

Abstract
Hemoglobin degradation during the asexual cycle of Plasmodium falciparum is an obligate process for parasite development and survival. It is established that hemoglobin is transported from the host erythrocyte to the parasite digestive vacuole (DV), but this biological process is not well characterized. Three-dimensional reconstructions made from serial thin-section electron micrographs of untreated, trophozoite-stage P. falciparum-infected erythrocytes (IRBC) or IRBC treated with different pharmacological agents provide new insight into the organization and regulation of the hemoglobin transport pathway. Hemoglobin internalization commences with the formation of cytostomes from localized, electron-dense collars at the interface of the parasite plasma and parasitophorous vacuolar membranes. The cytostomal collar does not function as a site of vesicle fission but rather serves to stabilize the maturing cytostome. We provide the first evidence that hemoglobin transport to the DV uses an actin-myosin motor system. Short-lived, hemoglobin-filled vesicles form from the distal end of the cytostomes through actin and dynamin-mediated processes. Results obtained with IRBC treated with N-ethylmaleimide (NEM) suggest that fusion of hemoglobin-containing vesicles with the DV may involve a soluble NEM-sensitive factor attachment protein receptor-dependent mechanism. In this report, we identify new key components of the hemoglobin transport pathway and provide a detailed characterization of its morphological organization and regulation.
INTRODUCTION
Malaria is a devastating disease that infects >300 million people each year, resulting in >1 million deaths, with the protozoan species Plasmodium falciparum being responsible for the majority of these deaths (1). The morbidity and mortality associated with the disease are largely the result of the parasite's asexual intraerythrocytic cycle (2). P. falciparum digests host cell hemoglobin to support parasite growth and asexual replication during the intraerythrocytic stage (3, 4). In order for the parasite to survive within the erythrocyte host, it degrades approximately 80% of the erythrocyte hemoglobin (5), with the majority of this digestion occurring during the trophozoite stage (18 to 32 h postinvasion) (3). The bulk of hemoglobin degradation occurs via a semiordered process by proteases contained within the parasite's digestive vacuole (DV) (6).
The transport of hemoglobin from the host erythrocyte cytosol to the parasite DV is a long-known but poorly understood biological process. The internalization of hemoglobin is thought to occur through an unusual structure, the cytostome. A cytostome is defined as a localized invagination of the parasite's outer membranes (the parasitophorous vacuolar membrane [PVM] and the parasite plasma membrane [PPM]), with a submembranous electron-dense collar associated with the neck of the cytostome (7, 8). It has traditionally been assumed that hemoglobin transport commences with the cytostome pinching off at the neck to form a double-membrane, hemoglobin-filled vesicle, similar to events in clathrin-mediated endocytosis (9). These vesicles are thought to be transported to the parasite's DV, where the outer membrane of the vesicle fuses with the DV plasma membrane, resulting in the delivery of a single-membrane, hemoglobin-filled vesicle into the DV. The hemoglobin and surrounding membrane are digested by resident DV proteases and lipases, respectively.
The main technique used to visualize this pathway is electron microscopy (EM), where a single thin section is typically 70 nm thick. Since an average intraerythrocytic parasite averages 5 μm in diameter, analysis based on single, thin-section images leaves open the possibility of misinterpretation of a complex morphology. However, recent technological advances provide the opportunity to better characterize parasite morphology by generating and analyzing three-dimensional (3D) models (10–13). While most groups agree that the cytostome is involved in hemoglobin trafficking, the precise morphological organization remains to be elucidated (7, 8, 10, 14).
In addition to the uncertainty surrounding the morphology of this pathway, there is also limited information on the mechanism(s) involved in the trafficking process. In eukaryotic cells, vesicles are trafficked by one of two mechanisms, an actin-myosin motor system or along microtubules with kinesin and dyneins (15–17). Microtubules are known to play important roles during schizogony and merozoite invasion in P. falciparum, but while tubulin is expressed at low levels in the trophozoite stage, organized microtubules do not form until the late schizont stage (18, 19). P. falciparum actin1 is expressed throughout the asexual stage, and its role in erythrocyte invasion has been investigated, but actin filaments have not been found associated with hemoglobin-filled vesicles. P. falciparum actin, however, is very distinct from its mammalian homologue in that it exists primarily in monomeric form and filaments that form are very short and unstable (20–23). Treatment of parasites with jasplakinolide (JAS), which stabilizes actin filaments and can promote filament nucleation (24), caused a redistribution of F-actin to the periphery of the parasite, morphological derangement of the cytostomal pathway and the inhibition of hemoglobin trafficking to the DV (14, 25, 26). Cytochalasin D (CytoD) prevents F-actin elongation by binding to the plus end of actin filaments, leading to the destabilization of actin filaments (27). With CytoD treatment, actin localization is not notably altered and hemoglobin delivery to the DV is not interrupted; however, double-membrane, hemoglobin-containing structures (HCs) appear to accumulate in the parasite cytosol (14, 26).
Actin has a role in maintaining and altering membrane structure in addition to the role it can play in vesicle trafficking (17). To determine which function(s) actin may have in hemoglobin trafficking, we investigated the role of actin's motor protein, myosin, in this process. P. falciparum encodes six myosins (P. falciparum MyoA, -B, -C, -D, -F, and -K) (28). Research to date has focused on myosin's roles in parasite invasion of host erythrocytes (28, 29). The role of P. falciparum MyoA during merozoite invasion of the erythrocyte has been well studied, but the role of the other myosins remains to be elucidated (29–32). To examine the possible involvement of an actin-myosin motor in the hemoglobin trafficking pathway, we used 2,3-butanedione monoxime (BDM) to inhibit myosin's ATPase activity (33, 34).
During the trafficking of hemoglobin to the DV, it is reasonable to propose that, at some point, a vesicle forms and fuses with the DV to allow efficient delivery of hemoglobin. Dynamin, a large GTPase, has a well-defined function as a pinchase in clathrin-mediated endocytosis (35, 36). The P. falciparum genome encodes two dynamin-like proteins, dynamin 1 (Dyn1) and Dyn2. In P. falciparum, Dyn1 is expressed during the trophozoite stage while Dyn2 is highly expressed during the schizont stage (37, 38). Treatment of trophozoite-stage parasites with Dynasore, a small-molecule inhibitor of dynamin's GTPase activity, inhibited parasite uptake of hemoglobin, suggesting that dynamin may be involved in hemoglobin trafficking (39). Following vesicle fission, vesicle fusion with a target membrane must take place for efficient vesicular transport to occur. In most cells, soluble N-ethylmaleimide (NEM)-sensitive factor (NSF) attachment protein receptors (SNAREs) play a major role in compartment recognition and vesicle fusion. The P. falciparum genome encodes several SNARE homologues (40). In addition, P. falciparum has a homolog of NSF, a cytoplasmic protein that interacts with SNAREs and is required for membrane fusion. We provide the first evidence that the fusion of a cytostome-derived vesicle with the DV may be a SNARE-mediated event.
In a previous report, we presented evidence for a new model of hemoglobin transport, wherein cytostomal tubes matured and extended to the DV to deliver hemoglobin (14). This model was developed after the examination of thousands of single sections of infected red blood cells (IRBC) and a limited number of serial sections. To further test our proposed model, we decided to compile an inventory of 3D reconstructions of IRBC made from electron micrographs of serial thin sections of untreated trophozoite-stage IRBC and IRBC treated with several pharmacological agents that modulate (cytoskeletal) proteins. This approach allows us to determine the basic morphology and functions of proteins that may be involved in this critical pathway.
