

Abstract
Despite significant improvements in diagnosis, understanding the pathophysiology and management of the patients with acute decompensated heart failure (ADHF), diuretic resistance, yet to be clearly defined, is a major hurdle. Secondary hyperaldosteronism is a pivotal factor in pathogenesis of sodium retention, refractory congestion in heart failure (HF) as well as diuretic resistance. In patients with decompensated cirrhosis who suffer from ascites, similar pathophysiological complications have been recognized. Administration of natriuretic doses of mineralocorticoid receptor antagonists (MRAs) has been well established in management of cirrhotic patients. However, this strategy in patients with ADHF has not been well studied. This article will discuss the potential use of natriuretic doses of MRAs to overcome the secondary hyperaldosteronism as an alternative diuretic regimen in patients with HF.
Keywords: Heart failure, Cirrhosis, Hyperaldosteronism, Mineralocorticoid receptor, antagonism, Refractory congestion
Introduction
Heart failure (HF) is a growing global healthcare problem. The prevalence of HF in the USA is 5.7 million with more than 1 million admissions for HF each year [1, 2]. Analysis of large clinical trials and data registries have shown that the main cause of hospitalizations for acute decompensated heart failure (ADHF) is due to signs and symptoms of venous congestion rather than a low cardiac output [3–8]. Although decongestion, mainly with loop diuretics, is an essential target of treatment in patients with ADHF, approximately 50 % of patients do not lose body weight during admission [3]. This treatment failure has major consequences. Refractory systemic congestion is a hemodynamic predictor of worsening renal function, rehospitalization and post-discharge mortality in patients hospitalized for ADHF [9–12]. On the other hand, higher doses of loop diuretics in HF patients may increase the risk of intravascular volume depletion, decline in cardiac preload and cardiac index, which subsequently causes arterial underfilling and further activation of neurohormonal axis including renin–angiotensin–aldosterone system (RAAS).
Notably, despite administration of angiotensin-converting enzyme inhibitor (ACE-I), aldosterone levels remain elevated in patients with ADHF [13]. Mineralocorticoid receptor antagonists (MRAs) have shown to improve mortality in HF patients [14, 15]. This is mainly due to blocking the non-genomic, non-natriuretic effects of aldosterone with spironolactone equivalent dose of 25 mg/day. In decompensated cirrhosis as complicated by ascites, natriuretic doses of spironolactone up to 400 mg/day have been recommended. However, despite pathophysiological similarities of congestive HF and decompensated cirrhosis, natriuretic doses of MRAs (i.e., ≥25 mg/day of spironolactone or ≥50 mg of eplerenone) have not been evaluated in HF.
This review will discuss the mechanism of action for aldosterone, the pathophysiology of sodium and water retention in decompensated cirrhosis and HF with a focus on hyperaldosteronism. Also the diuretic properties of high-dose MRAs in cirrhosis and HF, and their implications with respect to the new diuretic strategies for HF patients will be discussed.
Aldosterone: mechanism of action
Genomic versus non-genomic pathways
As a consequence of persistent stimulation of RAAS, plasma aldosterone level is elevated in patients with HF and decompensated cirrhosis with various deleterious effects, more specifically on cardiovascular and renal system [16, 17]. It has been proposed that aldosterone exerts its physiological effects via two major pathways, genomic and non-genomic.
The genomic pathway requires binding of aldosterone to the renal mineralocorticoid receptors (MRs) at the cytoplasmic level of collecting duct cells with transcriptional stimulation and protein synthesis [18]. This pathway will result in sodium retention and potassium loss. Considering the need for gene expression and new protein synthesis, the genomic regulations will take hours to days to process. In normal subjects, excessive aldosterone will lead to an initial decrease in urinary sodium excretion. However, with increased renal perfusion pressure, decreased proximal tubule sodium reabsorption and subsequent increase in sodium delivery to the distal nephron, the site of mineralocorticoid action, the initial increase in mineralocorticoid-induced sodium reabsorption is then negated. Further, with volume expansion, excretion of natriuretic peptides increases. This phenomenon is termed “aldosterone escape.” This compensatory mechanism is impaired in HF and decompensated cirrhosis [19].
On the other hand, the non-genomic effects are fast acting and occur within minutes presumably via the plasma membrane receptors [20]. These non-genomic effects of aldosterone may involve several organs such as cardiovascular, renal and hepatic system independent of the genomic mechanisms. For example, it has been shown that in patients with ESRD, spironolactone has beneficial effect on blood pressure management in the absence of any effect on sodium excretion [21]. The significant impact of non-genomic dosage of MRAs on cardiovascular morbidity and mortality of patients with HF has been well documented [14, 15, 22]. These mechanisms involve inhibitory effects of MRAs, resulting in reversal of aldosterone-induced coronary vasoconstriction, negative inotropic effects, oxidative stressor formations and fibrotic processes [23].
