

Abstract
Clustered, regularly interspaced, short palindromic repeat (CRISPR) loci and their associated (Cas) proteins provide adaptive immunity against viral infection in prokaryotes. Upon infection, short phage sequences known as spacers integrate between CRISPR repeats and are transcribed into small RNA guides that identify the viral targets (protospacers) of the Cas9 nuclease. Streptococcus pyogenes Cas9 cleavage of the viral genome requires the presence of an NGG protospacer adjacent motif (PAM) sequence immediately downstream of the target. It is not known if and how viral sequences with the correct PAM are chosen as new spacers. Here we show that Cas9 specifies functional PAM sequences during spacer acquisition. The replacement of cas9 with alleles that lack the PAM recognition motif or recognize an NGGNG PAM eliminated or changed PAM specificity during spacer acquisition, respectively. Cas9 associates with other proteins of the acquisition machinery (Cas1, Cas2 and Csn2), presumably to provide PAM-specificity to this process. These results establish a new function for Cas9 in the genesis of the prokaryotic immunological memory.
Introduction
Clustered, regularly interspaced, short palindromic repeat (CRISPR) loci and their CRISPR associated (Cas) proteins provide adaptive immunity to bacteria and archaea against their viruses1. To adapt to highly dynamic viral populations, CRISPR-Cas loci evolve rapidly, acquiring short phage sequences, known as spacers, that integrate between CRISPR repeats and constitute a memory record of infection2. Spacers are transcribed into small CRISPR RNAs (crRNAs) that identify viral targets (defined as protospacers) by direct Watson-Crick pairing with invasive DNA3. Based on their cas gene content, CRISPR-Cas systems can be classified into three distinct types, I, II and III4. Each CRISPR-Cas type possesses different mechanisms of crRNA biogenesis, target destruction and prevention of autoimmunity. In the type II CRISPR-Cas system present in Streptococcus pyogenes the Cas9 nuclease inactivates infective phages using crRNAs as guides to introduce double-strand DNA breaks into the viral genome5. Cas9 cleavage requires the presence of a protospacer adjacent motif (PAM) sequence immediately downstream of the protospacer6,7. This requirement avoids the cleavage of the spacer sequence within the CRISPR array, i.e. autoimmunity, since the adjacent repeat lacks a PAM sequence. The importance of the PAM sequence for target recognition and cleavage6–9 suggests the presence of a mechanism to ensure that newly acquired spacer sequences match protospacers flanked by a proper PAM sequence. For the type I-E CRISPR-Cas system of Escherichia coli, over-expression of cas1 and cas2 is sufficient for the acquisition of new spacers in the absence of phage infection. Reports indicate that spacers acquired in this fashion match preferentially (25–70%, depending on the study) to protospacers with the correct PAM (AWG, W=A/T)10–13, suggesting that Cas1 and Cas2 are sufficient for spacer acquisition and have some intrinsic ability to recognize protospacers with the right PAM. In the type II system of S. pyogenes the PAM sequence is NGG (and also NAG at a much lower frequency)3,6,14, where N is any nucleotide, and it is recognized and bound by a domain within the Cas9 tracrRNA:crRNA-guided nuclease during target cleavage7,15. How spacers are acquired in this system, particularly how spacers with correct PAM sequences are selected during this process, is not known.
Cas9 is required for spacer acquisition
To investigate the mechanisms of recognition of PAM-adjacent protospacers during spacer acquisition, we cloned the type II-A CRISPR-Cas locus of S. pyogenes (Fig. 1a) into the staphylococcal vector pC19416 and introduced the resulting plasmid [pWJ40 (ref.17)] into Staphylococcus aureus RN422018, a strain lacking CRISPR-Cas loci. We chose this experimental system because it facilitates the genetic manipulation of the S. pyogenes CRISPR-Cas system. We first tested the ability of the cells to mount adaptive CRISPR immunity by infecting them with the staphylococcal phage ϕNM4γ4, a lytic variant of ϕNM419 (see Methods for a description of ϕNM4γ4 isolation). Plate-based assays performed by mixing bacteria and phage in top agar allowed the selection of phage-resistant colonies that were checked by PCR to look for the expansion of the CRISPR array (Extended Data Fig. 1a). On average 50 % of the colonies acquired one or more spacers (8/13, 5/11 and 7/16 in three independent experiments), whereas the rest of the resistant colonies survived phage infection by a non-CRISPR mechanism, most likely including phage receptor mutations (Extended Data Fig. 2a). To maximize the capture of new spacer sequences, we performed the same assay in liquid and recovered surviving bacteria at the end of the phage challenge. These were analyzed by PCR of the CRISPR array and the amplification products of expanded loci were subjected to Illumina MiSeq sequencing to determine the extent of spacer acquisition. Analysis of 2.96 million reads detected protospacers adjacent to 2083 out of 2687 NGG sequences present in the viral genome, although with variation in the frequency of acquisition of each sequence (Extended Data Fig. 1b). The data revealed a prominent selection of spacers matching protospacers with downstream NGG PAM sequences (99.97 %, Extended Data Fig. 1c). The acquisition of new spacers by cells in liquid culture proved to be simple and highly efficient, providing the possibility to look at millions of new spacers in a single step. It was therefore implemented in the rest of our studies.
Figure 1. Cas9 is required for spacer acquisition.

a, Organization of the S. pyogenes type II CRISPR-Cas locus. Arrows indicate the annealing position of the primers used to check for the expansion of the CRISPR array. b, PCR-based analysis of liquid cultures to check for the acquisition of new spacer sequences in the presence or the absence of phage ϕNM4γ4 infection. Wild-type (WT) as well as different cas mutants were analyzed. Image is representative of three technical replicates. MOI; multiplicity of infection. c, Cultures over-expressing Cas1, Cas2 and Csn2 under the control of a tetracycline-inducible promoter were analyzed using PCR for spacer acquisition in the absence of phage infection. The strain was complemented with plasmids carrying either St or Sp Cas9 (see Extended Data Fig. 3), in the last case with or without the tracrRNA gene (Δtracr). Image is representative of three technical replicates. aTc; anhydrotetracycline.
To determine the genetic requirements for spacer acquisition we made individual deletions of cas1, cas2 or csn2 and challenged the mutant strains with phage ϕNM4γ4. Spacer acquisition was decreased to levels below our limit of detection in each of these mutants (Fig. 1b), corroborating previous experiments12,20. Therefore while Cas1, Cas2 and Csn2 are dispensable for anti-phage immunity in the presence of a pre-existing spacer (Extended Data Fig. 2b and c), they are required for spacer acquisition. To determine whether these genes are also sufficient for this process, we over-expressed cas1, cas2 and csn2 in the absence of cas9 using a tetracycline-inducible promoter in plasmid pRH223 and looked for the integration of new spacers in the absence of phage infection using a highly sensitive PCR assay (Extended Data Fig. 3). We were unable to detect new spacers even in the presence of the inducer (Fig. 1c). However, the addition of a second plasmid expressing tracrRNA (see below) and Cas9 from their native promoters (Fig. S4a) enabled spacer acquisition only in the presence of the inducer, with all the new spacers matching chromosomal or plasmid sequences (Fig. 1c and Extended Data Table 1). Although most likely the acquisition of such spacers causes cell death or plasmid curing, respectively, the acquisition event can still be detected in liquid culture using our highly sensitive PCR assay (Extended Data Fig. 3b and c). The tracRNA (Fig. 1a) is a small RNA bound by Cas9 that is required for crRNA processing3 and Cas9 nuclease activity6. We wondered if Cas9 involvement in spacer acquisition also required the presence of the tracrRNA. Deletion of the tracrRNA prevented spacer acquisition in the absence of phage infection (Fig. 1c), suggesting that apo-Cas9 is not sufficient to promote spacer acquisition and that association with its cofactor is also required. Altogether these data indicate that Cas1, Cas2 and Csn2 are necessary but not sufficient for the incorporation of new spacers and that tracrRNA/Cas9 is also required. This is in contrast to the type I-E CRISPR-Cas system of E. coli, where over-expression of Cas1 and Cas2 alone is sufficient for spacer acquisition10–13. It is important to note that the CRISPR array used in this assay consists of a single repeat, without pre-existing spacers (Extended Data Fig. 3). Therefore the Cas9 requirement is not a consequence of the phenomenon known as “primed” spacer acquisition. This refers to an increase in the frequency of spacer acquisition observed in type I CRISPR-Cas systems that relies on the presence of a pre-existing spacer with a partial match to the phage genome as well as the full targeting complex (Cascade)12,21,22.
