

Abstract
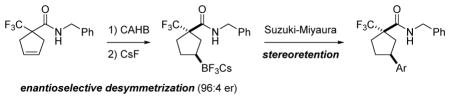
The rhodium-catalyzed enantioselective desymmetrization of symmetric γ,δ–unsaturated amides via carbonyl-directed catalytic asymmetric hydroboration (directed CAHB) affords chiral secondary organoboronates with up to 98% ee. The chiral γ–borylated products undergo palladium-catalyzed Suzuki-Miyaura cross-coupling via the trifluoroborate salt with stereoretention.
Chiral organoboronates are useful intermediates in organic synthesis,1,2 and consequently, a number of research groups are developing enantioselective methods for their preparation.3–10 We reported the rhodium-catalyzed carbonyl-directed catalytic asymmetric hydroborations (directed CAHBs) of certain (E)- and (Z)-disubstituted and -trisubstituted β,γ-unsaturated amides 1 and 1,1-disubstituted (i.e., methylidene) β,γ-unsaturated amides and tert-butyl esters 3.11 Simple chiral catalyst systems have been identified to give chiral β- and γ-borylated carbonyl compounds with high regio- and π-facial selectivity (Figure 1). We now report the efficient CAHB of symmetric γ,δ–unsaturated amides 5. For example, 5a undergoes carbonyl-directed CAHB with 4,4,6–trimethyl–1,3,2–dioxaborinane (tmdBH, (±)-B1) using Rh(nbd)2BF4 in conjunction with (BINOL)PN(Bn)Ph (L1b) to give the cis–γ–borylated amide (1R,3S)-6a in 80% yield and 94% ee (determined after oxidation) via enantioselective desymmetrization.12 Asymmetric desymmetrization is a widely used method for generating chiral substances;13 however, few examples employ CAHB.14
Figure 1.

Carbonyl-directed CAHB exploits π-facial selectivity for β,γ-unsaturated substrates 1 and 3 and re/si-site selectivity for the enantioselective desymmetrization of γ,δ–substrate 5a.
Varying the nature of the ligand used (i.e., L1–L5) for rhodium-catalyzed CAHB of 5a by tmdBH (B1) reveals a number of interesting effects on the sense and degree of enantioselectivity (Figure 2). BINOL-derived phosphoramidites L1a–b in combination with tmdBH (B1) give the most selective catalysts for the formation of (1R,3S)-6a (93–94% ee). Catalysts formed from TADDOL phenyl phosphites L2a–L5a are also quite selective (83–92% ee); small changes in the TADDOL backbone only incrementally affect enantioselectivity. In contrast to the TADDOL-derived phenyl phosphites L2a–L5a and BINOL-derived phosphoramidites L1a/b, TADDOL-derived phosphoramidites afford catalysts that are much less selective with tmdBH; the products obtained are near racemic for most combinations of L2–5 possessing substituents b–e.
Figure 2.