MATERIALS AND METHODS
P. falciparum parasite culture.
P. falciparum strains FCR3 and 3D7 were cultured by maintaining a 4% hematocrit with human O+ erythrocytes and RPMI 1640 supplemented with 5% AlbuMAX (Invitrogen) as described previously (41). To obtain synchronized trophozoite-stage parasites, cultures were subjected to serial sorbitol treatments (42).
EM.
After treatment as specified, P. falciparum cultures were placed in a fixative solution containing 0.1 M phosphate buffer (pH 7.4), 3% glutaraldehyde (Electron Microscopy Sciences [EMS]), and 1% tannic acid (EMS) as described previously (14). Cells were rinsed three times with 0.1 M phosphate buffer and postfixed for 1 h in 2% osmium tetroxide (EMS) in 0.1 M phosphate buffer (pH 7.4) at room temperature. Following four washes with deionized water, the samples were stained with 1% uranyl acetate at room temperature. Samples were then pelleted at 45°C in 3% ultralow-temperature-gelling agarose (Sigma, St. Louis, MO). The resulting pellet was dehydrated sequentially in graded steps of acetone (25 to 100%) and subsequently infiltrated with Spurr's low-viscosity embedding mixture (EMS) and polymerized at 65°C overnight. The resulting blocks were thin sectioned (∼80 nm thick) with a Diatome diamond knife on a Leica Ultracut UCT plus microtome. Serial thin sections or single thin sections were placed onto grids. Images were viewed with a FEI Techni12 electron microscope equipped with an AMTxr 111 digital camera.
Immuno-EM.
Immuno-EM was performed by Wandy Beatty at Washington University, St. Louis, MO. Briefly, IRBC were placed in a phosphate buffer fixative containing 4% paraformaldehyde and 0.05% glutaraldehyde for 1 h on ice and then washed with piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) buffer. The sample was then embedded in 10% gelatin and infiltrated with 20% polyvinylpyrrolidone–2.3 M sucrose overnight at 4°C. The gelatin block was cut and mounted on a cryo pin, and 50- to 70-nm sections were cut at −60°C and placed on Formvar/carbon Ni grids. The grids were incubated with a primary antibody against Toxoplasma gondii actin1, which reacts with Plasmodium but not human actin and a goat-anti-rabbit secondary antibody conjugated to 12-nm colloidal gold.
Treatment of parasites with JAS, CytoD, BDM, or Dynasore.
To perturb actin function, parasites were treated with JAS (Invitrogen) or CytoD (Calbiochem) as previously described (14). In brief, trophozoite-stage parasites at ∼5% parasitemia were treated with either 7 μM JAS or 10 μM CytoD for 3 h. To inhibit myosin activity, a 0.5 M stock solution of BDM was diluted to the desired concentrations in the parasite culture for various periods of time. To examine the role of SNAREs in hemoglobin transport to the DV, 4 mM NEM was added to the parasite culture for 15 min of incubation; this was followed by treatment with 8 mM dithiothreitol to inhibit NEM. Parasites were then immediately placed in EM fixative. To examine the role of P. falciparum Dyn1 in the formation of hemoglobin-containing vesicles (HCv), parasites were treated with 200 μM Dynasore for up to 3 h.
SLO treatment.
To selectively permeabilize the erythrocyte plasma membrane, IRBC were treated with streptolysin O (SLO; Sigma, St. Louis, MO). A total of 2 × 108 IRBC were resuspended in 500 μl of RPMI medium and 6 hemolytic units of SLO. The suspension was incubated for 5 min at 37°C. Cells were then washed with RPMI to remove hemoglobin and then immediately placed in EM fixative for further examination.
3D reconstructions.
3D reconstructions of electron micrograph serial sections were accomplished with the Reconstruct program. The analysis of the surface areas and volumes of these structures was done with data generated from the models by the program. Permission to use, copy, and redistribute Reconstruct is granted without a fee under the terms of the GNU General Public License version 2 as published by the Free Software Foundation.
Construction of a Plasmodium expression plasmid containing Dyn1.
P. falciparum Dyn1 was amplified from a pLN plasmid (kindly provided by Daniel Goldberg, Washington University, St. Louis, MO) and placed in a pLN vector containing an N-terminal hemagglutinin (HA) tag and FKBP12, which functions as a degradation domain. The pLN plasmid containing the FKBP-HA-tagged P. falciparum Dyn1 construct and a plasmid (pINT) to aid in integration were then prepared for transfection as previously described (43).
Transfection and selection of stably transfected parasites.
Transfection of malaria parasites with the expression construct pLN-FKBP-HA-Dyn and integrase construct pINT was performed by modification of the previously described erythrocyte loading technique (44). Briefly, erythrocytes (200 μl) were washed once with cold CytoMix and resuspended in 400 μl of CytoMix. Erythrocytes were then preloaded with 200 μg of DNA (100 μg of each plasmid) by electroporation. Parasites of strain 3D7attB were then allowed to invade the DNA-loaded erythrocytes. Parasites containing the plasmids were then selected by treatment with 2.5 μg/ml blasticidin and 125 μg/ml G418 for 3 weeks.
RESULTS
Characterization of the morphological organization of the hemoglobin transport pathway in P. falciparum trophozoites by EM.
It is impossible to differentiate among tubes, cisternae, vesicles, etc., in a single, thin-section electron micrograph. Structures that appear to be double-membrane HCv in a single section are almost always found to be cytostomes, which are invaginations of the PVM and PPM with an associated electron-dense collar, when sequentially viewed at different depths.
An electron micrograph of a single thin section (∼70-nm thickness) of a trophozoite-stage IRBC is depicted in Fig. 1A. We chose to focus on trophozoites (i.e., 20 to 34 h postinvasion), since this is where the majority of hemoglobin is digested (45). 3D reconstructions of serial thin sections of trophozoite-stage IRBC were generated with Reconstruct to investigate the morphology of HCs in the IRBC. In these reconstructed models, we identified three types of HCs: cytostomes, phagotrophs, and double-membrane HCv (Fig. 1B to D). Phagotrophs are defined as HCs that are open to the RBC cytosol, lack cytostomal collars, and have an opening with a diameter of >200 nm (Fig. 1C) (13). The only HCs to appear in every IRBC were cytostomes, with an average of 2.5 cytostomes per IRBC (Fig. 1E). In a typical untreated IRBC, there are one to four cytostomes, with volumes ranging from 0.013 to 0.272 fl and all having a similar-diameter neck opening of ∼100 nm. Electron-dense collars with no membrane invagination at the PVM-PPM interface surface were occasionally observed in <5% of the IRBC models, with the majority of the collars associated with cytostomes. The third subset of hemoglobin-containing structures are HCv, which differ from the first two structures in that they are not invaginations of the PVM-PPM interface but rather discrete vesicles in the parasite cytosol (Fig. 1D). For generated 3D models of untreated IRBC, see Fig. S1 in the supplemental material.
FIG 1.