Pathophysiology of sodium and fluid retention in decompensated cirrhosis and heart failure
Cirrhosis
Primary systemic arterial vasodilation hypothesis has been the core explanation for neurohormonal activation and subsequent hyperaldosteronism in decompensated cirrhosis [24]. As a consequence of increased circulatory prostacyclins and nitric oxide (NO) production, severe peripheral vasodilatation (i.e., mainly due to splanchnic vasodilatation) causes severe decrease in effective arterial blood volume (EABV) and arterial underfilling (Fig. 1). In an attempt to normalize EABV, plasma levels of renin, aldosterone, angiotensin II, norepinephrine and vasopressin will be elevated, resulting in an increase in cardiac output [25–27]. These hemodynamic alterations will lead to renal sodium and water retention and ultimate critical plasma volume expansion and edema formation [24].
Fig. 1.

Clinical conditions in which a decrease in cardiac output (a) and systemic arterial vasodilation (b) cause arterial underfilling with resultant neurohumoral activation and renal sodium and water retention. Adapted from ref. [73]
In a study by La Villa et al. [28], the crucial role of systemic vascular resistance has been illustrated. In their study among patients with compensated cirrhosis (no ascites) after administration of large doses of mineralocorticoid hormones, the patients with lower systemic vascular resistance developed ascites and exhibited impaired “aldosterone escape.” The severity of sodium retention in cirrhotic patients directly correlates with the severity of the decompensated status and ascites formation. In patients with large ascites, usually, the sodium excretion is <10 mEq/day and in mild to moderate cases above 10 mEq/day [29].
Sodium retention is more pronounced in patients with concurrent ascites and renal failure. It has been suggested that both proximal and distal tubules are involved in the increased sodium reabsorption in decompensated cirrhosis [30]. Activation of RAAS both at systemic and intrarenal level as well as sympathetic nervous system (SNS) has been shown in several experimental and clinical studies [31, 32]. Patients with decompensated cirrhosis have elevated plasma levels of aldosterone and an increased rate of urinary aldosterone [33]. Additionally, activation of SNS indirectly enhances the rate of sodium reabsorption by an elevation of β-adrenergic receptor stimulation and subsequent increase in renin secretion. Moreover, an increase in α-adrenergic activity will increase the rate of sodium reabsorption in proximal tubules [34, 35]. Elevated levels of arginine vasopressin (AVP) are the major contributing factor in the pathogenesis of solute-free water retention in decompensated cirrhosis. The arterial vasodilatation in cirrhosis unloads high-pressure baroreceptors which results in non-osmotic release of AVP and increase water retention in these patients.
Heart failure
It is hypothesized that atrial–renal reflexes are the main regulators of fluid balance in HF [36]. An increase in atrial pressure exerts inhibitory effects on both the non-osmotic AVP release and renal sympathetic tone. In addition, the secretion of atrial natriuretic peptide will be increased secondary to arterial stretch and dilatation. As a result, increase urinary sodium and water excretion will occur and prevent edema formation [37–39].
However, in advanced HF, additional pathophysiological events will contribute to edema formation (Fig. 1). Underfilling of the arterial circulation due to reduced cardiac output results in unloading of the arterial baroreceptors, which will increase the non-osmotic vasopressin release and activate SNS and RAAS. This adrenergic surge promotes additional stimulation of the RAAS, resulting in increased renal sodium and water retention. This volume expansion in conjunction with increased systemic vascular resistance via both sympathetic and RAAS stimulation tend to restore EABV [40–43]. Persistent hyperaldosteronism due to RAAS stimulation and failure of aldosterone escape leads to edema formation.
Increased sodium and water retention will also increase the cardiac preload and subsequently ventricular filling pressures, even before any clinical manifestations of HF occurs [6]. These early effects may be termed hemodynamic congestion. The elevated filling pressures result in increased ventricular wall stress and impaired cardiac venous drainage. On the other hand, considerable amount of volume expansion occurs on the venous side of the circulation and will cause leftward deviation of the inter-ventricular septum. This vicious cycle ultimately may contribute to diastolic dysfunction, which is more prominent in patients with preserved ejection fraction (HFpEF) [36].
Mineralocorticoid receptor antagonism as diuretics
Cirrhosis
As discussed above, inappropriate sodium retention is the underlying pathogenesis of ascites; therefore, pharmacological management should aim to improve the renal sodium excretion with dietary sodium restriction and diuretic administration.
The most efficient and accepted diuretic regimen in cirrhotic ascites consists of loop and distal diuretics in combination. Fogel et al. [44] reported combination therapy, with spironolactone and furosemide with daily doses of 100 and 40 mg, respectively, to be the most potent regimen resulting in prompt weight loss (17 % in combination group vs. 12 % in furosemide only group) and no recurrence of ascites. In this study, the group treated with only furosemide required frequent dose escalation to maintain an appropriate diuresis. Although, in healthy subjects, it has been shown that loop diuretics are more potent natriuretic agents than distal tubule diuretics, in cirrhotic patients, furosemide was less effective than spironolactone [45]. This finding is mainly due to altered furosemide pharmacokinetics in cirrhosis, which involves (1) decreased intraluminal secretion of furosemide [46], (2) exaggerated sodium uptake in proximal tubule with less distal delivery to the loop of Henle and, most importantly, (3) secondary hyperaldosteronism. Elevated plasma aldosterone levels not only cause an increase in distal sodium reabsorption but also induce an upregulation of aldosterone sensitive sodium transporters in the distal tubule leading to plasma volume expansion and edema formation [47].