Cas9 specifies the PAM sequence of newly acquired spacers
Given this newfound requirement in the CRISPR adaptation process and the well-established PAM recognition function of Cas9 during the surveillance and destruction of viral target sequences, we hypothesized that this nuclease could participate in the selection of PAM sequences during spacer acquisition. To test this we exchanged the cas9 genes of S. pyogenes (Sp) and S. thermophilus (St) CRISPR-Cas systems to create two chimeric CRISPR loci: tracrRNASp-cas9St-cas1Sp-cas2Sp-csn2Sp and tracrRNASt-cas9Sp-cas1St-cas2St-csn2St (Fig. 2a). We chose the type II-A CRISPR-Cas system of S. thermophilus (also known as CRISPR323) because it is an ortholog of the S. pyogenes system24. While the PAM sequence for the Sp CRISPR-Cas system is NGG, the PAM sequence for the St system is NGGNG23 (Fig. 2b and Extended Data Table 1). We infected each naïve strain with phage ϕNM4γ4, sequenced the newly acquired spacers, and obtained the PAM of the matching protospacers using WebLogo25. We found that each chimeric system acquired spacers with PAMs that correlated with the cas9, but not the tracrRNA, cas1, cas2 or csn2, allele present (Fig. 2b and Extended Data Table 1). To rule out the possibility that non-functional spacers are negatively selected during phage infection, i.e. they are acquired randomly and only those cells containing spacers with a correct PAM for Cas9 cleavage provide immunity and allow cell survival, we sequenced the PAMs of spacers acquired in the absence of phage infection (Fig. 1c and 2c). Either Cas9Sp or Cas9St were produced in cells overexpressing Cas1Sp, Cas2Sp and Csn2Sp. In this experiment, as explained above, spacers matching chromosomal or plasmid sequences will be acquired. The PCR products containing new spacers were cloned into a commercial vector from which they were sequenced (Extended Data Table 1). Expression of Cas9Sp led to the incorporation of spacers matching protospacers with an NGG PAM sequence, whereas the expression of Cas9St in the same cells shifted the composition of the PAM to NGGNG (Fig. 2d). These results demonstrate that Cas9 specifies PAM sequences to ensure the acquisition of functional spacers during CRISPR adaptation.
Figure 2. Cas9 determines the PAM sequence of acquired spacers.

a, c, Genetic composition of the CRISPR-Cas loci tested for spacer during phage infection (a), or in the absence of infection, with the experimental set up shown in Fig. S4 (c). b, d, Sequence logos obtained after the alignment of the 3’ flanking sequences of the protospacers matched by the newly acquired spacers in panels a and c, respectively. Numbers indicate the positions of the flanking nucleotides downstream from the spacer. n; number of sequences used in each alignment.
Cas9 associates with other Cas proteins involved in spacer acquisition
In type I CRISPR-Cas systems, Cas1 and Cas2 form a complex13 and the dsDNA nuclease activity of Cas1 has been implicated in the initial cleavage of the invading viral DNA to generate a new spacer26. The genetic analyses presented above suggest that in the type II S. pyogenes CRISPR-Cas system, the PAM-binding function of Cas9 observed in vitro7 could specify a PAM-adjacent site of cleavage for Cas1, or other members of the spacer acquisition machinery. This would guarantee that newly acquired spacers have the correct PAM needed for Cas9 activity later in this immune pathway. This hypothesis predicts an interaction between Cas9 and Cas1, Cas2 and/or Csn2. To test this we expressed the type II Cas operon in E. coli, using a histidyl tagged version of Cas9, and looked for other proteins that co-purify. We observed an abundant co-purifying protein with an apparent molecular weight close to 33 kDa, the expected size of Cas1 (Extended Data Fig. 4a). Mass spectrometry confirmed the identity of both of these proteins as well as the presence of Cas2 and Csn2 co-purifying with Cas9 (Extended Data Table 2). This result suggested the formation of a Cas9-Cas1-Cas2-Csn2 complex and therefore we explored other purification strategies to unequivocally determine its existence. We were able to isolate a Cas9-Cas1-Cas2-Csn2 complex when the histidyl tag was added to Csn2 (Fig. 3a and b). The identity of the purified proteins was confirmed by mass spectrometry (Extended Data Table 3). This demonstrates a biochemical link between the Cas9 nuclease and the other Cas proteins that function exclusively to acquire new spacers, supporting the role of Cas9 as a PAM specificity factor in the adaptation phase of CRISPR immunity.
Figure 3. Cas9Sp PAM recognition domain is required for the acquisition of spacers with an NGG PAM sequence.

a, Separation of the Cas9-Cas1-Cas2-Csn2 complex by ion exchange chromatography. b, SDS-PAGE of fraction 19 (peak) from the complex elution shown in panel a, representative of five technical replicates. The four proteins of the complex were individually purified and run alongside the purified fraction to identify each protein in the complex. c, Spacer acquisition was tested as in Fig. 1c in the presence or absence of different Cas1 or Cas9 activities. Image is representative of eight technical replicates. dCas1, nuclease-dead Cas1 (E220A mutation); dCas9, nuclease-dead Cas9 (D10A, H840A mutations); Cas9PAM lacks the PAM recognition function (R1333Q, R1335Q mutations). d, Sequence logos obtained after the alignment of the 3’ flanking sequences of the protospacers matched by the newly acquired spacers in panel c. Numbers indicate the positions of the flanking nucleotides downstream from the spacer. n; number of sequences used in each alignment.
The PAM binding motif of Cas9 is required for PAM selection
Within this complex the PAM-binding domain of Cas9 would specify a functional spacer (one adjacent to a correct PAM) and the nuclease activity of Cas1 and/or Cas9 would cleave the invading DNA to extract the spacer sequence. To test this we performed adaptation studies in the absence of phage selection as described in Extended Data Fig. 3 but using different combinations of wild-type Cas1, Cas1E220A (catalytically dead or dCas126), wild-type Cas9, Cas9D10A,H840A (catalytically dead or dCas96) and Cas9R1333Q,R1335Q (Cas9PAM, containing mutations in the PAM-binding motif that substantially reduces binding to target DNA sequences with NGG PAMs in vitro15). We observed that the nuclease activity of Cas1 is necessary for spacer acquisition (Fig. 3c). In contrast, the nuclease activity and PAM-binding function of Cas9 are dispensable for this process. Next we determined the PAM of the acquired spacers in the presence of mutated Cas9 (Fig. 3d). We found that whereas spacers acquired in the presence of dCas9 displayed correct PAMs, those acquired in the presence of Cas9PAM matched DNA regions without a conserved flanking sequence, i.e. without a PAM sequence. The same result was obtained with St dCas9 (Extended Data Fig. 5). Altogether these results indicate that Cas1 and Cas9 are part of a complex dedicated to spacer acquisition which requires Cas1 nuclease activity and Cas9 PAM-binding properties for the selection of new spacer sequences.
Discussion
The selection of new spacers with a correct PAM is fundamental for the survival of the infected host during CRISPR-Cas immunity. In the simplest scenario there is no active selection of PAM-flanked protospacers; any spacer sequence can be acquired but only those with the correct PAM allow Cas9 cleavage of the invader and survival. Bacteria that acquire spacers with ineffective flanking sequences are killed by the virus and as a consequence PAM-flanking spacers are enriched in the population. Here we show that even in the absence of phage selection, the type II CRISPR-Cas system acquires new spacers with correct PAMs, a result that rules out the possibility of random spacer selection with subsequent selection for functional spacers. How are PAM-flanked protospacers selected during type II CRISPR-Cas immunity? One possibility is that the proteins exclusively dedicated to spacer acquisition perform the PAM-selection function. The inability of cells over-expressing only cas1, cas2 and csn2 to expand the CRISPR array strongly suggest that none of the proteins encoded by these genes can recognize and select correct PAMs. Another possibility is that the known PAM recognition function of Cas915,27, essential for destroying the invading virus, could also be used during spacer acquisition to recognize PAM-flanking viral sequences. Experiments showing that the cas9 allele, but not the cas1-cas2-csn2 alleles, determine the PAM sequence of the newly acquired spacers, demonstrated that this scenario is likely correct. How does Cas9 select new spacers with the correct PAMs? Our experiments demonstrate that Cas9 forms a stable complex with Cas1, Cas2 and Csn2 that presumably participates in the selection of new spacers. The nuclease activity of Cas1, but not of Cas9, is required for spacer acquisition. The tracrRNA is also required, suggesting that the apo-Cas9 structure27, very different from holo-Cas915, does not have the correct conformation to participate in spacer acquisition. The key residues involved in Cas9 PAM recognition are not required for spacer acquisition, but they are necessary for the incorporation of new spacers with the correct PAM sequence. This suggests that the reported non-specific DNA binding property of Cas96,7 is sufficient for spacer acquisition, but not for the selection of functional spacers. There are currently two models for the incorporation of new spacers into the CRISPR array, one where the future spacer sequence is cut from the invading viral DNA, the “cut and paste” model, and another where this sequence is copied from the viral genome, the “copy and paste” model28. In the context of the first model, our data suggests that, at a low frequency that may reflect the dynamics of spacer acquisition, Cas1 cleaves the invading genome to extract a new spacer sequence. However, on its own, Cas1 nuclease activity is non-specific26. Therefore we propose that through the formation of the Cas9-Cas1-Cas2-Csn2 complex, Cas9 binding to PAM-adjacent sequences provides specificity to Cas1 endonuclease activity. In the “copy and paste” model, Cas1 nuclease activity is most likely necessary for downstream events, such as the cleavage of the repeat sequence that precedes spacer insertion, and Cas9 is required to “mark” sequences adjacent to GG motifs to be copied into the CRISPR array. In any case, following yet unknown processing and integration events, the selected DNA becomes a new functional spacer, i.e. its matching protospacer will have the correct PAM to license Cas9 cleavage (Extended Data Fig. 6). The molecular steps that take place after protospacer selection to incorporate it as a new spacer in the CRISPR array are still unknown. All genes of the type II-A CRISPR-Cas locus (tracrRNA, cas9, cas1, cas2 and csn2) are required for spacer acquisition, therefore most likely all the members of the Cas9-Cas1-Cas2-Csn2 complex participate in the process. Future work will address this and other aspects of the mechanisms of spacer integration in different CRISPR-Cas systems. The present work reveals a new function for Cas9 in CRISPR immunity. This nuclease is fundamental for both the execution of immunity, participating in the surveillance and destruction of infectious target viruses, and the generation of immunological memory, selecting the viral sequences that allow adaptation and resistance to viral predators.