Electron microscopic characterization of trophozoite-stage P. falciparum reveals three distinct HCs in IRBC. (A) Representative electron micrograph of an untreated trophozoite. The nucleus (Nuc), digestive vacuole (DV), mitochondria (Mito), and cytostome (Cyto) are indicated. (B to D) Typical 3D reconstructions of untreated trophozoites showing the parasite surface (dark blue), nucleus (light blue), DV (yellow), mitochondria (orange), hemoglobin-containing structures (red), and cytostomal collars (green). (B) 3D model of a parasite containing two cytostomes with collars (black arrowheads) (C) 3D model of a parasite with one cytostome and one phagotroph (black arrowhead) that contains hemoglobin but lacks a collar. (D) 3D model of a parasite with two cytostomes and an HCv (black arrowhead). (E) Bar graph illustrating the average number of various hemoglobin structures per parasite. Parasites have, on average, 2.65 cytostomes, 0.34 phagotrophs, and 0.25 hemoglobin vesicles. n = 15. (F) Graph comparing the percentages of single sections containing hemoglobin bodies in untreated parasites and permeabilized parasites that have been treated with SLO. HCv contain approximately 7% of the hemoglobin within a trophozoite. n = 600 sections. ***, P < 0.005. Scale bars, 500 nm.
To further confirm the existence of HCv in the parasite cytosol, IRBC were treated with SLO, which permeabilizes the host cell plasma membrane, but not the PVM or PPM, which removes any extraparasitic hemoglobin (46). This method allows discrimination between true intracellular vesicles and cytostomes and phagotrophs that are open to the RBC cytosol without the use of labor-intensive serial sectioning, greatly increasing the sample number from a few dozen to a few hundred observable parasites. HCv were observed in SLO-treated IRBC; however, they represented a small subset (<10%) of HCs in the parasite (Fig. 1F).
Actin is a component of the cytostomal collar.
The composition of the cytostomal collar is unknown, although we previously showed actin associated with the collar in JAS-treated parasites (14). In other eukaryotic cells, actin often plays a vital role in endocytic traffic and membrane stability. Immuno-EM with an antibody that specifically recognizes parasite actin (47) revealed that actin localizes with the cytostomal collar (Fig. 2A to C) and also with the cytostomal body (Fig. 2C).
FIG 2.

Actin localizes to the cytostome neck in IRBC. Representative immunoelectron micrographs of P. falciparum trophozoites probed with anti-T. gondii actin antibody. (A and B) Micrographs showing actin labeled with 12-nm gold particles (arrowheads) localized to electron-dense collars at the cytostome neck (dashed box). (C) Actin (arrowheads) associated with a hemoglobin-containing compartment in the parasite cytosol (dashed box). Scale bars, 250 nm.
JAS treatment causes the formation of hemoglobin-containing tubes.
Given actin's role in mammalian endocytic trafficking events, it seemed reasonable that actin could be playing a role in the trafficking of hemoglobin to the DV. Treatment with JAS produced an increase in the number of HCs compared to that in untreated IRBC, in agreement with previous results (14). In single sections, the HCs appeared to accumulate at the parasite periphery (Fig. 3A). In previous studies, it was thought that this accumulation of HCs was the result of vesicles unable to traffic properly to the DV (14, 26). In 3D reconstructions, it was apparent that what appeared in single sections to be multiple HCv were, in fact, long, hemoglobin-filled tubes (Fig. 3B). The tubes were localized at the parasite periphery. SLO treatment revealed that these tubes were open to the erythrocyte cytosol (Fig. 3D). Unlike typical cytostomes, these tubes did not have cytostomal collars. These tubes are not present in untreated parasites, and they are not a typical phenotype of compromised or dead parasites. The HCs in untreated IRBC are more spherical than the HCs in JAS-treated IRBC (Fig. 3C). For a library of 3D models of IRBC treated with JAS, see Fig. S2 in the supplemental material.
FIG 3.

Effects of JAS treatment on hemoglobin trafficking in a trophozoite-stage IRBC. (A) Representative single-section electron micrograph of an IRBC treated with 7 μM JAS for 3 h. Note the appearance of multiple HCs (arrows) at the parasite periphery. Nuc, nucleus; Hgb, hemoglobin; Cyto, cytoplasm; Mito, mitochondria. (B) Representative 3D reconstruction of a JAS-treated IRBC. The hemoglobin-containing compartments, which appeared to be vesicles in a single-section electron micrograph, are actually part of a single large, hemoglobin-filled tube located at the periphery of the parasite. The tube lacks a cytostomal collar and is open to the erythrocyte cytosol (arrowhead). The parasite surface (dark blue), nucleus (light blue), DV (yellow), mitochondria (orange), hemoglobin-containing structures (red), and cytostomal collars (green) are shown. Scale bars, 500 nm. (C) Graph comparing the shapes of the HCs in untreated and JAS-treated IRBC. Untreated IRBC contain more spherical hemoglobin shapes, while the JAS-treated parasites have more oval HCs. (n = 13). (D) JAS treatment increases the percentage of IRBC with HCs approximately 2-fold. However, the number of HCv (as indicated by the percentage of single sections with hemoglobin after SLO treatment) remains the same with JAS treatment. n = 600. ***, P < 0.005; *, P < 0.05.
HCv accumulate in the parasite cytosol as a result of CytoD treatment.
While filament stabilization as a result of JAS treatment inhibits hemoglobin trafficking to the DV, treatment with CytoD, which depletes the filament population, does not (14, 26). Single-section electron micrographs reveal an increase in the number of HCs in IRBC treated with CytoD (Fig. 4A). Analysis of the 3D reconstructions made it is clear that HCv had accumulated in the parasite cytosol (Fig. 4B). CytoD treatment causes an increase in the HCs and HCv populations within the parasite cytosol (Fig. 4B and C). Parasites that did not show an increase in HCv displayed enlarged phagotrophs (see Fig. S3 in the supplemental material). Comparison of CytoD models to untreated IRBC models showed a 4-fold higher average number of HCv per IRBC (Fig. 4C). An apparent increase in the HCv number in CytoD-treated IRBC was also evident when a large population of IRBC was examined by single-section analysis after SLO treatment (Fig. 4D). Through extensive analysis of >600 single thin sections, we were able to capture the fusion of an HCv with the DV (Fig. 4E).
FIG 4.

CytoD treatment causes HCv accumulation in IRBC. (A) Representative single-section electron micrograph of an IRBC incubated with 10 μM CytoD for 3 h showing an increased number of HCv in the parasite cytosol. Nuc, nucleus; Hgb, hemoglobin; Mito, mitochondria; DV, digestive vacuole. (B) Representative 3D reconstruction of a CytoD-treated IRBC illustrating HCv accumulation. The parasite surface (dark blue), nucleus (light blue), DV (yellow), mitochondria (orange), hemoglobin-containing structures (red), and cytostomal collars (green) are shown. (C) Comparison of the average numbers of hemoglobin-containing structures in untreated and CytoD-treated IRBC. The number of vesicles per cell increases 3-fold after CytoD treatment. n = 10. (D) CytoD treatment causes a doubling of the number of single EM sections with hemoglobin-containing structures. The percentage of single sections containing HCv increases 4-fold (compare SLO and CytoD + SLO), although HCv are still a low percentage (<5%) of the hemoglobin-containing compartments in IRBC. n = 600. *, P < 0.005. (E) Single-section electron micrograph of an IRBC that was treated with 10 μM CytoD for 3 h and then permeabilized by SLO treatment. Note the presence of an HCv fusing with the DV (arrowhead). Scale bars, 500 nm.