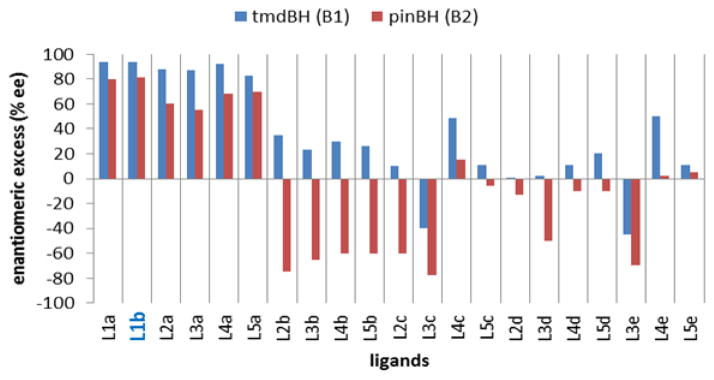
Varying the boranes and ligands employed strikingly affects the enantioselectivity for the CAHB of 5a.16
The enantioselectivity obtained for CAHB by pinBH (B2) with catalysts derived from BINOL-derived phosphoramidites L1a and b and the series of phenyl phosphites L2a–L5a is significantly lower (55–81% ee) than obtained with tmdBH (Figure 2). However, pinBH generally affords higher levels of enantioselectivity with TADDOL-derived phosphoramidites L2-5b-e. Furthermore, these phosphoramidites generally give the enantiomeric product compared to that obtained with corresponding phenyl phosphite; enantioselectivity as high as 78% ee for (1S,3R)-6a is found using L3c. The results are in qualitative agreement with our previous report of exceptionally high levels of enantioswitching15 with the acyclic substrate 1.11d
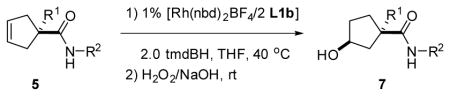
Using the most promising catalyst system identified in our brief ligand/borane survey (i.e., [(L1b)2Rh(nbd)BF4] with tmdBH), a series of γ,δ–unsaturated amides 5a–l varying in their amide- and α-substituents are converted via the γ-borylated intermediate to their chiral cyclopentanols 7a–l (Table 1 and Figure 3). Phenyl amides 5a–d, a series of substrates bearing α-H, -alkyl, -aryl, and -CF3 substituents, gave the respective γ-alcohol in good yield and high enantiomeric excess (i.e., 7a–d, 96:4 to 97:3 er) (entries 1–4). Substituting benzyl for phenyl as the amide substituent gave 7e–h in comparable yields but slightly lower enantioselectivity; enantiomer ratios ranged from 92:8 to 96:4 (entries 5–8). Chiral substrates 5i and 5j bear a chiral phenethyl substituent on the amide nitrogen and were included in the screening to confirm structural assignments (vide infra). These chiral substrates undergo CAHB with somewhat lower stereoselectivity compared to the corresponding phenyl and benzyl amides and exhibit a modest matched/mismatched diastereomer effect (entries 9 and 10).
Table 1.
Enantioselective desymmetrization via CAHB.
| |||||
|---|---|---|---|---|---|
| entry | 5 | R1 | R2 | yielda | er/drb |
| 1 | a | H | Ph | 80 | 97:3 |
| 2 | b | Me | Ph | 65 | 96:4 |
| 3 | c | Ph | Ph | 72 | 96:4 |
| 4 | d | CF3 | Ph | 78 | 97:3 |
| 5 | e | H | Bn | 69 | 94:6 |
| 6 | f | Me | Bn | 62 | 92:8 |
| 7 | g | Ph | Bn | 70 | 93:7 |
| 8 | h | CF3 | Bn | 71 | 96:4 |
| 9c | i | Ph | (R)-CH(Me)Ph | 76 | 88:12 |
| 10c | j | Ph | (S)-CH(Me)Ph | 74 | 20:80 |
Isolated yields, an average of three experiments generally exhibiting a spread of ±2%.
Enantiomer ratio (er) determined by chiral HPLC analysis or diastereomer ratio (dr) determined by 1H NMR.
2% catalyst loading.
Figure 3.

Group-selective enantioselective desymmetrization.
Two derivatives shown in Figure 3 highlight unusual group-selectivity in the CAHB. Substrates 5k and 5l each contain two alkene moieties ostensibly positioned equidistant from the carbonyl directing group, one endocyclic double bond and an unsaturated side chain substituent on the ring. The side-chain alkene is trans-disubstituted in the case of 5k and a methylidene derivative in the case of 5l. Both substrates undergo CAHB selectively with the endocyclic double bond to give the monounsaturated γ-alcohol [i.e., (1S,3S)-7k and 7l, respectively] in good yield (75–80%) and high enantioselectivity (99:1 and 97:3 er, respectively). We speculate that the preferred orientation of the carbonyl oxygen and/or somewhat greater reactivity for the endocyclic double bond might account for the observed product.
The CAHB of chiral amide 5i proved useful in unambiguously assigning the absolute configuration of the product and thereby the stereochemical course of the reaction. The intermediate organoboronate generated upon CAHB was converted to the cesium17 or potassium18 trifluoroborate by the reported methods. The latter gave crystals of the major diastereomer suitable for x-ray crystallographic analysis; the structure of (1R,3S)-8i (M = K) is shown in Figure 4.