With hyperaldosteronism as the cornerstone of decompensated cirrhosis, major guidelines in management of ascites recommend natriuretic doses of MRAs in conjunction with loop diuretics. The recommended regimen is spironolactone plus furosemide with starting dose of 100 and 40 mg/day, respectively [48]. The International Club of Ascites guidelines define diuretic resistance as an inadequate response to 400 mg of spironolactone and 160 mg of furosemide in cirrhotic patients with ascites [49]. Of note, to date, there are no clinical data on efficacy of eplerenone in the management of ascites.
Heart failure
Several studies have shown that despite maximal RAAS blockage, plasma aldosterone levels remain elevated in HF patients [18, 50, 51]. Also it has been shown that the aldosterone level will increase even more when diuretic therapy is intensified during decompensation [13]. In a study of stable chronic HF patients with reduced EF (<40 %) and standard medical therapy, Vittorio et al. [52] demonstrated no changes in plasma aldosterone levels after optimal ACE-I administration. The Randomized Aldosterone Evaluation Study (RALES) was the first large randomized trial to show a mortality benefit of spironolactone in patients with systolic HF and NYHA functional class III–IV [14]. Mineralocorticoid receptor antagonism in the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) improved survival in patients with systolic HF due to acute myocardial infarction [53]. This benefit was confirmed by the results of the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF) trial in patients with mild symptoms of HF [54]. In both studies, a remarkable reduction in all-cause mortality was documented (30 and 15 % in RALES and EPHESUS trial, respectively). On the other hand, MRA treatment in patients with HFpEF did not show similar beneficiary impact as described in the treatment of preserved cardiac function heart failure with an aldosterone antagonist (TOPCAT) trial [55]. In this international study of patients with HFpEF (LVEF ≥45 %), addition of spironolactone to current HF medical regimen (average dose of 25 mg/day) failed to have a beneficial impact on primary composite outcome of death from cardiovascular causes, aborted cardiac arrest or hospitalization for HF [55]. However, the spironolactone group had a nominally significant reduction in hospitalization rate (number of events in treated vs. placebo group were 206 versus 245, respectively).
The average dose of spironolactone in the RALES trial was 26 mg/day. A similar spironolactone equivalent dose of eplerenone (43.5 mg/day) was administered in the EPHESUS trial. The beneficiary impact of MRA in patients with systolic HF is considered to be due to non-genomic, non-natriuretic effects of aldosterone as the average daily dose of 25 mg in RALES did not increase urinary sodium excretion [15].
However, a few studies have shown higher doses of MRAs to promote substantial natriuresis in HF patients. Braunwald et al. [56] demonstrated the natriuretic effect of 100 mg spironolactone in three patients with HF with a significant increase in urinary sodium excretion (from 6 to 452, from 39 to 461 and from 435 to 676 mEq, respectively). In another non-randomized short-term study, administration of spironolactone 200 mg twice daily completely reversed sodium retention in advanced HF patients. There was no clinically significant hyperkalemia and mean body weight decreased by 2 kg over a 4-day period (Fig. 2) [57]. In a third study, 100 mg daily of spironolactone for seven consecutive days resulted in significant natriuresis with a 64 % decline in body overweight and symptomatic improvement in patients with decompensated HF and diuretic resistance [58].
Fig. 2.
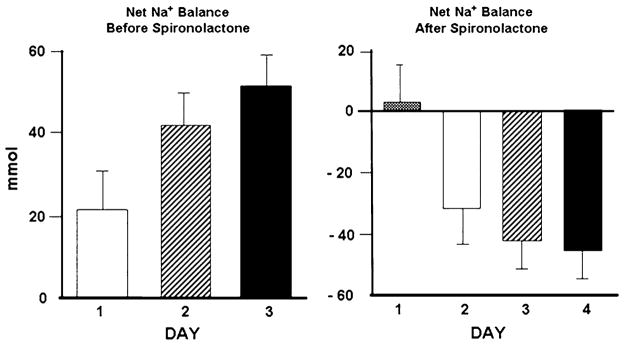
Reversal of sodium retention with aldosterone antagonism in patients with heart failure. Left positive sodium balance without spironolactone. Right negative sodium balance with spironolactone. Adapted from ref. [74]
Finally, a recent prospective single-center, single-blinded study of 100 patients with ADHF compared higher doses of spironolactone (50–100 mg daily) to standard acute HF regimen. There was no difference in use or dose of furosemide or ACE-I between the standard and treatment group. However, there was a significant improvement in sign and symptoms of congestion in spironolactone group. Moreover, a larger number of patients in the spironolactone group were transitioned to oral furosemide at day 3 (44 vs. 82 %; p <0.001). Also, a significant decrease in NT-proBNP as a surrogate of cardiac filling pressures occurred in the spironolactone group (Fig. 3). There was no incidence of hyper-kalemia in treatment group. Of note, patients with serum creatinine of>1.5 mg/dL or serum potassium>5.0 mmol/L were excluded from this study [59].
Fig. 3.