Inhibition of parasite myosin prevents hemoglobin trafficking to the DV and results in parasite death.
In general, vesicles are trafficked by one of two mechanisms, a microtubule-mediated mechanism or the action of an actin-myosin motor system. The results obtained with JAS and CytoD treatment strongly suggest a role for parasite actin in the hemoglobin trafficking pathway. Since parasites do not have any organized microtubules until the schizont stage, we hypothesized that hemoglobin transport to the DV may be mediated by an actin-myosin motor. To test this, IRBC were treated with the myosin inhibitor BDM. BDM at 25 mM was shown to inhibit the gliding motility of T. gondii (48). Myosin is a known component of the Plasmodium invasion machinery (49), but a functional role during the erythrocytic stage has not been described. Synchronous IRBC were incubated with increasing concentrations of BDM starting at the ring stage (6 to 18 h postinvasion), and the levels of parasitemia were determined at the trophozoite (32 to 44 h postinvasion) and schizont (48 to 60 h postinvasion) stages (Fig. 5A). BDM at 2.5 mM was without effect, but 25 and 50 mM had a parasiticidal effect, reducing parasitemia 3-fold compared to that in untreated IRBC (Fig. 5A, TP1). Since BDM inhibits parasite invasion of erythrocytes, cultures were washed at 32 to 44 h postinvasion to remove BDM and to determine if inclusion of BDM in the culture up to the time of washing was toxic to the parasites. Parasitemia was reduced by 50% by 2.5 mM BDM, whereas parasitemia was eliminated by 25 or 50 mM BDM (Fig. 5A, TP2). In IRBC treated with 25 mM BDM for 30 min, long, hemoglobin-filled tubes lacking a cytostomal collar were apparent (Fig. 5B). In IRBC where no tubes were observed, HCv accumulated (see Fig. S4 in the supplemental material).
FIG 5.

The myosin inhibitor BDM inhibits hemoglobin transport to the DV and parasite development. (A) IRBC were treated for 26 h with increasing concentrations of BDM beginning at 6 to 18 h postinvasion, and the level of parasitemia was determined at 32 to 44 h (TP1) or 48 to 56 h (TP2) postinvasion. BDM at 25 or 50 mM is parasiticidal (TP1). BDM strongly inhibits growth at 2.5 mM, whereas it essentially eliminates parasitemia at 25 or 50 mM. (B) 3D model of a parasite that was treated with 25 mM BDM for 30 min. Note the appearance of a long, hemoglobin-filled tube that lacks a cytostomal collar. (C) JAS and BDM treatments cause similar increases in the surface area-to-volume ratio of the hemoglobin-containing tubes. (D) Representative single-section electron micrograph of an IRBC that was treated for 3 h with the protease inhibitor E64. The asterisk indicates a DV swollen with undigested hemoglobin. (E) Representative single-section electron micrograph of an IRBC treated for 4 h with the protease inhibitor E64 and the myosin inhibitor BDM. The asterisk indicates a DV that is devoid of hemoglobin, showing that BDM treatment prevents hemoglobin from trafficking to the DV. Scale bars, 500 nm. (F) Graph comparing the percentages of hemoglobin-filled DV in IRBC treated with E64 and those treated with BDM and E64. n = 100.
BDM, like JAS, increases the volume of hemoglobin within the parasite cytosol. IRBC treated for 3 h with the protease inhibitor E64 to inhibit hemoglobin digestion showed a buildup of undigested hemoglobin in the DV (Fig. 5D). In contrast, IRBC incubated with E64 and BDM possessed DVs devoid of hemoglobin, indicating that BDM inhibited the transport of hemoglobin to the DV (Fig. 5E). These results, in combination with the results obtained with the actin-perturbing drugs, suggest for the first time that cytostomes use an actin-myosin motor to transport hemoglobin to the DV.
The pinchase dynamin is involved in hemoglobin trafficking to the DV.
Our new results clearly demonstrate the presence of HCv in the parasite cytosol. We do not know what structure(s) the vesicles originate from, but in order for them to form, they must pinch off from a parent membrane. To examine the role the pinchase dynamin may be playing in the formation of HCv, IRBC were treated with the specific dynamin inhibitor Dynasore (50). We empirically determined that >100 μM Dynasore was needed to perturb the morphology of the hemoglobin transport pathway. The 3D reconstruction of parasites treated with 200 μM Dynasore for 30 min showed a drastically altered morphology with narrow, hemoglobin-containing tubes apparent (Fig. 6A and B; see Fig. S5 in the supplemental material). The tubes were thin and tortuous, unlike those seen in IRBC treated with JAS or BDM. The tubes had a diameter of approximately 100 nm and were connected to the parasite's surface membranes but did not have an associated electron-dense collar. While cytostomal collars were also observed at the PVM-PPM interface in the Dynasore-treated models, there were no membrane invaginations (i.e., cytostomes) extending from them. This is the first report of a drug treatment that produced IRBC devoid of classical cytostomes.
FIG 6.

P. falciparum Dyn1 functions in the hemoglobin transport pathway. (A) Representative single-section electron micrograph of IRBC treated with 200 μM Dynasore for 30 min. The arrows indicate unusual tortuous, thin tubes containing hemoglobin that localize to the parasite periphery. Scale bar, 500 nm. (B) Representative 3D reconstruction of IRBC incubated with 200 μM Dynasore for 30 min. Arrowheads point to thin, elongated, hemoglobin-containing tubes that result from Dynasore treatment. (C) Western blot assay showing the expression of FKBP-HA-tagged P. falciparum Dyn1 with or without 0.5 μM Shld1 for 48 h. Whole-cell parasite lysate was probed with an anti-HA antibody. The same amount of protein (50 μg) was loaded into each lane. The third lane is lysate from untransfected parasites. The arrow indicates a size of 100 kDa. (D to G) Immunoelectron micrographs showing intraparasitic localization of FKBP-HA-tagged P. falciparum Dyn1. The arrows indicate gold particle labeling on membranous structures. (E) Black arrow indicating HCs labeling. (F and G) Representative images showing that P. falciparum Dyn1 is absent from cytostomal collars (arrowheads). Scale bar, 250 nm.
Generation of parasites expressing 3HA-tagged wild-type P. falciparum Dyn1 to investigate the role of P. falciparum Dyn1 in the hemoglobin transport pathway.