Figure 4.

Preparation and crystal structure of 8i (M = K).
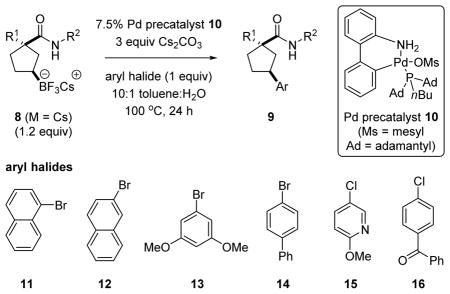
The cross-coupling reactions of chiral, secondary organoboronates have recently attracted a great deal attention.19 Several examples illustrating successful Suzuki-Miyaura cross-coupling of the benzyl and chiral phenethyl amide derivatives of (1R,3S)-8i (M = Cs) using Buchwald’s palladium-precatalyst 1020 are summarized in Table 2.21
Table 2.
Palladium-catalyzed cross-coupling of cesium tri-fluoroborate salt 8i.
| ||||||
|---|---|---|---|---|---|---|
| entry | 8 | R1 | R2 | aryl halide | 9 | yielda |
| 1b | 8g | Ph | Bn | 11 | a | 69 |
| 2b | 8h | CF3 | Bn | 11 | b | 63 |
| 3 | 8i | Ph | (R)-CH(Me)Ph | 11 | c | 86 |
| 4 | 8i | Ph | (R)-CH(Me)Ph | 12 | d | 66 |
| 5 | 8i | Ph | (R)-CH(Me)Ph | 13 | e | 66 |
| 6 | 8i | Ph | (R)-CH(Me)Ph | 14 | f | 71 |
| 7 | 8i | Ph | (R)-CH(Me)Ph | 15 | g | 75c |
| 8 | 8i | Ph | (R)-CH(Me)Ph | 16 | h | 70 |
Isolated yields based on limiting aryl halide, an average (±2%) of two runs.
Cross-coupling proceeds with high diastereoselectivity (ca. 94:6 dr).
5 equiv of CsOH replaces Cs2CO3.
Understanding the stereochemical course of the Suzuki-Miyaura reaction has recently attracted considerable attention.19 Spectral data obtained for products 9a and b indicate that the cross-coupling is highly diastereoselective. For example, the 19F NMR spectrum of 9b shows a single major resonance with no minor peak integrating for more than 6% abundance. As first demonstrated by Crudden,1g,2 Suzuki-Miyaura cross-couplings of α-chiral alkylboron compounds possessing adjacent π-systems proceed with stereoretention. However, substrates similar to those used here by Molander22 and Suginome,23 as well as other substrates reported by Biscoe,1a Morken4b and Hall,24 proceed with stereoinversion. For example, the borylated amides 17 and 19 undergo palladium-catalyzed cross-coupling with near complete stereoinversion (Figure 5). The intermediate γ–borylated amide (1R,3S)-8i was coupled with 2-methoxy-5-chloropyridine (15); product 9g was isolated and subsequently converted to its tetrafluoroborate salt 21 (66% overall). The latter gives crystals suitable for x-ray analysis which confirms the structure as (1R,3S)-21 and establishes that palladium-catalyzed cross-coupling proceeds with stereoretention.
Figure 5.

In contrast to recent examples of β-borylated amides, palladium-catalyzed cross-coupling of γ–borylated amide (1R,3S)-8i proceeds with stereoretention.