Changes in congestive sign and symptoms in control versus treated group at day 3. Adapted from ref. [59]
Adverse effects of mineralocorticoid antagonists
Hyperkalemia
Safety and tolerability of natriuretic doses of MRAs (>25 mg/day) have not been adequately studied. One major concern is the risk of hyperkalemia, which has led probably to underutilization of MRAs in even patients who are eligible to MRA treatment as per current HF guidelines [60]. In both RALES and EPHESUS trial, the incidence of clinically significant severe hyperkalemia (serum potassium>6 mEq/ L) was low. In the RALES trial, despite the fact that 95 % of patients were treated with an ACE-I or an angiotensin receptor blocker (ARB), the incidence of hyperkalemia was only 2 %. In EPHESUS trial, while 86 % of patients were on ACE-I or ARB, this incidence was 5.5 % in eplerenone group versus 3.9 % in placebo arm [14, 15] (Table 1).
Table 1.
Comparison of mineralocorticoid dose in major clinical trials in conjunction with ACE-I and β-blockers
| RALES | EPHESUS | EMPHASIS-HF | |
|---|---|---|---|
| Mean LVEF (%) | 25 | 33 | 26 |
| RF exclusion | sCr >2.5 mg/dL | sCr >2.5 mg/dL | eGFR <30 mL/min per 1.73 m2 |
| ACE-I/ARB (%) | 95 | 86 | 94 |
| β-Blocker (%) | 11 | 75 | 87 |
| Mean MRA dose (mg/day) | 26 | 43.5 | 39 |
| Serum K >6 meq/L (%) | 2 | 5.5 | 2.5 |
ACE-I angiotensin-converting enzyme inhibitor, ARB angiotensin receptor blocker, LVEF left ventricular ejection fraction, RF renal function, sCr serum creatinine, eGFR estimated glomerular filtration rate, MRA mineralocorticoid receptor blocker, serum K serum potassium
After the RALES was published, Juurlink et al. [61] reported an increase in spironolactone prescription as well as an increase rate of hospitalization and death associated with hyperkalemia (hyperkalemia defined as serum potassium >5.0 mEq/L). However, the authors considered all hospital admissions including a diagnosis of hyperkalemia. Therefore, it is unclear whether hyperkalemia was the primary reason for admission. Moreover, in this report, there was no indication of the renal function, which plays an important role in the incidence of adverse effects in MRA administration. On the other hand, a retrospective European study was unable to show a direct association between spironolactone administration and hospitalization due to MRA-induced hyperkalemia [62]. In this non-randomized cohort, the incidence of severe hyperkalemia (serum potassium >6 mEq/L) was 2.9 %, being patients with higher baseline serum creatinine or potassium at a higher risk for this complication.
In two recent subanalysis of EMPHASIS-HF trial, there was a statistically significant increased incidence of hyperkalemia with serum potassium >5.5 mEq/L in eplerenone group (11 vs. 6.8 %). Also worsening renal function (decline in eGFR >20 %) was higher in this group (30.1 vs. 24.4 %). However, the analysis did not report any significant severe hyperkalemia (serum potassium >6.0 mEq/L) or worsening renal function that was related to hospitalization or death. Moreover, eplerenone remained clinically beneficial on HF hospitalizations, cardiovascular death and all-cause mortality. These mortality benefits were independent of hyperkalemia (serum potassium >5.5 mEq/L) or worsening renal function (20–30 % decline in eGFR), even among patients at higher risk of developing hyperkalemia (i.e., age>75, baseline GFR<60, plasma potassium >4.5 mEq/L, hypertension, diabetes and antiarrhythmics drugs use) [63, 64].
The importance of close monitoring of renal function and electrolytes after initiation of MRAs especially in older patients with underlying comorbidities, i.e., diabetes, renal dysfunction, is important. A large clinical data registry analysis of elderly patients with HF (average age 77.6 years) and reduced ejection fraction (HFrEF) who were treated with aldosterone antagonists at the time of hospital discharge shows significant reduction in HF-associated readmission rate among patients who received MRAs (HR 0.87; 95 % CI 0.77, 0.98; p = 0.02). Although readmission due to hyperkalemia was higher in aldosterone antagonist group at 30 days and 1 year, the rate of hyperkalemia as the primary diagnosis for readmission was small [65].
Strategies to avoid hyperkalemia are discussed in 2013 ACCF/AHA Guideline for the management of HF and include close monitoring of serum potassium and creatinine: Potassium levels and renal function are most typically checked in 2–3 days and at 1 week after initiating therapy and at least monthly for the first 3 months [66].
In this regard, cautious initiation and titration of MRAs to natriuretic doses should be considered particularly in patients with eGFR of 30–49 mL/min/1.73 m2 and baseline serum potassium >4.5 mEq/L [67]. The ACCF/AHA Guideline recommends against administrations of MRAs in patients with eGFR <30 mL/min/1.73 m2 and/or baseline serum potassium >5.0 mEq/L.