To study the role of P. falciparum Dyn1 in hemoglobin trafficking, we generated parasites expressing a regulatable copy of endogenous P. falciparum Dyn1 with an HA tripeat (3HA) tag. To obtain rapid integration of DNA into parasites, we used an integrase system where parasites containing an integrase site (3D7attB) are cotransfected with a plasmid containing the recombinant protein (pLN) and a plasmid encoding a mycobacteriophage integrase (pINT) (51, 52). The calmodulin promoter in the pLN plasmid was replaced with a 1-kb region of P. falciparum DNA upstream of the predicted P. falciparum Dyn1 start site. Regulation of the protein was achieved by the addition of a degradation domain (ddFKBP). ddFKBP the promotes degradation of the attached protein; however, in the presence of ddFKBP ligand (i.e., Shld1), the degradation of the protein is inhibited, allowing the protein to accumulate within the parasite (53). In the absence of Shld1, the tagged protein is expected to be degraded. We observed, however, that in the absence of Shld1, there were detectable levels of ddFKBP-3HA-tagged wild-type P. falciparum Dyn1 (Fig. 6C). We also attempted to generate parasites expressing a dominant negative copy of the gene (P. falciparum DynK44A). It is likely that the presence of undegraded P. falciparum DynK44A, in the absence of Shld1, inhibited parasite growth, which prevented the generation of parasites expressing this form of the protein. These results support previous findings that dynamin may play an essential role in blood-stage parasite development (39).
Immuno-EM was performed to investigate P. falciparum Dyn1 localization. P. falciparum Dyn1 was absent from cytostomal collars (Fig. 6F and G). Consistent with a previous publication, the protein localized to the parasites peripheral membranes (Fig. 6D and E) (42). In one IRBC, P. falciparum Dyn1 was found associated with HCs in the parasite (Fig. 6E).
SNAREs play a role in HCv fusion with the DV.
We have strong evidence that vesicles are involved in hemoglobin trafficking. The final step in vesicle-mediated transport is fusion of the vesicle with a target membrane. This process usually involves SNARE-mediated fusion in eukaryotic cells. NEM inhibits the NSF protein, which is a crucial component of SNARE recycling. Treatment of parasites with NEM for 15 min led to the formation of hemoglobin-filled tubes (Fig. 7B), which were an indicator of inhibited hemoglobin trafficking in IRBC treated with JAS or BDM. This is the first evidence to suggest that SNAREs are involved in P. falciparum hemoglobin trafficking to the DV.
FIG 7.
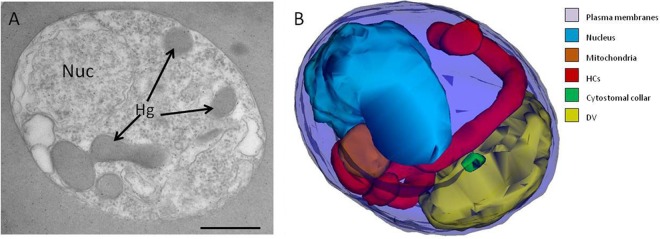
NEM disrupts the hemoglobin transport pathway. (A) Representative single-section electron micrograph depicting a trophozoite-stage IRBC from a culture incubated with 4 mM NEM for 15 min. Nuc, nucleus; Hg, hemoglobin. (B) Typical 3D reconstruction of a NEM-treated trophozoite revealing the presence of a long, hemoglobin-filled tube without an electron-dense collar (red). Note the presence of a collar that is not associated with a cytostome (green). Scale bar, 500 nm.
DISCUSSION
Until recently, the consensus was that hemoglobin trafficking to the DV in IRBC was achieved by an unusual, vesicle-mediated process. HCv were thought to form from the cytostome at the PPM-PVM interface, using the cytostomal collar to pinch off the HCv from the parasite surface membranes. These double-membrane HCv were then assumed to traffic to the DV by an undefined process and subsequently fuse with the DV, delivering a single-membrane HCv to the DV for digestion. This pathway was widely accepted because it had features in common with eukaryotic endocytic pathways, including clathrin-mediated endocytosis and autophagy. However, with the application of new technologies, evidence has emerged to provide a more accurate and detailed view of this key parasite pathway.
Previously, we described a model of hemoglobin trafficking in which no HCv were involved in hemoglobin transport (14). We proposed that cytostomes matured, elongated, and directly interacted with the DV. This conclusion was drawn after extensive analysis of single EM sections and a limited number of serial sections. While our data were consistent with this model, we were unable to construct a plausible model of the membrane complexity that would allow hemoglobin transfer from the elongated, double-membrane cytostomal tube to the DV while preserving the permeability properties of the DV. We decided to use whole-cell modeling techniques to determine if we might have failed to capture HCv because of the limited number of serial sections of IRBC in référence 14. Here we present new HCv evidence that requires us to refine our prior model.
Upon examination of the 3D reconstructions of untreated IRBC, certain features became apparent. Every IRBC contained at least one cytostome, with an average of 2.5 cytostomes per IRBC (Fig. 1E). HCv and phagotrophs were observed in 10% of the IRBC modeled (Fig. 1E). If vesicles were the main route of hemoglobin transport, we would expect them to be more abundant and possibly be present in every cell. Since the number of 3D models we could reasonably generate was relatively small, we decided to use another method to allow us to look for vesicles in a larger population of IRBC. Permeabilization of IRBC with SLO removed hemoglobin from structures that remain open to the RBC cytosol (cytostomes and phagotrophs) while leaving HCv within the parasite cytosol intact. Analysis of approximately 600 single-section electron micrographs of IRBC confirmed that there are HCv present within the parasite cytosol; however, most of the hemoglobin present in the parasite is contained in open structures, namely, either cytostomes or phagotrophs (Fig. 1F). Combining the SLO results with the information gathered from the models indicates that hemoglobin is likely trafficked to the DV by cytostomes; vesicles are a part of the pathway, and they appear to be transient. These conclusions are also supported by other reports (10, 13, 26, 45). Although here we conclude that the cytostome is the major mechanism of hemoglobin transport, we cannot exclude the possibility that phagotrophs also contribute. However, given their infrequency during the metabolically active trophozoite stage, ∼0.25 per cell (Fig. 1E), this seems unlikely and agrees with a model proposed by Tilley and coworkers (45).
Several investigations have provided evidence that actin plays a role in the hemoglobin trafficking pathway (10, 14, 26). Actin dynamics were shown to be important for delivery of HCv to the DV, with CytoD increasing the HCv pool without inhibiting parasite growth, whereas JAS disrupted the cytostomal morphology and inhibited hemoglobin transport to the DV. However, the mechanistic details remained to be elucidated. Regardless of the various models, no evidence was provided to explain how HCv were directionally trafficked to the DV. Since microtubules do not form in asexual parasites until the late schizont stage, a hitherto unidentified actin-myosin motor might be involved. IRBC treated with the myosin inhibitor BDM exhibited dose-dependent growth inhibition (Fig. 5A). These parasites were also unable to transport hemoglobin to the DV (Fig. 5D and E). BDM-treated IRBC displayed morphological alterations similar to those of JAS-treated IRBC, characterized by the appearance of elongated, hemoglobin-containing tubes (Fig. 5B and C). It has been suggested that JAS treatment may cause the formation of an actin meshwork that prevents hemoglobin from going beyond the periphery of the parasite (26). However, we suggest that hemoglobin-filled tubes form because of a disruption of an actin-myosin motor, not as a result of the formation of an actin meshwork, as the tubes are also seen upon myosin inhibition by BDM, where no peripheral actin meshwork is formed (see Fig. S6 in the supplemental material). These results strongly suggest that an actin-myosin motor is required for hemoglobin trafficking to the DV. This is the first evidence of a role for an actin-myosin motor in the hemoglobin transport process.