Substrate reactivity is another aspect of the cross-coupling chemistry that has attracted recent attention.25 For example, Molander19 proposed that intramolecular hemi-labile π-complexation of palladium by a suitably disposed benzyl substituent was a key element facilitating cross-coupling with stereoretention. In contrast to the corresponding benzyl and phenethyl amides, phenyl amide 22 (M = K or Cs), gives little or no cross-coupling product under the conditions used in Table 2. For example, the attempted cross-coupling of phenyl amide 22 with 1-bromonaphthalene gives only 20% (based on limiting aryl bromide) of the cross-coupled product 23; in addition, 7c is isolated in 85% yield (based on the amount of 22) from the reaction mixture after oxidation with Oxone® (Figure 6). The reproducible, low yield of cross-coupled product initially suggested that the greater rotational freedom and reach available to benzyl amides was a necessary feature for efficient cross-coupling. However, the direct competition of equal amounts of phenyl amide 22 and 8i (R1 = Ph, R2 = (R)-CH(Me)Ph) for a limiting amount of 1-bromonaphthalene 11 gave surprising results and raise doubt about that explanation (Figure 6). The total yield of cross-coupled products is high (90% based on the limiting aryl bromide) and a near 1:1 mixture of 23 (48%) and 9c (42%) is obtained along with commensurate amounts of the respective alcohols 7c and 7i resulting from oxidation of the two residual starting materials.
Figure 6.

Unexpected influence of the amide substituent on the facility of palladium-catalyzed cross-coupling.
In summary, carbonyl-directed CAHB of γ,δ–unsaturated substrate 5 proceeds with efficient π-facial discrimination to introduce boron cis with respect to the amide functional group consistent with two-point binding of the substrate as described in a prior computational study;12 efficient re/si-site selectivity by the chiral catalyst controls enantioselectivity. The ligand and the borane employed have striking effects on the level and sense of enantioinduction including in some cases enantioswitching. Unusual group-selectivity is seen in the CAHBs of the doubly γ,δ–unsaturated substrates 5k and 5l for which the endocyclic alkene preferentially undergoes reaction. The chiral γ–trifluoroborates produced via CAHB undergo palladium-catalyzed Suzuki-Miyaura cross-coupling with stereoretention. The amide substituent influences the efficiency of the cross-coupling reaction under the conditions examined, although the reasons are not clear. Further studies are in progress.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support for these studies from the NIH (GM100101). The crystal structures reported herein were determined by V.W. Day at the KU Small-Molecule X-ray Crystallography Lab using instrumentation purchased with funds from the NSF (CHE-0923449) and the University of Kansas.
Footnotes
Experimental procedures and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Li L, Zhao S, Joshi-Pangu A, Diane M, Biscoe MR. J Am Chem Soc. 2014;136:14027–14030. doi: 10.1021/ja508815w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Matthew SC, Glasspoole BW, Eisenberger P, Crudden CM. J Am Chem Soc. 2014;136:5828–5831. doi: 10.1021/ja412159g. [DOI] [PubMed] [Google Scholar]; (c) Buesking AW, Ellman JA. Chem Sci. 2014;5:1983–1987. [Google Scholar]; (d) Bonet A, Odachowski M, Leonori D, Essafi S, Aggarwal VK. Nat Chem. 2014;6:584–589. doi: 10.1038/nchem.1971. [DOI] [PubMed] [Google Scholar]; (e) Zhang C, Yun J. Org Lett. 2013;15:3416–3419. doi: 10.1021/ol401468v. [DOI] [PubMed] [Google Scholar]; (f) Mlynarski SN, Karns AS, Morken JP. J Am Chem Soc. 2012;134:16449–16451. doi: 10.1021/ja305448w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Imao D, Glasspoole BW, Laberge VS, Crudden CM. J Am Chem Soc. 2009;131:5024–2025. doi: 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]
- 2.(a) Leonori D, Aggarwal VK. Angew Chem, Int Ed. 2015;54:1082–1096. doi: 10.1002/anie.201407701. [DOI] [PubMed] [Google Scholar]; (b) Swift EC, Jarvo ER. Tetrahedron. 2013;69:5799–5817. doi: 10.1016/j.tet.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stoichiometric reagents: Brown JM, Nguyen BN. Stereoselective hydroboration and diboration of carbon-carbon double bonds; Vol. 1. Georg Thieme Verlag; 2011. pp. 295–324.