Methods
Bacterial strains and growth conditions
Cultivation of S. aureus RN4220 (ref.18), was carried out in brain-heart infusion (BHI) or heart infusion (HI) media (BD) at 37 °C. Whenever applicable, media were supplemented with chloramphenicol at 10 µg/ml or erythromycin at 5 µg/ml to ensure pC194- (ref.16) and pE194-derived29 plasmid maintenance, respectively.
On-plate spacer acquisition assay
To detect individual adapted colonies on a plate, cells from overnight cultures were mixed with phage at MOI = 1 in top agar containing appropriate antibiotic and 5 mM CaCl2. The mixture was poured on BHI plates with antibiotic and incubated at 37°C overnight. Subsequently, colonies that survived phage infection were restreaked on fresh BHI plates in order to remove contaminating virus and dead cells. Plates were incubated at 37°C overnight. To check for spacer acquisition, individual colonies were resuspended in lysis buffer (250 mM KCl, 5 mM MgCl2 50 mM Tris-HCl at pH 9.0, 0.5% Triton X-100), treated with 50 ng/µl lysostaphin and incubated at 37°C for 5 minutes, then 98°C for 5 minutes. Following centrifugation (13,200 rpm), a sample of the supernatant was used as template for TopTaq PCR amplification with primers L400 and H050. The PCR reactions were analyzed on 2% agarose gels (Fig. 1a).
In-liquid spacer acquisition assay
Overnight cultures launched from single colonies were diluted 1:1000 into a fresh 10-ml culture of BHI containing appropriate antibiotic and 5 mM CaCl2. When the cultures reached OD600 of 0.4, depending on the experiment, they were either infected with phage MOI = 1 (Fig. 1b) or induced with 1 µg/ml anhydrotetracycline (Fig. 1c). After 16 hours, plasmids carrying the CRISPR systems were extracted using a slightly modified QIAprep Spin Miniprep Kit protocol: the pelleted bacterial cells were resuspended in 250 µl buffer P1 containing 50 ng/µl lysostaphin and incubated at 37°C for 1 h, followed by the standard QIAprep protocol. 100 ng of plasmid DNA was used to amplify the CRISPR locus using Phusion DNA Polymerase (New England Biolabs) with the following primer mix: 3 parts JW8 and 1 part each of JW3, JW4 and JW5. The following cycling conditions were used: (1) 98 °C for 30 s; (2, 30 times) 98 °C for 10 s, 64 °C for 20 s, 72 °C for 10 s; (3) 72 °C for 5 min. The PCR reactions were analyzed on 2% agarose gels. To sequence individual spacers, the adapted bands were extracted, gel-purified and cloned via Zero Blunt TOPO PCR Cloning Kit (Invitrogen). CRISPR loci of individual clones were checked for expansion of the arrays by PCR using the primers listed above and sent for sequencing.
Phage Adsorption Assay
The phage adsorption assay was performed as described previously30 with minor modifications. Cells were grown in BHI and 10 mM CaCl2 to an OD600 of 0.4. The phage solution was prepared at 106 pfu/ml and 100 µl of this was added to 900 µl of cells. The mixture was incubated for 10 min at 37°C to allow adsorption of the phage to the cellular membrane. The mixture was centrifuged for 1 min at 13,600 rpm and the number of phage particles left in the supernatant was determined by phage titer assay.
Phage titer assay
Serial dilutions of the phage stock were prepared in triplicate and spotted on fresh top agar lawns of RN4220 in HI agar supplemented with the appropriate antibiotic and 5 mM CaCl2. Plates were incubated at 37°C overnight (Fig. S2).
High-throughput sequencing
Plasmid DNA was extracted from adapted cultures using the in-liquid spacer acquisition assay described above. 100 ng of plasmid DNA was used as template for Phusion PCR to amplify the CRISPR locus with primers H182 and H183. Following gel extraction and purification of the adapted bands, samples were subject to Illumina MiSeq Sequencing.
Plasmid construction
Construction of pWJ40 was described elsewhere17. For the construction of pC194- and pE194-derived plasmids, cloning was performed using chemically competent S. aureus cells, as described previously17. The Δcas1 (pRH059), Δcas2 (pRH061) and Δcsn2 (pRH063) mutants were constructed by one-piece Gibson assembly31 from pWJ40 using the pairs of primers H016-H017, H018-H019, H020-H021, respectively. Plasmid pRH087 containing the wild type cas genes of S. pyogenes was obtained by inserting the first spacer of S. pyogenes (annealed primers H049 and H050 containing compatible BsaI overhangs) in pDB184 using BsaI cloning32. BsaI cloning was also used to construct pRH079 and pRH233 by inserting a φNM4γ4 targeting spacer (annealed primers H029 and H030) into pDB114 and pDB184, respectively. Plasmid pRH200 harbors the wild type CRISPR3 system from S. thermophilus LMD-9 amplified with H168 and H169 from genomic DNA. The fragment was inserted on pE194 via Gibson assembly using H166 and H167. pRH213 was constructed by replacing Cas9Sp on pRH087 with Cas9St from pRH200 using the primer pairs H232-H233 and H231-H234, respectively. pRH214 was constructed by replacing Cas9St on pRH200 with Cas9Sp from pRH087 using the primer pairs H227-H230 and H228-H229, respectively. pGG32 was created by reducing the CRISPR locus of pWJ40 to a single repeat. This was accomplished by 'round the horn PCR33 using primers oGG82 and oGG83, followed by blunt ligation. pRH228 was constructed by replacing Cas9Sp on pGG32 with Cas9St from pRH200 using the primer pairs H232-H233 and H231-H234, respectively. pRH223 was constructed as a three-piece Gibson assembly combining TetR+ptet from pKL55-iTet (primers B534 and B616), pE194 (primers B532 and B617) and the cas1, cas2, csn2 genes and the array from pGG32 (primers H176-H177). pRH231 was constructed from pGG32 by one piece Gibson assembly with primers H289-H290. pRH234 contains Cas1 E220A and was constructed via one-piece Gibson assembly from pRH223, respectively, using the primer pair H312-H313. pRH227 was constructed from pGG32 via two sequential single-piece Gibson assemblies: first, D10A was introduced with B337-B338 and second, H840A was introduced with B339-B340. pRH229 was constructed via one-piece Gibson assembly from pGG32 using the primer pair H276-H277. Plasmids pRH240, pRH241, pRH242, pRH243 and pRH244 were constructed by one-piece Gibson assembly with primers H237-H238 from pGG32, pRH228, pRH227, pRH229 and pRH231, respectively. pRH245 was constructed from pRH241 via two sequential single-piece Gibson assemblies: first, D10A was introduced with H336-H337 and second, H847A was introduced with H338-H339.
Isolation and sequencing of φNM4γ4
For the initial isolation of φNM4, supernatants from overnight cultures of S. aureus Newman were filtered and used to infect soft agar lawns of TB4::φNM1,2 double lysogens19. A single plaque was picked and then plaque-purified in two additional rounds of infection using TB4 soft agar lawns, and subsequently used to lysogenize TB4. For the resultant lysogen, specific primers were used to verify the presence of φNM4 and the absence of φNM1,2 by colony PCR. High titer lysates of φNM4 (~1011 p.f.u. ml−1) were then prepared from this lineage and used for infection of TB4/pGG9 soft agar lawns harboring spacer 2B17. An escaper plaque was picked and then plaque-purified in two additional rounds of infection using TB4/pGG9 soft agar lawns. The resultant φNM4γ4 phage exhibited a clear plaque phenotype and was used to prepare a high titer lysate from which DNA was purified, deep sequenced, and assembled as described previously17. The full sequence of the ϕNM4γ4 has been deposited on Genebank under accession number KP209285, and includes a 2784 bp deletion encompassing the C-terminal 80% of the φNM4 cI-like repressor gene.