One of the striking features of the elongated tubes in BDM- and JAS-treated IRBC is the absence of a cytostomal collar, even though they remain connected to the parasite's outer membrane. The absence of a collar is not a defect in the ability to form collars, as normal collars and cytostomes are also observed in JAS- and BDM-treated cells (Fig. 3B and 5B). It was proposed that collars disassociate from mature cytostomes and that this disassembly may result in the formation of phagotrophs (8). Taking this into consideration, it is possible that the elongated tubes are a result of the disruption of the later stages of hemoglobin transport to the DV. It is likely that cytostomal maturation normally requires a functional actin-myosin motor system to form HCv, and when the motor is inhibited, elongated tubes form. An alternative explanation may be that cytostomes need to have Rabs or other accessory proteins associate with them in order to initiate the process of vesicle scission, in a process similar to endosomal maturation. When that association is prevented by inhibition of the actin-myosin motor, cytostomes are rendered nonproductive and eventually their collars disassemble, producing the elongated-tube phenotype observed in JAS- and BDM-treated IRBC. Another reason we believe the elongated, hemoglobin-containing tubes are derived from the cytostomes is that they have a restricted opening at the PVM-PPM interface that is very similar to a normal cytostome (∼100 nm).
Actin's role in the hemoglobin transport pathway is not limited to actin-myosin motor activity. We have conclusive evidence that actin is a component of the cytostomal collar (Fig. 2). Destabilization of actin filaments by CytoD treatment caused an increase in the number of HCv but did not abrogate hemoglobin transport to the DV or markedly affect parasite development (14, 26). This suggests that actin dynamics are important in the formation, directional trafficking, and/or fusion of HCv with the DV. We propose that actin filaments are required to maintain cytostome stability, and when the filament population is depleted by CytoD treatment, an increase in vesiculation of the cytostome is observed (Fig. 4). Since we did not quantitatively compare the hemoglobin content of the DV in CytoD-treated IRBC with that in untreated IRBC, we cannot rule out the possibility that the increased HCv population is due to the disruption of directional transport to the DV or the inhibition of fusion with the DV.
It has long been assumed that the purpose of the cytostomal collar was to aide in fission of the cytostome from the PVM-PPM interface to create an HCv. It was therefore reasonable to propose that the pinchase dynamin might play a role in cytostomal pinching from the collar. Using immuno-EM, we found that dynamin was conspicuously absent from the collar (Fig. 6F and G). This suggests that the collar does not function to aid in the fission of the cytostome from the membrane but may serve to stabilize the elongating cytostome. While dynamin was absent from the collar, it did appear to localize to membranous structures within the parasite, including HCs (Fig. 6E). Treatment of IRBC with Dynasore, an inhibitor of dynamin, resulted in the appearance of thin, tortuous tubes, confirming that dynamin plays a role in the hemoglobin transport pathway (Fig. 6B) (39). These tubes may be a result of dynamin constricting the cytostomes or a result of aberrant trafficking that results when the cytostomes are unable to pinch vesicles from their terminal end (54). We were unable to generate parasites expressing a regulatable dominant negative dynamin, further indicating a crucial role for dynamin in the asexual stages of development. We propose that dynamin functions to form vesicles from the cytostome, but these vesicles originate from the terminal end of the cytostome rather than from the collar at the PVM-PPM interface.
We have now established that vesicles are part of the hemoglobin trafficking pathway. In all other eukaryotic organisms, SNAREs are involved in vesicle fusion events. Here we used NEM treatment to investigate the involvement of SNAREs in HCv fusion with the DV. NEM altered the morphology of HCs within IRBC (Fig. 7). Because of the broad effects of NEM, it is difficult to make any conclusive statements about SNARE involvement in HCv-to-DV fusion; however, it is likely that novel SNARE complexes are involved in HCv-to-DV fusion (40).
Combining our results with other published data allows us to propose a refined model of hemoglobin trafficking in P. falciparum trophozoites (Fig. 8). Hemoglobin transport likely begins with the invagination of the parasite's outer membranes, the PVM-PPM interface, from a preexisting cytostomal collar. The details of what factors initiate cytostome collar formation and its protein composition are unknown. The collar then functions to stabilize the cytostome as it extends into the parasite cytosol in an actin-myosin-dependent manner. The cytostome expands until it reaches a certain size or location, receives a specific signal, or is bound by accessory proteins. At that point, a vesicle is generated from the distal end of the cytostome in a dynamin-dependent process. In addition to dynamin, dynamic actin could play a role in HCv generation or transport, as we saw an increase in these structures in CytoD-treated IRBC. The HCv then fuses its outer membrane with the DV, likely using parasite SNARE proteins, resulting in the delivery of a single-membrane HCv to the DV. Once inside the DV, the single-membrane HCv is digested in the acidic environment of the DV by resident lipases and proteases.
FIG 8.
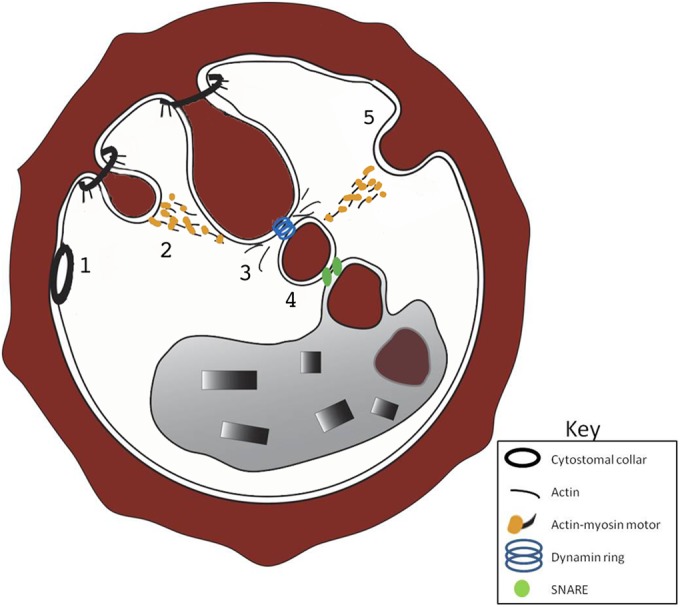
Refined model of hemoglobin trafficking in trophozoite-stage IRBC. Point 1 shows an electron-dense collar at the PVM-PPM interface. Point 2 shows how cytostomes form from a preexisting electron-dense collar and are trafficked to the DV by an actin-myosin motor. Point 3 shows the creation of an HCv from the distal end of the cytostome. Actin dynamics and dynamin pinching play key roles in HCv formation. Point 4 shows that fusion of the HCv with the DV likely uses a SNARE-dependent mechanism. Point 5 shows phagotrophs in a low percentage (<20%) of IRBC. If they contribute hemoglobin to the DV, they use an actin-myosin motor.