Hyperkalemia appears to be less frequent in cirrhotic patients treated with spironolactone in comparison with HF population, while both groups routinely receive potassium-losing loop diuretics. The underlying mechanism of this observation is not adequately studied. This could be explained partially by more prevalent risk factors of renal dysfunction in HF patients, including: older age, underlying diabetes, history of prolonged hypertension and frequent incidences of acute kidney injury in the setting of decompensation episodes. The lower frequency of ACEI administration in cirrhotic patients should be also considered. On the contrary, cirrhotic patients may receive non-selective β-blockers that can induce hyperkalemia, especially in light of RAAS suppression.
Gynecomastia
Gynecomastia is an uncommon adverse effect of spironolactone with an incidence of 4 % in clinical trials [68]. Spironolactone may also cause sexual dysfunction [69]. Since eplerenone is a selective inhibitor of aldosterone receptor, there are significantly lower androgenic side effects [53, 70, 71].
Conclusions
In summary, persistent neurohormonal activation in decompensated cirrhosis and HF increases plasma aldosterone level with detrimental genomic and non-genomic effects. In a setting of congestion, escalating doses of loop diuretics may further promote neurohormonal activation as well as renal tubular hypertrophy and, ultimately, diuretic resistance. Addition of natriuretic doses of MRAs, which is currently recommended in decompensated cirrhosis, may offer an alternative to intensification of loop diuretic regimen in treating congestion in HF patients, specifically in hyper- and normotensive profile [72]. However, this hypothesis needs to be tested in prospective randomized studies with cautious patient selection with respect to subgroups at high risk of developing hyperkalemia and renal failure.
Footnotes
Conflict of interest The authors do not have any conflict of interests.
Contributor Information
Amirali Masoumi, Email: am4052@columbia.edu, Division of Cardiology, New York-Presbyterian Hospital, Columbia University College of Physicians and Surgeons, New York, NY, USA.
Fernando Ortiz, Department of Medicine, Columbia University College of Physicians and Surgeons, New York, NY, USA.
Jai Radhakrishnan, Email: jr55@columbia.edu, Division of Nephrology, New York-Presbyterian Hospital, Columbia University College of Physicians and Surgeons, New York, NY, USA.
Robert W. Schrier, Email: Robert.Schrier@ucdenver.edu, Division of Nephrology, University of Colorado School of Medicine, Aurora, CO, USA
Paolo C. Colombo, Email: pcc2001@cumc.columbia.edu, Division of Cardiology, New York-Presbyterian Hospital, Columbia University College of Physicians and Surgeons, New York, NY, USA
References
- 1.Lloyd-Jones D, et al. Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119(3):480–486. doi: 10.1161/CIRCULATIONAHA.108.191259. [DOI] [PubMed] [Google Scholar]
- 2.Ambrosy AP, et al. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol. 2014;63(12):1123–1133. doi: 10.1016/j.jacc.2013.11.053. [DOI] [PubMed] [Google Scholar]
- 3.Gheorghiade M, et al. Acute heart failure syndromes: current state and framework for future research. Circulation. 2005;112(25):3958–3968. doi: 10.1161/CIRCULATIONAHA.105.590091. [DOI] [PubMed] [Google Scholar]
- 4.Felker GM, et al. The problem of decompensated heart failure: nomenclature, classification, and risk stratification. Am Heart J. 2003;145(2 Suppl):S18–S25. doi: 10.1067/mhj.2003.150. [DOI] [PubMed] [Google Scholar]
- 5.Fonarow GC, et al. Influence of a performance-improvement initiative on quality of care for patients hospitalized with heart failure: results of the organized program to initiate lifesaving treatment in hospitalized patients with heart failure (OPTIMIZE-HF) Arch Intern Med. 2007;167(14):1493–1502. doi: 10.1001/archinte.167.14.1493. [DOI] [PubMed] [Google Scholar]
- 6.Gheorghiade M, et al. Congestion in acute heart failure syndromes: an essential target of evaluation and treatment. Am J Med. 2006;119(12 Suppl 1):S3–S10. doi: 10.1016/j.amjmed.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Fonarow GC, et al. Temporal trends in clinical characteristics, treatments, and outcomes for heart failure hospitalizations, 2002 to 2004: findings from Acute Decompensated Heart Failure National Registry (ADHERE) Am Heart J. 2007;153(6):1021–1028. doi: 10.1016/j.ahj.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 8.Allen LA, et al. Improvements in signs and symptoms during hospitalization for acute heart failure follow different patterns and depend on the measurement scales used: an international, prospective registry to evaluate the evolution of measures of disease severity in acute heart failure (MEASURE-AHF) J Card Fail. 2008;14(9):777–784. doi: 10.1016/j.cardfail.2008.07.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lucas C, et al. Freedom from congestion predicts good survival despite previous class IV symptoms of heart failure. Am Heart J. 2000;140(6):840–847. doi: 10.1067/mhj.2000.110933. [DOI] [PubMed] [Google Scholar]