Protein purification
Cas9
pMJ806 (WT Cas9) plasmid was obtained from Addgene. Both the proteins were purified as described before6 with minor modifications as follows. The proteins were expressed in E. coli BL21 Rosetta 2(DE3) codon plus cells (EMD Millipore). Cultures (2 L) were grown at 37°C in Terrific Broth medium containing 50 µg/ml kanamycin and 34 µg/ml chloramphenicol until the A600 reached 0.6. The cultures were supplemented with 0.2 mM isopropyl-1-thio-β-d-galactopyranoside and incubation was continued for 16 h at 16 °C with constant shaking. The cells were harvested by centrifugation and the pellets stored at −80 °C. All subsequent steps were performed at 4 °C. Thawed bacteria were resuspended in 30 ml of buffer A (50 mM Tris–HCl pH 7.5, 500 mM NaCl, 200 mM Li2SO4, 10% sucrose, 15 mM Imidazole) supplemented with complete EDTA free protease inhibitor tablet (Roche). Triton X-100 and lysozyme were added to final concentrations of 0.1 % and 0.1 mg/ml, respectively. After 30 min, the lysate was sonicated to reduce viscosity. Insoluble material was removed by centrifugation for 1 hr at 15,000 rpm in a Beckman JA-3050 rotor. The soluble extract was bound in batch to mixed for 1 hr with 5 ml of Ni2+-Nitrilotriacetic acid-agarose resin (Qiagen) that had been pre-equilibrated with buffer A. The resin was recovered by centrifugation, and then washed extensively with buffer A. The bound protein was eluted step-wise with aliquots of IMAC buffer (50 mM Tris-HCl pH 7.5, 250 mM NaCl, 10% glycerol) containing increasing concentrations of imidazole. The 200 mM imidazole elutes containing the His6-MBP tagged Cas9 polypeptide was pooled together. The His6-MBP affinity tag was removed by cleavage with TEV protease during overnight dialysis against 20 mM Tris-HCl pH 7.5, 150 mM KCl, 1 mM TCEP and 10% glycerol. The tagless Cas9 protein was separated from the fusion tag by using a 5 ml SP Sepharose HiTrap column (GE Life Sciences). The protein was further purified by size exclusion chromatography using a Superdex 200 10/300 GL in 20 mM Tris HCl pH 7.5, 150 mM KCl, 1 mM TCEP, and 5% glycerol. The elution peak from the size exclusion was aliquoted, frozen and kept at −80 °C.
Cas1
Plasmid pKW01 (Cas1-WT) was constructed by through amplification of pWJ40 as a template for polymerase chain reactions (PCRs) to clone Cas1 into pET28b-His10Smt3 using the primers PS192 and PS193. Full sequencing of cloned DNA fragment confirmed perfect matches to the original sequence. The pKW01 plasmid was transformed into E. coli BL21 (DE3) Rosetta 2 cells (EMD Millipore). Cultures were grown and protein was purified by Ni- affinity chromatography step, as mentioned before in Cas9 purification. The 200 mM imidazole elutes containing the His10-Smt3 tagged Cas1 polypeptide was pooled together. The His10-Smt3 affinity tag was removed by cleavage with SUMO protease during overnight dialysis against 50 mM Tris-HCl pH 7.5, 250 mM NaCl, 20 mM and 10% glycerol. The tagless Cas1 protein was separated from the fusion tag by using a second Ni-NTA affinity step. The protein was further purified by size exclusion chromatography using a Superdex 200 10/300 GL in 20 mM Tris HCl pH 7.5, 500 mM KCl, 1 mM TCEP, and 5% glycerol. The elution peak from the size exclusion was aliquoted, frozen and kept at −80 °C.
Cas2
The sequence encoding Cas2 was PCR amplified with primers PS334 and PS335 from pWJ40 and inserted into a pET-His6 MBP TEV cloning vector (Addgene Plasmid # 29656) using ligation independent cloning (LIC). Sequencing of the resultant plasmid (pPS059) confirmed the matches to the wild type sequence. The protein was expressed and purified following the same procedure as that for Cas9.
Csn2
Plasmid pPS060 was constructed by through amplification of pWJ40 as a template for polymerase chain reactions (PCRs) to clone Csn2 into pET28b-His10Smt3 using the primers PS336 and PS337. Full sequencing of cloned DNA fragment confirmed perfect matches to the original sequence. Csn2 was expressed and purified following the same method as that of Cas1. Protein concentrations for all the purifications were determined by using the Bradford dye reagent with BSA as the standard. Previously Csn2 was shown to form a tetramer34.
Cas9-Cas1-Cas2-Csn2 complex
pKW07 (His10-Cas9-Cas1-Cas2-Csn2) was constructed by amplification of pWJ40 with primers PS199/PS202 and pET16b (Novagen) with primers PS200/PS203, followed by Gibson assembly of the fragments. Plasmid pPS061 (His10Cas9-Cas1) was created by amplification of pWJ40 with primers PS202/PS355 and pET16b (Novagen) with primers PS203/PS354, followed by Gibson assembly of the fragments. Full sequencing of cloned DNA fragment was done to confirm perfect matches to the original sequence. The proteins were expressed in E. coli BL21 Rosetta 2(DE3) codon plus cells (EMD Millipore). Cultures were grown and protein was purified by Ni- affinity chromatography step, as mentioned before in Cas9 purification with minor modifications. The 200 mM imidazole eluates were dialyzed overnight against 20 mM Tris-HCl pH 7.5, 150 mM KCl, 1 mM TCEP and 10% glycerol and subjected to mass spectrometry for the identification of the co-purifying proteins. pKW06 (Cas9-Cas1-Cas2-Csn2-His6) was constructed by amplification of pWJ40 with primers PS204/PS205 and pET23a (Novagen) with primers PS206/PS207, followed by Gibson assembly of the fragments. Full sequencing of cloned DNA fragment was done to confirm perfect matches to the original sequence. The proteins were expressed in E. coli BL21 Rosetta 2(DE3) codon plus cells (EMD Millipore). Cultures were grown and protein was purified by Ni- affinity chromatography step, as mentioned before in Cas9 purification with minor modifications. The 200 mM imidazole eluates were dialyzed overnight against 20 mM Tris-HCl pH 7.5, 150 mM KCl, 1 mM TCEP and 10% glycerol. The proteins were further purified using a 5 ml SP Sepharose HiTrap column (GE Life Sciences), eluting with a linear gradient of 150 mM – 1 M KCl.
Extended Data
Extended Data Figure 1. The S. pyogenes type II CRISPR-Cas system displays a strong bias for the acquisition of spacers matching viral protospacers with NGG PAMs.

a, Analysis of bacteriophage-insensitive mutant colonies using PCR and agarose gel electrophoresis, representative of five technical replicates. Bacteria and phage were mixed in top agar and incubated overnight. DNA was isolated from individual colonies resistant to phage infection and used as template for a PCR reaction with primers (arrows) H182 and H183 (Extended Data Table 2), which amplify the 5’ end of the S. pyogenes CRISPR array. The size of the PCR band indicates the number of new spacers (shown at the top of the gel). Cells without additional spacers resist infection by a CRISPR-independent mechanisms, presumably envelope resistance. b, Analysis of acquired spacers during phage infection of a population of bacteria carrying the S. pyogenes type II CRISPR-Cas system. Liquid cultures of bacteria were infected with phage, surviving cells were collected at the end of the infection, DNA extracted and used as template for a PCR reaction as described above. Amplification products were separated by agarose gel electrophoresis and the DNA of the bands corresponding to products with additional spacers was extracted and sent for Mi-Seq next generation sequencing. Reads corresponding to newly acquired spacers were plotted according to their position in the phage ϕNM4γ4 genome (x-axis) and their abundance (y-axis). Each dot represents a unique spacer sequence; blue and red dots indicate a corresponding protospacer with an NGG or non-NGG PAM. Top and bottom plots indicate protospacers in the top and bottom strands of the ϕNM4γ4 DNA. The map as well as the different functions of the phage genes are indicated in between the plots. The raw data used to make this graph is in the Supplementary file. c, Weblogo showing the conservation of the 5’ flanking sequences of 10,000 protospacers randomly selected from the experiment shown in b. Absolute conservation of the NGG PAM was observed.
Extended Data Figure 2.

a, Analysis of bacteriophage-resistant mutants that do not acquire a new spacer. Three colonies that survived phage infection in our in-plate adaptation assay (Fig. S1a) were subjected to phage adsorption assay. Briefly, surviving colonies as well as the wild-type S. aureus RN4220 control were grown in liquid and mixed with bacteriophage. After a brief incubation, cells were pelleted by centrifugation and the phages present in the supernatant (unable to bind and infect cells) were counted on a lawn of sensitive cells. The number of plaque-forming units (pfu) of a control experiment in the absence of host cells were used to determine the 100% free-phage, or 0% adsorption, value. No plaques were observed in the control experiment using wild-type cells and this value was used to set the 100% adsorption limit. The three CRISPR-independent, bacteriophage-resistant mutants displayed a marked defect in phage adsorption (about 50 %), indicating that most likely they carry envelope resistance mutations. Error bars: mean ± s.d. (n=3). b, cas1, cas2 and csn2 are not required for the execution of immunity using previously acquired spacers. Position within the phage ϕNM4γ4 genome of the type II CRISPR-Cas target used in this experiment. The protospacer sequence is in the bottom strand (shown in 3’–5’ direction) and flanked by a TGG PAM (in green). c, Comparison of immunity provided by a type II CRISPR-Cas system programmed to target the sequence shown in panel a in the presence (wild-type, wt) or absence (Δcas1,Δcas2, Δcsn2) of cas1, cas2 and csn2. Immunity is measured as the plaque forming units (pfu) of a φNM4γ4 phage lysate spotted on top agar lawns of S. aureus RN4220 cells containing no CRISPR system (−), a wild type S. pyogenes CRISPR-Cas type II system (wt, pRH233), or the same CRISPR-Cas systems with a deletion of cas1, cas2 and csn2 genes (Δcas1,Δcas2, Δcsn2, pRH079). Error bars: mean ± s.d. (n=3).