The role of phagotrophs in the hemoglobin transport pathway remains uncertain. We imagine four possibilities: (i) the phagotroph is a transient, nonproductive invagination of the PVM and PPM; (ii) the phagotroph is a participant in the hemoglobin transport pathway and produces HCv; (iii) the phagotroph may be a remnant of a cytostomal tube that retracts back to the PVM-PPM interface after an HCv pinches off; or (iv) the formation of a phagotroph may be the first step in the formation of a cytostome. If phagotrophs deliver hemoglobin to the DV and this contribution is important for parasite viability and development, it is actin-myosin mediated since both JAS and BDM treatments blocked hemoglobin transport to the DV and led to parasite death. We do not favor the notion that the phagotroph is a precursor to a cytostome, since we observed cytostome-free, electron-dense collars at the PVM-PPM interface in untreated IRBC. Our data do not allow us to estimate the relative contributions of the cytostomes and phagotrophs to the delivery of hemoglobin to the DV in trophozoites. We currently favor a model wherein hemoglobin transport to the DV in trophozoites is primarily through the cytostomes, with a possible secondary contribution from the phagotrophs. A major factor in reaching this conclusion is the low frequency of phagotrophs (∼0.25 per IRBC) compared to that of cytostomes (1 to 4 per IRBC) at the parasite developmental stage, where the majority of hemoglobin degradation occurs.
The working model proposed here presents the most complete picture of hemoglobin trafficking to date, illustrating not only the morphology of the transport pathway but also some of the key proteins and regulatory mechanisms involved. We recognize the possible limitations of inhibitor studies generating potential off-target effects. Future investigations will use a more direct approach to validate the model.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant AI090158 from the National Institute of Allergy and Infectious Diseases.
We thank Jessica Gutierrez for assistance with manuscript preparation.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00267-14.
REFERENCES
- 1.Waitumbi JN, Gerlach J, Afonina I, Anyona SB, Koros JN, Siangla J, Ankoudinova I, Singhal M, Watts K, Polhemus ME, Vermeulen NM, Mahoney W, Steele M, Domingo GJ. 2011. Malaria prevalence defined by microscopy, antigen detection, DNA amplification and total nucleic acid amplification in a malaria-endemic region during the peak malaria transmission season. Trop Med Int Health 16:786–793. doi: 10.1111/j.1365-3156.2011.02773.x. [DOI] [PubMed] [Google Scholar]
- 2.Bledsoe GH. 2005. Malaria primer for clinicians in the United States. South Med J 98:1197–1204. doi: 10.1097/01.smj.0000189904.50838.eb. [DOI] [PubMed] [Google Scholar]
- 3.Ginsburg H. 1990. Some reflections concerning host erythrocyte-malarial parasite interrelationships. Blood Cells 16:225–235. [PubMed] [Google Scholar]
- 4.Sherman IW. 1977. Amino acid metabolism and protein synthesis in malarial parasites. Bull World Health Organ 55:265–276. [PMC free article] [PubMed] [Google Scholar]
- 5.Francis SE, Sullivan DJ Jr, Goldberg DE. 1997. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol 51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- 6.Goldberg DE, Slater AF, Beavis R, Chait B, Cerami A, Henderson GB. 1991. Hemoglobin degradation in the human malaria pathogen Plasmodium falciparum: a catabolic pathway initiated by a specific aspartic protease. J Exp Med 173:961–969. doi: 10.1084/jem.173.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aikawa M, Hepler PK, Huff CG, Sprinz H. 1966. The feeding mechanism of avian malarial parasites. J Cell Biol 28:355–373. doi: 10.1083/jcb.28.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slomianny C. 1990. Three-dimensional reconstruction of the feeding process of the malaria parasite. Blood Cells 16:369–378. [PubMed] [Google Scholar]
- 9.Klemba M, Beatty W, Gluzman I, Goldberg DE. 2004. Trafficking of plasmepsin II to the food vacuole of the malaria parasite Plasmodium falciparum. J Cell Biol 164:47–56. doi: 10.1083/jcb200307147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elliott DA, McIntosh MT, Hosgood HD III, Chen S, Zhang G, Baevova P, Joiner KA. 2008. Four distinct pathways of hemoglobin uptake in the malaria parasite Plasmodium falciparum. Proc Natl Acad Sci U S A 105:2463–2468. doi: 10.1073/pnas.0711067105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanssen E, Goldie KN, Tilley L. 2010. Ultrastructure of the asexual blood stages of Plasmodium falciparum. Methods Cell Biol 96:93–116. doi: 10.1016/S0091-679X(10)96005-6. [DOI] [PubMed] [Google Scholar]
- 12.Hanssen E, McMillan PJ, Tilley L. 2010. Cellular architecture of Plasmodium falciparum-infected erythrocytes. Int J Parasitol 40:1127–1135. doi: 10.1016/j.ijpara.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Tilley L, Hanssen E. 2008. A 3D view of the host cell compartment in P. falciparum-infected erythrocytes. Transfus Clin Biol 15:72–81. doi: 10.1016/j.tracli.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 14.Lazarus MD, Schneider TG, Taraschi TF. 2008. A new model for hemoglobin ingestion and transport by the human malaria parasite Plasmodium falciparum. J Cell Sci 121:1937–1949. doi: 10.1242/jcs.023150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doherty GJ, McMahon HT. 2009. Mechanisms of endocytosis. Annu Rev Biochem 78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 16.Lanzetti L. 2007. Actin in membrane trafficking. Curr Opin Cell Biol 19:453–458. doi: 10.1016/j.ceb.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 17.Murray JW, Wolkoff AW. 2003. Roles of the cytoskeleton and motor proteins in endocytic sorting. Adv Drug Deliv Rev 55:1385–1403. doi: 10.1016/j.addr.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 18.Bannister LH, Mitchell GH. 1995. The role of the cytoskeleton in Plasmodium falciparum merozoite biology: an electron-microscopic view. Ann Trop Med Parasitol 89:105–111. [DOI] [PubMed] [Google Scholar]
- 19.Delves CJ, Alano P, Ridley RG, Goman M, Holloway SP, Hyde JE, Scaife JG. 1990. Expression of alpha and beta tubulin genes during the asexual and sexual blood stages of Plasmodium falciparum. Mol Biochem Parasitol 43:271–278. doi: 10.1016/0166-6851(90)90151-B. [DOI] [PubMed] [Google Scholar]
- 20.Sattler JM, Ganter M, Hliscs M, Matuschewski K, Schüler H. 2011. Actin regulation in the malaria parasite. Eur J Cell Biol 90:966–971. doi: 10.1016/j.ejcb.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 21.Schmitz S, Grainger M, Howell S, Calder LJ, Gaeb M, Pinder JC, Holder AA, Veigel C. 2005. Malaria parasite actin filaments are very short. J Mol Biol 349:113–125. doi: 10.1016/j.jmb.2005.03.056. [DOI] [PubMed] [Google Scholar]