- 4.Group-selective cross-coupling of diboranes: Mlynarski SN, Schuster CH, Morken JP. Nature. 2014;505:386–90. doi: 10.1038/nature12781.Sun C, Potter B, Morken JP. J Am Chem Soc. 2014;136:6534–6537. doi: 10.1021/ja500029w.Dewhurst RD, Marder TB. Nat Chem. 2014;6:279–280. doi: 10.1038/nchem.1905.
- 5.Enantioselective B-H bond insertion: Cheng Q, Zhu S, Zhang Y, Xie X, Zhou Q. J Am Chem Soc. 2013;135:14094–14097. doi: 10.1021/ja408306a.
- 6.Conjugate borylation: Radomkit S, Hoveyda AH. Angew Chem, Int Ed. 2014;53:3387–3391. doi: 10.1002/anie.201309982.Luo Y, Roy ID, Madec AGE, Lam HW. Angew Chem, Int Ed. 2014;53:4186–4190. doi: 10.1002/anie.201310380.Calow ADJ, Batsanov AS, Pujol A, Sole C, Fernandez E, Whiting A. Org Lett. 2013;15:4810–4813. doi: 10.1021/ol4022029.Callow ADJ, Whiting A. Org Biomol Chem. 2012;10:5485–5497. doi: 10.1039/c2ob25908g.
- 7.From vinylboronates: Verendel JJ, Pamies O, Dieguez M, Andersson PG. Chem Rev. 2014;114:2130–2169. doi: 10.1021/cr400037u.Boese D, Niesobski P, Luebcke M, Pietruszka J. J Org Chem. 2014;79:4699–4703. doi: 10.1021/jo5004168.Lee JCH, Hall DG. J Am Chem Soc. 2010;132:5544–5545. doi: 10.1021/ja9104057.
- 8.Catalytic asymmetric diboration: Yu Z, Ely RJ, Morken JP. Angew Chem, Int Ed. 2014;53:9632–9636. doi: 10.1002/anie.201405455.Coombs JR, Haeffner F, Kliman LT, Morken JP. J Am Chem Soc. 2013;135:11222–11231. doi: 10.1021/ja4041016.Ferris GE, Hong K, Roundtree IA, Morken JP. J Am Chem Soc. 2013;135:2501–2504. doi: 10.1021/ja400506j.Kliman LT, Mlynarski SN, Ferris GE, Morken JP. Angew Chem, Int Ed. 2012;51:521–524. doi: 10.1002/anie.201105716.Schuster CH, Li B, Morken JP. Angew Chem, Int Ed. 2011;50:7906–7909. doi: 10.1002/anie.201102404.Burks HE, Morken JP. Chem Commun. 2007:4717–4725. doi: 10.1039/b707779c.
- 9.Enantioselective aminoborylation: Matsuda N, Hirano K, Satoh T, Miura M. J Am Chem Soc. 2013;135:4934–4937. doi: 10.1021/ja4007645.Hong K, Morken JP. J Am Chem Soc. 2013;135:9252–9254. doi: 10.1021/ja402569j.
- 10.Enantioselective hydroboration of dienes and vinylarenes: He Z, Zhao Y, Tian P, Wang C, Dong H, Lin G. Org Lett. 2014;16:1426–1429. doi: 10.1021/ol500219e.Feng X, Jeon H, Yun J. Angew Chem, Int Ed. 2013;52:3989–3992. doi: 10.1002/anie.201208610.Kubota K, Yamamoto E, Ito H. Adv Synth Catal. 2013;355:3527–3531.Moteki SA, Toyama K, Liu Z, Ma J, Holmes AE, Takacs JM. Chem Commun. 2012;48:263–265. doi: 10.1039/c1cc16146f.Sasaki Y, Zhong C, Sawamura M, Ito H. J Am Chem Soc. 2010;132:1226–1227. doi: 10.1021/ja909640b.Carroll A, O’Sullivan TP, Guiry PJ. Adv Synth Catal. 2005;347:609–631.