Extended Data Figure 3. Generation of an experimental system for the overexpression of cas1, cas2 and csn2 and the detection of spacer acquisition in the absence of phage infection.

a, Plasmids used in the spacer acquisition experiments presented in Fig. 1c and Fig. 2c–d. pRH223 contains cas1, cas2 and csn2 from S. pyogenes under a tetracycline-inducible promoter. Cells containing this plasmid only acquired spacers when a second plasmid expressing cas9 was introduced, pRH240 or pRH241, containing the tracrRNA gene, the leader and first repeat from the S. pyogenes type II CRISPR-Cas system as well as cas9 from S. pyogenes (cas9Sp) or S. thermophilus (cas9St), respectively. The leader is a short, AT rich sequence immediately upstream of the first repeat that contains the promoter for the transcription of the CRISPR array. b, Highly sensitive PCR assay to enrich for amplification products of adapted CRISPR loci. Arrows indicate primer annealing position and direction. The forward primer (JW8) anneals on the leader. For the reverse primer, a cocktail of JW3, JW4 and JW5 was used. The three reverse primers anneal on the repeat and differ only in their 3’-end nucleotide that never matches the last nucleotide of the leader (red arrowhead). Because this nucleotide is critical for the annealing of the primers, loci that acquire spacers ending in A, C or T are preferentially amplified over unadapted loci. c, To quantify the sensibility of this technique, we mixed pGG32 (one repeat, unadapted) with pRH087 (repeat-spacer-repeat, adapted) in known ratios. The amplification of adapted plasmid was detected even when it represented 0.01% (104) of the total plasmid template, representative of three technical replicates. This highly sensitive PCR assay is not required to detect acquisition during phage infection, as in this case adapted cells survive and are enriched within the population, making their detection much easier.
Extended Data Figure 4. Purification of a Cas9-Cas1-Cas2-Csn2 complexes.
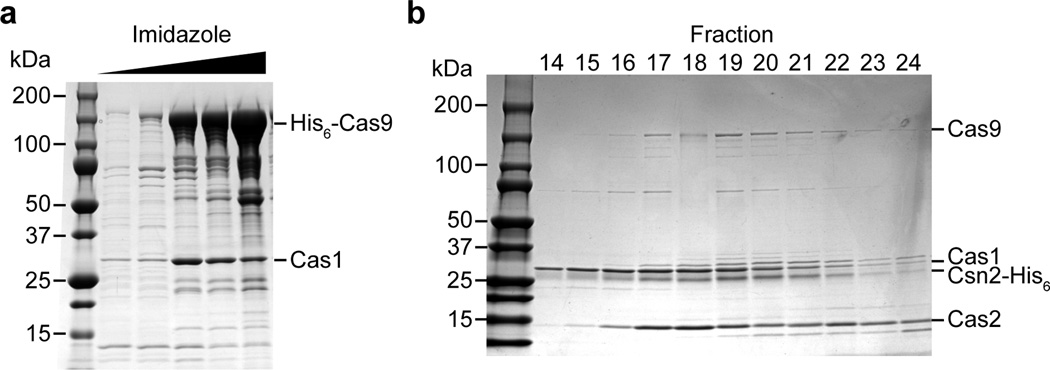
a, The cas9-cas1-cas2-csn2 operon of S. pyogenes SF370 was cloned into the pET16b vector (generating pKW07) to add an N-terminal histidyl tag to Cas9 and express all proteins in E. coli. Purification was performed using Ni-NTA affinity chromatography. SDS-PAGE followed by Coomassie stain of the purified proteins revealed a co-purifying protein that was identified as Cas1 by mass spectrometry, representative of five technical replicates. Mass spectrometry identification of all the eluted proteins co-purifying with Cas9 is shown in Extended Data Table 2. b, The cas9-cas1-cas2-csn2 operon of S. pyogenes SF370 was cloned into the pET23a vector (generating pKW06) to add an C-terminal histidyl tag to Csn2 and express all proteins in E. coli. Purification was performed using Ni-NTA affinity chromatography followed by ion exchange chromatography. The elution fractions that constituted the peak containing the complex (Fig. 3a) were separated by SDS-PAGE and visualized by Coomassie staining, representative of three technical replicates.
Extended Data Figure 5. dCas9St can also support spacer acquisition.

A plasmid derived from pRH241 containing mutations in the active site of St Cas9 (D10A, H847A; dCas9St) was used to characterize spacer acquisition in the absence of phage infection. Upon over-expression of Cas1, Cas2 and Csn2 using anydrotetracycline (aTc), we were able to detect spacer acquisition. Sequencing of spacers and alignment of the protospacer flanking sequences demonstrated the selection of an NGGNG PAM. Image is representative of three technical replicates.
Extended Data Figure 6. A model for the selection of PAM-flanking spacers by Cas9.

After injection of the phage DNA an adaptation complex formed by Cas9, Cas1, Cas2 and Csn2 uses the Cas9 PAM binding domain to specify functional protospacers, i.e., that are followed by the correct PAM. It is not known how the protospacer sequence is extracted from the viral DNA to become a spacer. In the “cut and paste” model, a nuclease, possibly Cas1, cuts the viral DNA to generate the spacer. In the “copy and paste” model the protospacer sequence is copied first. Once loaded with the selected protospacer sequence, this complex promotes the integration of this sequence into the CRISPR array, thus becoming a new spacer. Previous studies demonstrated that Cas1 dimerizes and interacts with Cas2 (ref.13); Csn2 has been determined to form a tetramer34.
Extended Data Table 1.
Sequences of the spacers analyzed in the sequence logos of Figs. 2, 3 and Extended Data Fig. 5.