- 22.Schmitz S, Schaap IA, Kleinjung J, Harder S, Grainger M, Calder L, Rosenthal PB, Holder AA, Veigel C. 2010. Malaria parasite actin polymerization and filament structure. J Biol Chem 285:36577–36585. doi: 10.1074/jbc.M110.142638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schüler H, Mueller AK, Matuschewski K. 2005. Unusual properties of Plasmodium falciparum actin: new insights into microfilament dynamics of apicomplexan parasites. FEBS Lett 579:655–660. doi: 10.1016/j.febslet.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 24.Holzinger A. 2001. Jasplakinolide. An actin-specific reagent that promotes actin polymerization. Methods Mol Biol 161:109–120. [DOI] [PubMed] [Google Scholar]
- 25.Mizuno Y, Makioka A, Kawazu S, Kano S, Kawai S, Akaki M, Aikawa M, Ohtomo H. 2002. Effect of jasplakinolide on the growth, invasion, and actin cytoskeleton of Plasmodium falciparum. Parasitol Res 88:844–848. doi: 10.1007/s00436-002-0666-8. [DOI] [PubMed] [Google Scholar]
- 26.Smythe WA, Joiner KA, Hoppe HC. 2008. Actin is required for endocytic trafficking in the malaria parasite Plasmodium falciparum. Cell Microbiol 10:452–464. doi: 10.1111/j.1462-5822.2007.01058.x. [DOI] [PubMed] [Google Scholar]
- 27.Sampath P, Pollard TD. 1991. Effects of cytochalasin, phalloidin, and pH on the elongation of actin filaments. Biochemistry 30:1973–1980. doi: 10.1021/bi00221a034. [DOI] [PubMed] [Google Scholar]
- 28.Foth BJ, Goedecke MC, Soldati D. 2006. New insights into myosin evolution and classification. Proc Natl Acad Sci U S A 103:3681–3686. doi: 10.1073/pnas.0506307103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones ML, Kitson EL, Rayner JC. 2006. Plasmodium falciparum erythrocyte invasion: a conserved myosin associated complex. Mol Biochem Parasitol 147:74–84. doi: 10.1016/j.molbiopara.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 30.Baum J, Gilberger TW, Frischknecht F, Meissner M. 2008. Host-cell invasion by malaria parasites: insights from Plasmodium and Toxoplasma. Trends Parasitol 24:557–563. doi: 10.1016/j.pt.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Heintzelman MB. 2003. Gliding motility: the molecules behind the motion. Curr Biol 13:R57–R59. doi: 10.1016/S0960-9822(02)01428-8. [DOI] [PubMed] [Google Scholar]
- 32.Pinder JC, Fowler RE, Dluzewski AR, Bannister LH, Lavin FM, Mitchell GH, Wilson RJ, Gratzer WB. 1998. Actomyosin motor in the merozoite of the malaria parasite, Plasmodium falciparum: implications for red cell invasion. J Cell Sci 111(Pt 13):1831–1839. [DOI] [PubMed] [Google Scholar]
- 33.Herrmann C, Wray J, Travers F, Barman T. 1992. Effect of 2,3-butanedione monoxime on myosin and myofibrillar ATPases. An example of an uncompetitive inhibitor. Biochemistry 31:12227–12232. [DOI] [PubMed] [Google Scholar]
- 34.Ostap EM. 2002. 2,3-Butanedione monoxime (BDM) as a myosin inhibitor. J Muscle Res Cell Motil 23:305–308. doi: 10.1023/A:1022047102064. [DOI] [PubMed] [Google Scholar]
- 35.Danino D, Hinshaw JE. 2001. Dynamin family of mechanoenzymes. Curr Opin Cell Biol 13:454–460. doi: 10.1016/S0955-0674(00)00236-2. [DOI] [PubMed] [Google Scholar]
- 36.Lenz M, Morlot S, Roux A. 2009. Mechanical requirements for membrane fission: common facts from various examples. FEBS Lett 583:3839–3846. doi: 10.1016/j.febslet.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 37.Charneau S, Bastos IM, Mouray E, Ribeiro BM, Santana JM, Grellier P, Florent I. 2007. Characterization of PfDYN2, a dynamin-like protein of Plasmodium falciparum expressed in schizonts. Microbes Infect 9:797–805. doi: 10.1016/j.micinf.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 38.Li H, Han Z, Lu Y, Lin Y, Zhang L, Wu Y, Wang H. 2004. Isolation and functional characterization of a dynamin-like gene from Plasmodium falciparum. Biochem Biophys Res Commun 320:664–671. doi: 10.1016/j.bbrc.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Zhou HC, Gao YH, Zhong X, Wang H. 2009. Dynamin like protein 1 participated in the hemoglobin uptake pathway of Plasmodium falciparum. Chin Med J (Engl) 122:1686–1691. [PubMed] [Google Scholar]
- 40.Ayong L, Pagnotti G, Tobon AB, Chakrabarti D. 2007. Identification of Plasmodium falciparum family of SNAREs. Mol Biochem Parasitol 152:113–122. doi: 10.1016/j.molbiopara.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 42.Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65:418–420. doi: 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- 43.Wu Y, Sifri CD, Lei HH, Su XZ, Wellems TE. 1995. Transfection of Plasmodium falciparum within human red blood cells. Proc Natl Acad Sci U S A 92:973–977. doi: 10.1073/pnas.92.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deitsch K, Driskill C, Wellems T. 2001. Transformation of malaria parasites by the spontaneous uptake and expression of DNA from human erythrocytes. Nucleic Acids Res 29:850–853. doi: 10.1093/nar/29.3.850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abu Bakar N, Klonis N, Hanssen E, Chan C, Tilley L. 2010. Digestive-vacuole genesis and endocytic processes in the early intraerythrocytic stages of Plasmodium falciparum. J Cell Sci 123:441–450. doi: 10.1242/jcs.061499. [DOI] [PubMed] [Google Scholar]
- 46.Ansorge I, Benting J, Bhakdi S, Lingelbach K. 1996. Protein sorting in Plasmodium falciparum-infected red blood cells permeabilized with the pore-forming protein streptolysin O. Biochem J 315(Pt 1):307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobrowolski JM, Niesman IR, Sibley LD. 1997. Actin in the parasite Toxoplasma gondii is encoded by a single copy gene, ACT1 and exists primarily in a globular form. Cell Motil Cytoskeleton 37:253–262. doi:. [DOI] [PubMed] [Google Scholar]
- 48.Dobrowolski JM, Carruthers VB, Sibley LD. 1997. Participation of myosin in gliding motility and host cell invasion by Toxoplasma gondii. Mol Microbiol 26:163–173. doi: 10.1046/j.1365-2958.1997.5671913.x. [DOI] [PubMed] [Google Scholar]
- 49.Frénal K, Polonais V, Marq JB, Stratmann R, Limenitakis J, Soldati-Favre D. 2010. Functional dissection of the apicomplexan glideosome molecular architecture. Cell Host Microbe 8:343–357. doi: 10.1016/j.chom.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 50.Kirchhausen T, Macia E, Pelish HE. 2008. Use of dynasore, the small molecule inhibitor of dynamin, in the regulation of endocytosis. Methods Enzymol 438:77–93. doi: 10.1016/S0076-6879(07)38006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crabb BS, Gilson PR. 2007. A new system for rapid plasmid integration in Plasmodium parasites. Trends Microbiol 15:3–6. doi: 10.1016/j.tim.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 52.Nkrumah LJ, Muhle RA, Moura PA, Ghosh P, Hatfull GF, Jacobs WR Jr, Fidock DA. 2006. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat Methods 3:615–621. doi: 10.1038/nmeth904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herm-Götz A, Agop-Nersesian C, Munter S, Grimley JS, Wandless TJ, Frischknecht F, Meissner M. 2007. Rapid control of protein level in the apicomplexan Toxoplasma gondii. Nat Methods 4:1003–1005. doi: 10.1038/nmeth1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takei K, McPherson PS, Schmid SL, De Camilli P. 1995. Tubular membrane invaginations coated by dynamin rings are induced by GTP-gamma S in nerve terminals. Nature 374:186–190. doi: 10.1038/374186a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.