10.1038/bjc.2012.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Corkery B., Crown J., Clynes M., O'Donovan N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Annals of Oncology. 2009;20(5):862–867. doi: 10.1093/annonc/mdn710. [DOI] [PubMed] [Google Scholar]
- 92.Davis A. A., Kaklamani V. G. Metabolic syndrome and triple-negative breast cancer: a new paradigm. International Journal of Breast Cancer. 2012;2012:10. doi: 10.1155/2012/809291.809291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jain R., Strickler H. D., Fine E., Sparano J. A. Clinical studies examining the impact of obesity on breast cancer risk and prognosis. Journal of Mammary Gland Biology and Neoplasia. 2013;18(3-4):257–266. doi: 10.1007/s10911-013-9307-3. [DOI] [PubMed] [Google Scholar]
- 94.Sturtz L. A., Melley J., Mamula K., Shriver C. D., Ellsworth R. E. Outcome disparities in African American women with triple negative breast cancer: a comparison of epidemiological and molecular factors between African American and Caucasian women with triple negative breast cancer. BMC Cancer. 2014;14(1, article 62) doi: 10.1186/1471-2407-14-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pierobon M., Frankenfeld C. L. Obesity as a risk factor for triple-negative breast cancers: a systematic review and meta-analysis. Breast Cancer Research and Treatment. 2013;137(1):307–314. doi: 10.1007/s10549-012-2339-3. [DOI] [PubMed] [Google Scholar]
- 96.Creighton C. J., Casa A., Lazard Z., et al. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. Journal of Clinical Oncology. 2008;26(25):4078–4085. doi: 10.1200/jco.2007.13.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Litzenburger B. C., Creighton C. J., Tsimelzon A., et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clinical Cancer Research. 2011;17(8):2314–2327. doi: 10.1158/1078-0432.ccr-10-1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Castaño Z., Marsh T., Tadipatri R., et al. Stromal EGF and IGF-I together modulate plasticity of disseminated triple-negative breast tumors. Cancer Discovery. 2013;3(8):922–935. doi: 10.1158/2159-8290.CD-13-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kuperwasser C., Chavarria T., Wu M., et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(14):4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Janzen D. M., Rosales M. A., Paik D. Y., et al. Progesterone receptor signaling in the microenvironment of endometrial cancer influences its response to hormonal therapy. Cancer Research. 2013;73(15):4697–4710. doi: 10.1158/0008-5472.CAN-13-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]