| Primer | Sequence |
|---|---|
| B337 | gacgctatttgtgccgatagctaagcctattgagtatttc |
| B338 | gaaatactcaataggcttagctatcggcacaaatagcgtc |
| B339 | ggaaactttgtggaacaatggcatcgacatcataatcact |
| B340 | agtgattatgatgtcgatgccattgttccacaaagtttcc |
| B532 | ctttttccgtgatggtaactgttcatatttatcagagctcgtg |
| B534 | gagctctgataaatatgaacagttaccatcacggaaaaaggttatg |
| B616 | ttattttaattatgctctatcaa |
| B617 | gagtgatcgttaaatttatactgc |
| H016 | aggaggtgactgatgggagttcctgaatttaggatatgag |
| H017 | taaattcaggaactcccatcagtcacctcctagctgactc |
| H018 | ttaggatatgagtgaggcttttgatgaatcttaatttttc |
| H019 | ttcatcaaaagcctcactcatatcctaaattcaggaactc |
| H020 | tttgatgaatcttaataaaaatatggtataatactcttaa |
| H021 | ttataccatatttttattaagattcatcaaaagcctcccc |
| H029 | aaacaaaaatgttttaacacctattaacgtagtatg |
| H030 | aaaacatactacgttaataggtgttaaaacattttt |
| H049 | aaactgcgctggttgatttcttcttgcgctttttg |
| H050 | aaaacaaaaagcgcaagaagaaatcaaccagcgca |
| H166 | gaaatgtgagaagggacctctgataaatatgaacatgatgagtgatcg |
| H167 | ggactcttttatctctactcgtgctataattatactaattttataaggagg |
| H168 | agtataattatagcacgagtagagataaaagagtcctttggatgattcc |
| H169 | tgttcatatttatcagaggtcccttctcacatttcaatactagactc |
| H176 | ttgatagagcataattaaaataagatgccactcttatccatcaatcc |
| H177 | gcagtataaatttaacgatcactctaaaacctctccaactacctccc |
| H182 | nnnnncagcaaaattttttagacaaaaatagtc |
| H183 | nnnnncagaagaagaaatcaaccagcgc |
| H227 | taatggcaggttggagaacagtagtc |
| H228 | actactgttctccaacctgccattagtcacctcctagctgactc |
| H229 | agatttttcaaataaggagaaatgtttgaaatcatcaaactcattatggatttaatttaaactttttattttagg |
| H230 | acatttctccttatttgaaaaatctaaatttatagaaattattatacgc |
| H231 | aactttttattttaggaggcaaaaagcgtataataatttctataaatttagatttttcaaataagg |
| H232 | ttttgcctcctaaaataaaaagtttaaattaaatccataatgag |
| H233 | tgatggctggttggcgtac |
| H234 | caacagtacgccaaccagccatcaaccctctcctagtttggc |
| H237 | ggcgtactgatgaagattatttcttaataactaaaaatatgg |
| H238 | tttagttattaagaaataatcttcatcagtacgccaaccagcc |
| H276 | ttgatcaaaaacaatatacgtctacaaaagaag |
| H277 | tagacgtatattgtttttgatcaattgttgtatcaa |
| H289 | agcgcttgggagaaattcaaagaaatttatcagcc |
| H290 | tttctttgaatttctcccaagcgctttcaaaacgc |
| H312 | gatattatggcaccatttaggcctttagtgg |
| H313 | aaaggcctaaatggtgccataatatcgctagc |
| H336 | catactcaattggacttgctattggaacgaatagtgttgg |
| H337 | cgttccaatagcaagtccaattgagtatggcttagtc |
| H338 | gtaattatgatattgatgctattattcctcaagc |
| H339 | gaggaataatagcatcaatatcataattacttaatc |
| JW3 | aaaacagcatagctctaaaacg |
| JW4 | aaaacagcatagctctaaaaca |
| JW5 | aaaacagcatagctctaaaact |
| JW8 | ggcttttcaagactgaagtctag |
| L400 | cgaaattttttagacaaaaatagtc |
| oGG82 | aacattgccgatgataacttgag |
| oGG83 | gttttgggaccattcaaaacagcatagctctaaaacctcgtag |
| PS192 | CGCGGATCCATGGCTGGTTGGCGTACTGTTGTGG |
| PS193 | CGCCTCGAGTCATATCCTAAATTCAGGAACTCC |
| PS199 | CGAGCATATGACGACCTTCGATATGATCGGCAATGTTGAATGGAGACCATTC |
| PS200 | GAATGGTCTCCATTCAACATTGCCGATCATATCGAAGGTCGTCATATGCTCG |
| PS202 | CATCATCATCATCATCACAGCAGCGGCATGGATAAGAAATACTCAATAGG |
| PS203 | CCTATTGAGTATTTCTTATCCATGCCGCTGCTGTGATGATGATGATGATG |
| PS204 | CGACAAGCTTGCGGCCGCACTCGAGCTTTTTATTTTAGGAGGCAAAAATG |
| PS205 | GGATCTCAGTGGTGGTGGTGGTGGTGTACCATATTTTTAGTTATTAAGAAATAATC |
| PS206 | GATTATTTCTTAATAACTAAAAATATGGTACACCACCACCACCACCACTGAGATCC |
| PS207 | CATTTTTGCCTCCTAAAATAAAAAGCTCGAGTGCGGCCGCAAGCTTGTCG |
| PS284 | GCTAGCGATATTATGGcACCATTTAGGCCTTTAG |
| PS285 | CTAAAGGCCTAAATGGTgCCATAATATCGCTAGC |
| PS334 | TACTTCCAATCCAATGCAATGAGCTATCGCTATATG |
| PS335 | TTATCCACTTCCAATGTTATTATTAGCTTTCATCAAAGGC |
| PS336 | CGCGGATCCATGAACCTGAACTTTAGCCTGCTGG |
| PS337 | CGCCTCGAGTTACACCATATTTTTGGTAATCAG |
| PS354 | GTTCCTGAATTTAGGATATGAAACATTGCCGATCATATCGAAGG |
| PS355 | CCTTCGATATGATCGGCAATGTTTCATATCCTAAATTCAGGAAC |
Extended Data Table 2.
Mass spectrometry analysis of proteins purified through Ni-NTA shown in Extended Data Fig. 4a.
| Figure | Spacer | Sequence PAM | Target |
|---|---|---|---|
| 2b | 1 | gcaacaatgggaaccaagctatgttgatag aGGgt | phage |
| 1st logo | 2 | gagaacaaaaccatcctacccggtaataaa tGGta | phage |
| 3 | aatagagatactttatctaacatgatacac gGGag | phage | |
| 4 | ccattttagatttcaaaagtttagtatctat aGGca | phage | |
| 5 | agtattggaatctgatgaatattcatctct cGGta | phage | |
| 6 | agaaaatttatacattgattattcaccaac aGGca | phage | |
| 7 | acatactccaaacaattgatggatttgtgt aGGtg | phage | |
| 8 | gctaagactgtgaagcataatactgctact aGGta | phage | |
| 9 | ttttaagctattcattttaaaaggtcatat gGGca | phage | |
| 10 | acttatgccgtttctatacttcactacagca tGGtc | phage | |
| 11 | atgaatggattgaagagaacacagacgaac aGGac | phage | |
| 12 | ccacaaatagaaatagagctagggagtttaa cGGta | phage | |
| 13 | attagttactccacaaatagaaatagagct aGGga | phage | |
| 14 | ggagtaactaatatctgaattgttatcagt tGGtt | phage | |
| 15 | tagttttttgagtatgcttactttttcttg tGGtt | phage | |
| 16 | tgaacgaattgtcagtatgtacagattaat aGGaa | phage | |
| 17 | cattacggacgtagtagaagcaattagaaa tGGaa | phage | |
| 18 | tggatatgacgaccaagatttagcgtttta aGGtg | phage | |
| 19 | cgacataacgctaatacatgtttgtcatag tGGtt | phage | |
| 20 | acaaacttaacaatagtggttttttcaaga gGGag | phage | |
| 2b | 1 | agagtacaatattgtcctcattggagacac tGGgG | phage |
| 2nd logo | 2 | tgtttgggaaaccgcagtagccatgattaa gGGtG | phage |
| 3 | ctcatattcgttagttgcttttgtcataaa aGGtG | phage | |
| 4 | tttatgtctatatactcaaagtaatcatttt cGGaG | phage | |
| 5 | taatatcaacggtatgtgggtgtctggtga cGGtG | phage | |
| 6 | aataagtctaaaaaaccaacgtttaatgat tGGgG | phage | |
| 7 | gttgatattacgttcatagaacatacctga tGGtG | phage | |
| 8 | tcaatgtttggtacaagttggtcacagata tGGaG | phage | |
| 9 | ttagttactccacaaatagaaatagagcta gGGaG | phage | |
| 10 | caattgtttttcttggaaatcatatttata cGGcG | phage | |
| 11 | tatctaagtttgccaattattacattaaagc tGGtG | phage | |
| 12 | taggacatagagatgaaaaaacgactataa aGGtG | phage | |
| 13 | tgaagaaatgattcaagaaacacaaaagag tGGcG | phage | |
| 14 | tcggactgttagggtacgcgaagggcaaaa aGGaG | phage | |
| 15 | aatactttcttctaaaaaacctaagtcaac aGGaG | phage | |
| 16 | taatccaattacaacattaaaaattaatga cGGaG | phage | |
| 17 | acaatgttaagcaaccagcacattacacata cGGcG | phage | |
| 18 | ggattttaaaataaaagtaaatgttgatac tGGcG | phage | |
| 19 | caggcaatgttattttatcggattttaaaaa cGGcG | phage | |
| 20 | agaatctttattattagctgacttacaaga aGGtG | phage | |
| 21 | aaaaccccaatatcttttaaaaataaagtt aGGtG | phage | |
| 22 | tagggcaatgattgaagaatttgatgataa cGGaG | phage | |
| 2b | 1 | aaaggcaacatatttgaatcatcacatttat tGGaG | phage |
| 3rd logo | 2 | ttggaatggaattaaacaataaaactttta tGGaG | phage |
| 3 | atattcatcagattccaatactacgttaat aGGtG | phage | |
| 4 | acaatttaaaaattagaaatgtaaatgtag aGGtG | phage | |
| 5 | cagaatgaactatgaaacaggggtccaact aGGtG | phage | |
| 6 | acataacatcaaaaccctttctgaagaaat tGGtG | phage | |
| 7 | taagttgtttgaaatgtacgagatggaagg aGGaG | phage | |
| 8 | atacgtgtaaagacatattagatcgagtca aGGaG | phage | |
| 9 | tgtgcaggagctacgttcaataaatgtgaa aGGaG | phage | |
| 10 | ttaagaaagttattgtcatcgagcttaaat tGGtG | phage | |
| 11 | acacacatactaaacctgaacgattaagga gGGgG | phage | |
| 12 | tttaccaacatccttagttgatagattttt aGGcG | phage | |
| 13 | gtttgaatacgttccgtttctgatacccagt aGGcG | phage | |
| 14 | aagttaaaaagaatttaaagtcaagaagta tGGgG | phage | |
| 15 | attctcagaagatagcgaagatgggagaaa aGGaG | phage | |