- 10.Gheorghiade M, et al. Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure: a randomized controlled trial. JAMA. 2004;291(16):1963–1971. doi: 10.1001/jama.291.16.1963. [DOI] [PubMed] [Google Scholar]
- 11.Nohria A, et al. Cardiorenal interactions: insights from the ESCAPE trial. J Am Coll Cardiol. 2008;51(13):1268–1274. doi: 10.1016/j.jacc.2007.08.072. [DOI] [PubMed] [Google Scholar]
- 12.Damman K, et al. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol. 2009;53(7):582–588. doi: 10.1016/j.jacc.2008.08.080. [DOI] [PubMed] [Google Scholar]
- 13.Aronson D, Burger AJ. Neurohormonal prediction of mortality following admission for decompensated heart failure. Am J Cardiol. 2003;91(2):245–248. doi: 10.1016/s0002-9149(02)03119-3. [DOI] [PubMed] [Google Scholar]
- 14.Pitt B, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341(10):709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 15.The RALES Investigators. Effectiveness of spironolactone added to an angiotensin-converting enzyme inhibitor and a loop diuretic for severe chronic congestive heart failure (the Randomized Aldactone Evaluation Study [RALES]) Am J Cardiol. 1996;78(8):902–907. doi: 10.1016/s0002-9149(96)00465-1. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt BM, et al. Rapid nongenomic effects of aldosterone on the renal vasculature in humans. Hypertension. 2006;47(4):650–655. doi: 10.1161/01.HYP.0000205224.58715.cc. [DOI] [PubMed] [Google Scholar]
- 17.Farquharson CA, Struthers AD. Spironolactone increases nitric oxide bioactivity, improves endothelial vasodilator dysfunction, and suppresses vascular angiotensin I/angiotensin II conversion in patients with chronic heart failure. Circulation. 2000;101(6):594–597. doi: 10.1161/01.cir.101.6.594. [DOI] [PubMed] [Google Scholar]
- 18.Struthers A, Krum H, Williams GH. A comparison of the aldosterone-blocking agents eplerenone and spironolactone. Clin Cardiol. 2008;31(4):153–158. doi: 10.1002/clc.20324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schrier RW. Aldosterone ‘escape’ vs ‘breakthrough’. Nat Rev Nephrol. 2010;6(2):61. doi: 10.1038/nrneph.2009.228. [DOI] [PubMed] [Google Scholar]
- 20.Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150(11):776–783. doi: 10.7326/0003-4819-150-11-200906020-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gross E, et al. Effect of spironolactone on blood pressure and the renin–angiotensin–aldosterone system in oligoanuric hemodialysis patients. Am J Kidney Dis. 2005;46(1):94–101. doi: 10.1053/j.ajkd.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 22.Pitt B, et al. Serum potassium and clinical outcomes in the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) Circulation. 2008;118(16):1643–1650. doi: 10.1161/CIRCULATIONAHA.108.778811. [DOI] [PubMed] [Google Scholar]
- 23.Chai W, et al. Nongenomic effects of aldosterone in the human heart: interaction with angiotensin II. Hypertension. 2005;46(4):701–706. doi: 10.1161/01.HYP.0000182661.98259.4f. [DOI] [PubMed] [Google Scholar]
- 24.Schrier RW, et al. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8(5):1151–1157. doi: 10.1002/hep.1840080532. [DOI] [PubMed] [Google Scholar]
- 25.Vallance P, Moncada S. Hyperdynamic circulation in cirrhosis: a role for nitric oxide? Lancet. 1991;337(8744):776–778. doi: 10.1016/0140-6736(91)91384-7. [DOI] [PubMed] [Google Scholar]
- 26.Iwakiri Y, Groszmann RJ. The hyperdynamic circulation of chronic liver diseases: from the patient to the molecule. Hepatology. 2006;43(2 Suppl 1):S121–S131. doi: 10.1002/hep.20993. [DOI] [PubMed] [Google Scholar]
- 27.Guarner C, et al. Renal prostaglandins in cirrhosis of the liver. Clin Sci (Lond) 1986;70(5):477–484. doi: 10.1042/cs0700477. [DOI] [PubMed] [Google Scholar]
- 28.La Villa G, et al. Mineralocorticoid escape in patients with compensated cirrhosis and portal hypertension. Gastroenterology. 1992;102(6):2114–2119. doi: 10.1016/0016-5085(92)90340-5. [DOI] [PubMed] [Google Scholar]
- 29.Gines P, et al. Pathogenesis of ascites in cirrhosis. Semin Liver Dis. 1997;17(3):175–189. doi: 10.1055/s-2007-1007196. [DOI] [PubMed] [Google Scholar]
- 30.Gines P, et al. Ascites and renal dysfunction in liver disease: pathogenesis, diagnosis, and treatment. 2. Wiley; Malden: 2005. [Google Scholar]
- 31.Henriksen JH, et al. Estimated central blood volume in cirrhosis: relationship to sympathetic nervous activity, beta-adrenergic blockade and atrial natriuretic factor. Hepatology. 1992;16(5):1163–1170. [PubMed] [Google Scholar]