- 11.(a) Smith SM, Hoang GL, Pal R, Khaled MOB, Pelter LSW, Zeng XC, Takacs JM. Chem Commun. 2012;48:12180–12182. doi: 10.1039/c2cc36199j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Smith SM, Uteuliyev M, Takacs JM. Chem Commun. 2011;47:7812–7814. doi: 10.1039/c1cc11746g. [DOI] [PubMed] [Google Scholar]; (c) Smith SM, Takacs JM. J Am Chem Soc. 2010;132:1740–1741. doi: 10.1021/ja908257x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smith SM, Takacs JM. Org Lett. 2010;12:4612–4615. doi: 10.1021/ol101932q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Smith SM, Thacker NC, Takacs JM. J Am Chem Soc. 2008;130:3734–3735. doi: 10.1021/ja710492q. [DOI] [PubMed] [Google Scholar]
- 12.Related computational study, see: Yang Z, Pal R, Hoang GL, Zeng XC, Takacs JM. ACS Catal. 2014;4:763–773. doi: 10.1021/cs401023j.
- 13.Fernandez-Perez H, Etayo P, Lao JR, Nunez-Rico J, Vidal-Ferran A. Chem Commun. 2013;49:10666–10675. doi: 10.1039/c3cc45466e. [DOI] [PubMed] [Google Scholar]
- 14.(a) Rubina M, Rubin M, Gevorgyan V. J Am Chem Soc. 2003;125:7198–7199. doi: 10.1021/ja034210y. [DOI] [PubMed] [Google Scholar]; (b) Perez Luna A, Bonin M, Micouin L, Husson H. J Am Chem Soc. 2002;124:12098–12099. doi: 10.1021/ja026714m. [DOI] [PubMed] [Google Scholar]; (c) Brunel J, Buono G. Tetrahedron Lett. 1999;40:3561–3564. [Google Scholar]; (d) Burgess K, Ohlmeyer MJ. J Org Chem. 1988;53:5178–9. [Google Scholar]
- 15.Escorihuela J, Burguete MI, Luis SV. Chem Soc Rev. 2013;42:5595–1617. doi: 10.1039/c3cs60068h. [DOI] [PubMed] [Google Scholar]
- 16.Conditions: 1% [L2Rh(nbd)BF4], 2 equiv B1 or B2, 40 °C, THF; % ee determined by chiral HPLC analysis after oxidation and is an average of two experiments exhibiting a spread of ±1% ee.
- 17.Lennox AJJ, Lloyd-Jones G. Angew Chem, Int Ed. 2012;51:9385–9388. doi: 10.1002/anie.201203930. [DOI] [PubMed] [Google Scholar]
- 18.Molander GA, Shin I, Jean-Gerard L. Org Lett. 2010;12:4384–4387. doi: 10.1021/ol101865e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molander GA, Wisniewski SR. J Am Chem Soc. 2012;134:16856–16868. doi: 10.1021/ja307861n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bruno NC, Tudge MT, Buchwald SL. Chem Sci. 2013;4:916–920. doi: 10.1039/C2SC20903A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The cesium salt gives higher overall yields in the CAHB/Suzuki-Miyaura cross-coupling sequence, mostly due to higher yields in forming the salt. Data on the on the corresponding potassium salts are given in the Supporting Information, Figure S1.
- 22.Sandrock DL, Jean-Gerard L, Chen C, Dreher SD, Molander GA. J Am Chem Soc. 2010;132:17108–17110. doi: 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Awano T, Ohmura T, Suginome M. J Am Chem Soc. 2011;133:20738–20741. doi: 10.1021/ja210025q. [DOI] [PubMed] [Google Scholar]; (b) Ohmura T, Awano T, Suginome M. J Am Chem Soc. 2010;132:13191–13193. doi: 10.1021/ja106632j. [DOI] [PubMed] [Google Scholar]
- 24.Lee JC, McDonald R, Hall DG. Nat Chem. 2011;3:894–899. doi: 10.1038/nchem.1150. [DOI] [PubMed] [Google Scholar]
- 25.Lennox AJJ, Lloyd-Jones G. Chem Soc Rev. 2014;43:412–443. doi: 10.1039/c3cs60197h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.