| 16 | tgagcgactgctgggtgtgcttcgaatagtt tGGcG | phage | |
| 17 | taatatatgctcatacttaattgaattgtc tGGtG | phage | |
| 18 | atcttcttttttaatacgtccatcaacaag cGGtG | phage | |
| 19 | cgatattggcggtgtgaataataactttaa aGGaG | phage | |
| 20 | caacgagctggcaacaacataagatgacag aGGcG | phage | |
| 2b | 1 | taaactactacgacttaagcaggtgccata tGGca | phage |
| 4th logo | 2 | gacaaatgctattcaacattcagttaaaga aGGta | phage |
| 3 | acaattattaattgaacaagcgcaagctaa cGGct | phage | |
| 4 | cacatcaattagtaagacgccaaaagtaac aGGta | phage | |
| 5 | aaacgatgagtacacaaaatacaaaatcta cGGca | phage | |
| 6 | gtaataatatttttaataacctcaacatct tGGtc | phage | |
| 7 | tcatgaaaaagtgaattgctagtagtgtgt tGGtc | phage | |
| 8 | tacgctatcgcaaaagcagtcaaagctaaa gGGca | phage | |
| 9 | agggaatcttacagttattaaataactatt tGGat | phage | |
| 10 | aaaacgagcaaattaagtggtacgtagaca aGGgt | phage | |
| 11 | ctaaatgttgccatttcgttatctcctttc tGGta | phage | |
| 12 | actggatgacattgaacaaagcaccgaata tGGcc | phage | |
| 13 | taaatatttgataacaacattatacacgaa aGGag | phage | |
| 14 | cacatcaattagtaagacgccaaaagtaac aGGta | phage | |
| 15 | aaggtgatgacggcgaatggtacacaacata tGGtc | phage | |
| 16 | taacgacggtacttattccgtcgttgctac tGGtg | phage | |
| 17 | ataaataaaaaagttactactcacacacta aGGca | phage | |
| 18 | tctaggttcgaactcttctttaaatttaat aGGca | phage | |
| 19 | ctcatcaatatcattctgattggttatttt gGGat | phage | |
| 20 | tctctttgataaataactttatccacataa aGGtg | phage | |
| 21 | ttagacttttactttccattacttaaatca tGGtc | phage | |
| 22 | aatttgttcttgcgcttcaatagtgatagt aGGgt | phage | |
| 23 | ataagtctaaaaaaccaacgtttaatgatt gGGga | phage | |
| 2d | 1 | acatgttatgcatatcgtaagtgaagtcac aGGta | chromosome |
| 1st logo | 2 | agatcaaattgtaacaactaatcctattgc aGGta | chromosome |
| 3 | gtttcagcaatatatctcttagtgcatcac cGGtt | chromosome | |
| 4 | tacaatgtaggctgctctacacctagcttc tGGgc | pRH223 | |
| 5 | tttgattacaatggcacatgtacttatgcc tGGat | chromosome | |
| 6 | catttgtcttagcacatgaattaggtcatgc aGGtc | chromosome | |
| 7 | catgattgcacccattgttgcacctagtac aGGtt | chromosome | |
| 8 | taccaataacttaagggtaactagcctcgc cGGca | chromosome | |
| 9 | gagtatgtttgcgcgtgaagtggttgtgtc tGGat | pRH223 | |
| 10 | agaatggttagatttatggcgtgatgtaac gGGca | chromosome | |
| 11 | ctgcttccatgataactggaccatcagcaac cGGat | chromosome | |
| 12 | gattgaagctacaatacctgatgttgctgc gGGaa | chromosome | |
| 13 | cgaaatacttggctaagcacgacgaggcct tGGtg | pRH223/240 | |
| 14 | gctctacacctagcttctgggcgagtttac gGGtt | pRH223 | |
| 15 | atatggaagttacattttttggaacgagtgc aGGtt | chromosome | |
| 16 | ttatgaagcgttacgtcaacaagattttcc aGGat | chromosome | |
| 17 | ttatcgaagtatacgagttcacagaagaac aGGct | chromosome | |
| 18 | ccagttcttgttgttttggtgctttagtca aGGtt | chromosome | |
| 19 | tggatgatcttgtctttcatgtgtacctgt tGGaa | chromosome | |
| 20 | caggatttagttttcctagcggtcatgcta tGGga | chromosome | |
| 2d | 1 | ttgactatcaaatgtctttttcaatgtttc gGGtG | chromosome |
| 2nd logo | 2 | atccgttctgcagaagagattgtttcttgc aGGcG | pRH223 |
| 3 | tgaacatttcgattatgtattaatgagtgc tGGtG | chromosome | |
| 4 | catctttaggacgaatgccagcacgttctgc tGGaG | chromosome | |
| 5 | caccatgttaaaaatacctccatcatcacc aGGaG | pRH223/240 | |
| 6 | tcgtgagacagttcggtccctatccgtcgt gGGcG | chromosome | |
| 7 | tttgcgcagtcggcttaaaccagttttcgc tGGtG | pRH223 | |
| 8 | aaagaagtcataagtaccatgacttgagtt tGGtG | chromosome | |
| 9 | ctaatttttcttcttcaacaccatctatggc tGGcG | chromosome | |
| 10 | ccaagtattcaaagttggaacgggtggtct aGGtG | chromosome | |
| 11 | atccgttctgcagaagagattgtttcttgc aGGcG | pRH223 | |
| 12 | tttgcgcagtcggcttaaaccagttttcgc tGGtG | pRH223 | |
| 13 | aacgcgtatacatagcaagcgttctcatgt tGGaG | chromosome | |
| 14 | agtttgggagtcaattatcggctttttaac tGGcG | chromosome | |
| 3d | 1 | tgacttctctgaagagccatctttttgcact tGGaa | chromosome |
| 1st logo | 2 | ggtcagatgcaattcgacatgtggacggac tGGtt | pRH223 |
| 3 | atcttttctagcttttctccaagcacagac aGGac | chromosome | |
| 4 | gttggtctaattgtttcaatagttccacct tGGtc | chromosome | |
| 5 | tgccggttggggtggctgagacggcaccct aGGaa | chromosome | |
| 6 | tgagtatgtttgcgcgtgaagtggttgtgtc tGGat | pRH223 | |
| 7 | ttgagttagaaaacggtcgtaaacggatgc tGGct | pRH242 | |
| 8 | agtttgggagtcaattatcggctttttaac tGGcg | chromosome | |
| 9 | aattaagaaatcttctaaccaactgattgc tGGaa | chromosome | |
| 10 | aacagaaagaataggaaggtatccgactgc tGGta | pRH223/240 | |
| 11 | tggtattgtaggcgttattttaggtattcc gGGat | chromosome | |
| 12 | aaatctcagcaggacaagctggtacaggtgc tGGtt | chromosome | |
| 13 | ctcaagagatttggagcatccaatcaatgc aGGtc | pRH223 | |
| 14 | ctaaggtggcaccacggtaacgcgtccttac aGGta | chromosome | |
| 15 | tgattaaacttaaaaatgtattacctagtgc aGGta | chromosome | |
| 16 | atttgagtcagctaggaggtgactgatggc tGGtt | pRH223 | |
| 17 | ataagagaagatgctagacgtataagttcac tGGtc | chromosome | |
| 18 | acgttttatctgtatttgcgacaatcgttg gGGta | chromosome | |
| 19 | ataacatacgccgagttatcacataaaagc gGGaa | pRH223 | |
| 20 | gcattttaaacaaaaaaagatagacagcac tGGca | pRH223 | |
| 21 | aatcccagttagaacaaacgctaaaatggc gGGcc | chromosome | |
| 22 | taccaataacttaagggtaactagcctcgc cGGca | chromosome | |
| 3d | 1 | gaagtctagctgagacaaatagtgcgatta caaaa | pRH223/240 |
| 2nd logo | 2 | agcatagctctaaaacctcgtagactattt ttgtc | pRH223/240 |
| 3 | aaattttttagacaaaaatagtctacgag gtttt | pRH223/240 | |
| 4 | aagtcgaacttcataatcatcgctttcgg catat | chromosome | |
| 5 | ccaatttctacagacaatgcaagttggggt gtggg | chromosome | |
| 6 | gttatttctgaaatgcccgttacatcacgc cataa | pRH243 | |
| 7 | tgtttgccctccaaatatgaaaacatggcc cggta | chromosome | |
| 8 | atgagatgaggcgataaaagaacgtcgcta aaacg | chromosome | |
| 9 | tactacttcaaggaattctatagaacctac tatat | chromosome | |
| 10 | gtaccacagtgccacatgttggcaattggc gagac | chromosome | |
| 11 | taaagctggtgaagcgattaacactgtacc aagta | chromosome | |
| 12 | atttcttcgttattagaaatataaaattgc gttgt | chromosome | |
| 13 | attttttatgattaagccatatggggttaa gcaag | pRH223/240 | |
| 14 | aaagactgggatccaaaaaaatatggtggt tttga | pRH243 | |
| 15 | attttcaaatgcataaaaactgtttctcaac gatat | chromosome | |
| 16 | ttttgtattggaatggcattttttgctatc aaggt | chromosome | |
| 17 | taaaacaggaccacttgtcatgtaagcttt aagtt | pRH223/240 | |
| 18 | tgtatcttgtggtttcatctgtgctaacttt ggcag | chromosome | |
| 19 | agaggatgcagaacgtgcaatcttagctgc aagac | chromosome | |
| 20 | ttcaaacgagaataattatggcgttggttta ggtat | chromosome | |
| 21 | gcgaatacactcattaaaacaattgcatcc tgatt | chromosome | |
| 22 | tcttatcttgataataagggtaactattgc cgatg | chromosome | |
| 23 | caaataaaggtgcgttattaataacagtgc caggc | chromosome | |
| 24 | acaacagtacgccaaccagccatcagtcac ctcct | pRH223 | |
| S6 | 1 | cagctaacaatgccatgattggtcggctga gGGaG | pRH223 |
| 2 | tggtaaatttacagaagatgctgaagatgc tGGtG | chromosome | |
| 3 | gagtcagctaggaggtgactgatggctggt tGGcG | pRH223 | |
| 4 | caaataagtctagacatattagctcgttatc aGGtG | chromosome | |
| 5 | acgaccttgttgcaacatagcgccccactc tGGtG | chromosome | |
| 6 | acatgttatgcatatcgtaagtgaagtcac aGGtG | chromosome | |
| 7 | atccgttctgcagaagagattgtttcttgc aGGcG | pRH223 | |
| 8 | agatgcttgttgtgttgtttgtgttgatgc cGGtG | chromosome | |
| 9 | atccgttctgcagaagagattgtttcttgc aGGcG | pRH223 | |
| 10 | agtttgggagtcaattatcggctttttaac tGGcG | chromosome | |
| 11 | aaaaagttatctcgtagacattacactggc tGGgG | pRH245 | |
Extended Data Table 3.