- 32.Arroyo V, et al. Plasma renin activity and urinary sodium excretion as prognostic indicators in nonazotemic cirrhosis with ascites. Ann Intern Med. 1981;94(2):198–201. doi: 10.7326/0003-4819-94-2-198. [DOI] [PubMed] [Google Scholar]
- 33.Trevisani F, et al. Circadian variation in renal sodium and potassium handling in cirrhosis. The role of aldosterone, cortisol, sympathoadrenergic tone, and intratubular factors. Gastroenterology. 1989;96(4):1187–1198. doi: 10.1016/0016-5085(89)91640-5. [DOI] [PubMed] [Google Scholar]
- 34.Henriksen JH, et al. The sympathetic nervous system in liver disease. J Hepatol. 1998;29(2):328–341. doi: 10.1016/s0168-8278(98)80022-6. [DOI] [PubMed] [Google Scholar]
- 35.Esler M, Kaye D. Increased sympathetic nervous system activity and its therapeutic reduction in arterial hypertension, portal hypertension and heart failure. J Auton Nerv Syst. 1998;72(2–3):210–219. doi: 10.1016/s0165-1838(98)00107-6. [DOI] [PubMed] [Google Scholar]
- 36.Schrier RW. Role of diminished renal function in cardiovascular mortality: marker or pathogenetic factor? J Am Coll Cardiol. 2006;47(1):1–8. doi: 10.1016/j.jacc.2005.07.067. [DOI] [PubMed] [Google Scholar]
- 37.Henry JP, Gauer OH, Reeves JL. Evidence of the atrial location of receptors influencing urine flow. Circ Res. 1956;4(1):85–90. doi: 10.1161/01.res.4.1.85. [DOI] [PubMed] [Google Scholar]
- 38.Linden RJ, Kappagoda CT. Atrial receptors. Monogr Physiol Soc. 1982;39:1–363. [PubMed] [Google Scholar]
- 39.Packer M. Neurohormonal interactions and adaptations in congestive heart failure. Circulation. 1988;77(4):721–730. doi: 10.1161/01.cir.77.4.721. [DOI] [PubMed] [Google Scholar]
- 40.Schrier RW. Body fluid volume regulation in health and disease: a unifying hypothesis. Ann Intern Med. 1990;113(2):155–159. doi: 10.7326/0003-4819-113-2-155. [DOI] [PubMed] [Google Scholar]
- 41.Schrier RW. Water and sodium retention in edematous disorders: role of vasopressin and aldosterone. Am J Med. 2006;119(7 Suppl 1):S47–S53. doi: 10.1016/j.amjmed.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 42.Schrier RW. Decreased effective blood volume in edematous disorders: what does this mean? J Am Soc Nephrol. 2007;18(7):2028–2031. doi: 10.1681/ASN.2006111302. [DOI] [PubMed] [Google Scholar]
- 43.Schrier RW, Abraham WT. Hormones and hemodynamics in heart failure. N Engl J Med. 1999;341(8):577–585. doi: 10.1056/NEJM199908193410806. [DOI] [PubMed] [Google Scholar]
- 44.Fogel MR, et al. Diuresis in the ascitic patient: a randomized controlled trial of three regimens. J Clin Gastroenterol. 1981;3(Suppl 1):73–80. doi: 10.1097/00004836-198100031-00016. [DOI] [PubMed] [Google Scholar]
- 45.Perez-Ayuso RM, et al. Randomized comparative study of efficacy of furosemide versus spironolactone in nonazotemic cirrhosis with ascites. Relationship between the diuretic response and the activity of the renin–aldosterone system. Gastroenterology. 1983;84(5 Pt 1):961–968. [PubMed] [Google Scholar]
- 46.Pinzani M, et al. Altered furosemide pharmacokinetics in chronic alcoholic liver disease with ascites contributes to diuretic resistance. Gastroenterology. 1987;92(2):294–298. doi: 10.1016/0016-5085(87)90120-x. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez-Llama P, et al. Sodium retention in cirrhotic rats is associated with increased renal abundance of sodium transporter proteins. Kidney Int. 2005;67(2):622–630. doi: 10.1111/j.1523-1755.2005.67118.x. [DOI] [PubMed] [Google Scholar]
- 48.Runyon BA. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology. 2013;57(4):1651–1653. doi: 10.1002/hep.26359. [DOI] [PubMed] [Google Scholar]
- 49.Gines P, Schrier RW. Renal failure in cirrhosis. N Engl J Med. 2009;361(13):1279–1290. doi: 10.1056/NEJMra0809139. [DOI] [PubMed] [Google Scholar]
- 50.Pitt B. Aldosterone blockade in patients with chronic heart failure. Cardiol Clin. 2008;26(1):15–21. doi: 10.1016/j.ccl.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 51.McKelvie RS, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation. 1999;100(10):1056–1064. doi: 10.1161/01.cir.100.10.1056. [DOI] [PubMed] [Google Scholar]
- 52.Vittorio TJ, et al. Comparison of high- versus low-tissue affinity ACE-inhibitor treatment on circulating aldosterone levels in patients with chronic heart failure. J Renin Angiotensin Aldosterone Syst. 2007;8(4):200–204. doi: 10.3317/jraas.2007.025. [DOI] [PubMed] [Google Scholar]
- 53.Pitt B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348(14):1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 54.Zannad F, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364(1):11–21. doi: 10.1056/NEJMoa1009492. [DOI] [PubMed] [Google Scholar]