Mass spectrometry analysis of protein bands from the purified Cas9-Cas1-Cas2-Csn2 complex shown in Extended Data Fig. 4b.
| Accession | Protein | % Coverage |
Unique Peptides |
Total peak area |
|---|---|---|---|---|
| Cas9 | 83.26 | 170 | 9.7×1010 | |
| Cas1 | 91.35 | 40 | 1.9×1010 | |
| Cas2 | 84.07 | 13 | 1.9×109 | |
| Csn2 | 91.82 | 18 | 2.9×109 | |
| P77398 | Bifunctional polymyxin resistance protein ArnA (arnA) | 85.76 | 43 | 8.2×108 |
| P60422 | 50S ribosomal protein L2 (rplB) | 67.40 | 24 | 1.9×109 |
| P17169 | Glucosamine--fructose-6-phosphate aminotransferase (glmS) | 79.31 | 38 | 1.8×108 |
| P0AA43 | Ribosomal small subunit pseudouridine synthase A (rsuA) | 85.71 | 17 | 8.9×108 |
| P0A9K9 | FKBP-type peptidyl-prolyl cis-trans isomerase (slyD) | 68.88 | 7 | 3.7×109 |
| P0ACJ8 | Catabolite gene activator (crp) | 82.86 | 18 | 5.4×108 |
| P45395 | Arabinose 5-phosphate isomerase (kdsD) | 73.17 | 21 | 1.2×108 |
| P0A6F5 | 60 kDa chaperonin (groL) | 83.94 | 38 | 2.8×108 |
| P0A9A9 | Ferric uptake regulation protein (fur) | 78.38 | 8 | 1.2×109 |
| P08622 | Chaperone protein DnaJ (dnaJ) | 72.07 | 19 | 1.4×109 |
| P00393 | NADH dehydrogenase (ndh) | 59.22 | 16 | 3.6×108 |
Extended Data Table 4.
Oligonucleotides used in this study.
| Protein | % Coverage | Unique Peptides | Total peak area |
|---|---|---|---|
| Cas1 | 67.82 | 26 | 3.4×108 |
| Cas2 | 90.27 | 13 | 1.2×109 |
| Cas9 | 68.49 | 111 | 4.1×108 |
| Csn2 | 82.27 | 19 | 4.1×108 |
Acknowledgements
We thank members of the lab for critical discussion of the experiments and their results, Alexey Zaytsev for help with the deep sequencing data analysis and Andrew Sherlock for help with plasmid construction. R.H. is the recipient of a Howard Hughes International Student Research Fellowship. P.S. is supported by a Helmsley Postdoctoral Fellowship for Basic and Translational Research on Disorders of the Digestive System at The Rockefeller University. J.W.M is a Fellow of The Jane Coffin Childs Memorial Fund for Medical Research. D.B. is supported by a Harvey L. Karp Discovery Award and the Bettencourt Schuller Foundation. L.A.M is supported by the Rita Allen Scholars Program, an Irma T. Hirschl Award, a Sinsheimer Foundation Award and a NIH Director’s New Innovator Award (1DP2AI104556-01).
Footnotes
Author Contributions. RH, PS, DB and LAM conceived the study and designed experiments. RH and PS executed the experimental work with help from CW. JWM set up the experimental system to detect spacer acquisition in the absence of phage infection. GWG isolated and characterized phage ϕNM4γ4 and constructed the pGG32 plasmid. DB analyzed MiSeq data. LAM wrote the paper with the help of the rest of the authors.
The authors have no conflicting financial interests.
References
- 1.Barrangou R, Marraffini LA. CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol. Cell. 2014;54:234–244. doi: 10.1016/j.molcel.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 3.Deltcheva E, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makarova KS, et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garneau JE, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 6.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szczelkun MD, et al. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. U.S.A. 2014;111:9798–9803. doi: 10.1073/pnas.1402597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diez-Villasenor C, Guzman NM, Almendros C, Garcia-Martinez J, Mojica FJ. CRISPR-spacer integration reporter plasmids reveal distinct genuine acquisition specificities among CRISPR-Cas I-E variants of Escherichia coli. RNA Biol. 2013;10:792–802. doi: 10.4161/rna.24023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yosef I, Goren MG, Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datsenko KA, et al. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 2012;3:945. doi: 10.1038/ncomms1937. [DOI] [PubMed] [Google Scholar]
- 13.Nunez JK, et al. Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014;21:528–534. doi: 10.1038/nsmb.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anders C, Niewoehner O, Duerst A, Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–573. doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horinouchi S, Weisblum B. Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J. Bacteriol. 1982;150:815–825. doi: 10.1128/jb.150.2.815-825.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldberg GW, Jiang W, Bikard D, Marraffini LA. Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature. 2014;514:633–637. doi: 10.1038/nature13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kreiswirth BN, et al. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983;305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 19.Bae T, Baba T, Hiramatsu K, Schneewind O. Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol. Microbiol. 2006;62:1035–1047. doi: 10.1111/j.1365-2958.2006.05441.x. [DOI] [PubMed] [Google Scholar]
- 20.Sapranauskas R, et al. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011;39:9275–9282. doi: 10.1093/nar/gkr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li M, Wang R, Zhao D, Xiang H. Adaptation of the Haloarcula hispanica CRISPR-Cas system to a purified virus strictly requires a priming process. Nucleic Acids Res. 2014;42:2483–2492. doi: 10.1093/nar/gkt1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richter C, et al. Priming in the Type I-F CRISPR-Cas system triggers strand-independent spacer acquisition, bi-directionally from the primed protospacer. Nucleic Acids Res. 2014;42:8516–8526. doi: 10.1093/nar/gku527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horvath P, et al. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 2008;190:1401–1412. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fonfara I, et al. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42:2577–2590. doi: 10.1093/nar/gkt1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiedenheft B, et al. Structural basis for DNase activity of a conserved protein implicated in CRISPR-mediated genome defense. Structure. 2009;17:904–912. doi: 10.1016/j.str.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 27.Jinek M, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arslan Z, Hermanns V, Wurm R, Wagner R, Pul U. Detection and characterization of spacer integration intermediates in type I-E CRISPR-Cas system. Nucleic Acids Res. 2014;42:7884–7893. doi: 10.1093/nar/gku510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods references
- 29.Horinouchi S, Weisblum B. Nucleotide sequence and functional map of pE194, a plasmid that specifies inducible resistance to macrolide, lincosamide, and streptogramin type B antibodies. J. Bacteriol. 1982;150:804–814. doi: 10.1128/jb.150.2.804-814.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duplessis M, Moineau S. Identification of a genetic determinant responsible for host specificity in Streptococcus thermophilus bacteriophages. Mol. Microbiol. 2001;41:325–336. doi: 10.1046/j.1365-2958.2001.02521.x. [DOI] [PubMed] [Google Scholar]
- 31.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 32.Bikard D, et al. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014;32:1146–1150. doi: 10.1038/nbt.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore SD, Prevelige PE., Jr A P22 scaffold protein mutation increases the robustness of head assembly in the presence of excess portal protein. J. Virol. 2002;76:10245–10255. doi: 10.1128/JVI.76.20.10245-10255.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arslan Z, et al. Double-strand DNA end-binding and sliding of the toroidal CRISPR-associated protein Csn2. Nucleic Acids Res. 2013;41:6347–6359. doi: 10.1093/nar/gkt315. [DOI] [PMC free article] [PubMed] [Google Scholar]