- 55.Pitt B, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370(15):1383–1392. doi: 10.1056/NEJMoa1313731. [DOI] [PubMed] [Google Scholar]
- 56.Braunwald E, Plauth WH, Jr, Morrow AG. A method for the detection and quantification of impaired sodium excretion. Results of an oral sodium tolerance test in normal subjects and in patients with heart disease. Circulation. 1965;32:223–231. doi: 10.1161/01.cir.32.2.223. [DOI] [PubMed] [Google Scholar]
- 57.Hensen J, et al. Aldosterone in congestive heart failure: analysis of determinants and role in sodium retention. Am J Nephrol. 1991;11(6):441–446. doi: 10.1159/000168356. [DOI] [PubMed] [Google Scholar]
- 58.van Vliet AA, et al. Spironolactone in congestive heart failure refractory to high-dose loop diuretic and low-dose angiotensin-converting enzyme inhibitor. Am J Cardiol. 1993;71(3):21a–28a. doi: 10.1016/0002-9149(93)90241-4. [DOI] [PubMed] [Google Scholar]
- 59.Ferreira JP, et al. Mineralocorticoid receptor antagonism in acutely decompensated chronic heart failure. Eur J Intern Med. 2014;25(1):67–72. doi: 10.1016/j.ejim.2013.08.711. [DOI] [PubMed] [Google Scholar]
- 60.Chamsi-Pasha MA, et al. Utilization pattern of mineralocorticoid receptor antagonists in contemporary patients hospitalized with acute decompensated heart failure: a single-center experience. J Card Fail. 2014;20(4):229–235. doi: 10.1016/j.cardfail.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 61.Juurlink DN, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med. 2004;351(6):543–551. doi: 10.1056/NEJMoa040135. [DOI] [PubMed] [Google Scholar]
- 62.Wei L, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768. doi: 10.1136/bmj.c1768. [DOI] [PubMed] [Google Scholar]
- 63.Rossignol P, et al. Incidence, determinants, and prognostic significance of hyperkalemia and worsening renal function in patients with heart failure receiving the mineralocorticoid receptor antagonist eplerenone or placebo in addition to optimal medical therapy: results from the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF) Circ Heart Fail. 2014;7(1):51–58. doi: 10.1161/CIRCHEARTFAILURE.113.000792. [DOI] [PubMed] [Google Scholar]
- 64.Eschalier R, et al. Safety and efficacy of eplerenone in patients at high risk for hyperkalemia and/or worsening renal function: analyses of the EMPHASIS-HF study subgroups (Eplerenone in Mild Patients Hospitalization And Survival Study in Heart Failure) J Am Coll Cardiol. 2013;62(17):1585–1593. doi: 10.1016/j.jacc.2013.04.086. [DOI] [PubMed] [Google Scholar]
- 65.Hernandez AF, et al. Associations between aldosterone antagonist therapy and risks of mortality and readmission among patients with heart failure and reduced ejection fraction. JAMA. 2012;308(20):2097–2107. doi: 10.1001/jama.2012.14795. [DOI] [PubMed] [Google Scholar]
- 66.Yancy CW, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):e240–e327. doi: 10.1161/CIR.0b013e31829e8776. [DOI] [PubMed] [Google Scholar]
- 67.Schrier RW, Gheorghiade M. Challenge of rehospitalizations for heart failure: potential of natriuretic doses of mineralocorticoid receptor antagonists. Am Heart J. 2011;161(2):221–223. doi: 10.1016/j.ahj.2010.10.039. [DOI] [PubMed] [Google Scholar]
- 68.Ezekowitz JA, McAlister FA. Aldosterone blockade and left ventricular dysfunction: a systematic review of randomized clinical trials. Eur Heart J. 2009;30(4):469–477. doi: 10.1093/eurheartj/ehn543. [DOI] [PubMed] [Google Scholar]
- 69.Jeunemaitre X, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol. 1987;60(10):820–825. doi: 10.1016/0002-9149(87)91030-7. [DOI] [PubMed] [Google Scholar]
- 70.Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol. 2004;217(1–2):27–31. doi: 10.1016/j.mce.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 71.Dimitriadis G, Papadopoulos V, Mimidis K. Eplerenone reverses spironolactone-induced painful gynaecomastia in cir-rhotics. Hepatol Int. 2011;5(2):738–739. doi: 10.1007/s12072-010-9235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Albaghdadi M, Gheorghiade M, Pitt B. Mineralocorticoid receptor antagonism: therapeutic potential in acute heart failure syndromes. Eur Heart J. 2011;32(21):2626–2633. doi: 10.1093/eurheartj/ehr170. [DOI] [PubMed] [Google Scholar]
- 73.Bansal S, Lindenfeld J, Schrier RW. Sodium retention in heart failure and cirrhosis: potential role of natriuretic doses of mineralocorticoid antagonist? Circ Heart Fail. 2009;2(4):370–376. doi: 10.1161/CIRCHEARTFAILURE.108.821199. [DOI] [PubMed] [Google Scholar]
- 74.Schrier RW, Masoumi A, Elhassan E. Aldosterone: role in edematous disorders, hypertension, chronic renal failure, and metabolic syndrome. Clin J Am Soc Nephrol. 2010;5(6):1132–1140. doi: 10.2215/CJN.01410210. [DOI] [PubMed] [Google Scholar]